

Abstract
It is well known that genotyping errors lead to loss of power in gene-mapping studies and underestimation of the strength of correlations between trait- and marker-locus genotypes. In two-point linkage analysis, these errors can be absorbed in an inflated recombination-fraction estimate, leaving the test statistic quite robust. In multipoint analysis, however, genotyping errors can easily result in false exclusion of the true location of a disease-predisposing gene. In a companion article, we described a “complex-valued” extension of the recombination fraction to accommodate errors in the assignment of trait-locus genotypes, leading to a multipoint LOD score with the same robustness to errors in trait-locus genotypes that is seen with the conventional two-point LOD score. Here, a further extension of this model to “hypercomplex-valued” recombination fractions (hereafter referred to as “hypercomplex recombination fractions”) is presented, to handle random and systematic sources of marker-locus genotyping errors. This leads to a multipoint method (either “model-based” or “model-free”) with the same robustness to marker-locus genotyping errors that is seen with conventional two-point analysis but with the advantage that multiple marker loci can be used jointly to increase meiotic informativeness. The cost of this increased robustness is a decrease in fine-scale resolution of the estimated map location of the trait locus, in comparison with traditional multipoint analysis. This probability model further leads to algorithms for the estimation of the lower bounds for the error rates for genomewide and locus-specific genotyping, based on the null-hypothesis distribution of the LOD-score statistic in the presence of such errors. It is argued that those genome scans in which the LOD score is 0 for >50% of the genome are likely to be characterized by high rates of genotyping errors in general.
Introduction
Marker-locus genotyping errors occur in every gene-mapping project in every laboratory—sometimes with an alarmingly high frequency (Lathrop et al. 1983; Lincoln and Lander 1992; Brzustowicz et al. 1993)—and they are difficult to detect, unless they lead to Mendelian inconsistencies in the data (Ehm et al. 1996). It is well known that such errors can seriously deflate the power and can lead to inflated recombination-fraction estimates in two-point linkage analysis (Smith 1937; Terwilliger et al. 1990; Buetow 1991). In linkage-disequilibrium analysis, such errors may pose an even bigger problem, since a single genotyping error can then destroy evidence of many nonrecombinant meioses in the past (see Terwilliger et al. 1997; de la Chapelle and Wright 1998; Göring et al. 1997; Terwilliger, in press). In multipoint analysis, their effects can be further magnified as the marker-locus density increases (Shields et al. 1991), leading to increased potential for false exclusion of the true disease locus. Multipoint analysis can likewise become less robust as the number of loci analyzed jointly increases when marker-locus parameters—such as allele frequencies, linkage disequilibrium, locus order, and interlocus genetic distances—are misspecified (Ott 1992; Daw et al. 1998; Göring and Terwilliger 2000b). As more and more marker loci are used, it is likely that more—rather than fewer—genotyping errors will occur, especially since the techniques used for automation of such genotyping are new and are not yet well tested. In addition, as marker loci become individually less polymorphic (e.g., single-nucleotide polymorphisms) and as the individual pedigrees become smaller and smaller (e.g., sib pairs or even singletons), genotyping errors become more difficult to eliminate through detection of Mendelian inconsistencies (Holmans and Craddock 1997).
Since marker-locus genotyping will never be completely error free, we propose a method for the computation of pedigree likelihoods that allows for marker-locus genotyping errors explicitly in the probability model. As an extension of the model in the companion article (Göring and Terwilliger 2000a), additional “imaginary” components will be added to the recombination fraction, ultimately leading to definition of recombination fractions (with four components) in the hypercomplex number system. Furthermore, we demonstrate how this model allows for the estimation of rates of genotyping errors in a genomewide and locus-specific sense. Throughout this article, as in the companion articles (Göring and Terwilliger 2000a, 2000b, 2000c), our use of the terms “frequency” and “probability” matches that of Walley (1991) and Jaynes (1996).
Probability Model for Marker-Locus Genotyping Errors under Misclassification Symmetry
Let us assume for now that the two types of misclassification of recombination status that result from marker-locus genotyping errors—misclassification of a true recombinant as a nonrecombinant and misclassification of a true nonrecombinant as a recombinant—have the same probability, denoted as γ=P(Nobs|Rtrue)=P(Robs|Ntrue), which is analogous to the parameter ε introduced in the companion article (Göring and Terwilliger 2000a). If one assumes, for the moment, that there is an absence of errors in the disease-locus genotypes, then the probability model shown in figure 1 obtains. The expected frequency of an observed recombinant can be seen to be 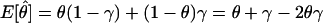 . When γ>0 and θ<0.5, the estimate of the recombination fraction is thus biased upward. If one also allows for misclassification of the recombination status as a result of errors in the trait-locus genotype, then the definition of the recombination fraction can be expanded to Θ=θ+εi+γj, represented as a vector in the hypercomplex number system H1. A graphical representation of Θ is given in figure 2. The real-valued frequency of an apparent recombination event is equal to the length of the vector Θ, which is defined—according to the theta-summing (“ts”) mode (see the companion article by Göring and Terwilliger [2000a])—as
. When γ>0 and θ<0.5, the estimate of the recombination fraction is thus biased upward. If one also allows for misclassification of the recombination status as a result of errors in the trait-locus genotype, then the definition of the recombination fraction can be expanded to Θ=θ+εi+γj, represented as a vector in the hypercomplex number system H1. A graphical representation of Θ is given in figure 2. The real-valued frequency of an apparent recombination event is equal to the length of the vector Θ, which is defined—according to the theta-summing (“ts”) mode (see the companion article by Göring and Terwilliger [2000a])—as 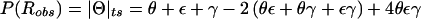 , which is analogous to the formula for adding three recombination fractions (θ, ε, and γ) under the assumption of no interference (Haldane 1919).
, which is analogous to the formula for adding three recombination fractions (θ, ε, and γ) under the assumption of no interference (Haldane 1919).
Figure 1.

Probability model for misclassification of recombination status resulting from marker-locus genotyping errors under the assumption of misclassification symmetry. The parental marker-locus genotypes and the disease-locus genotypes in both parent and child are assumed to be error free. P(Robs)=θ(1-γ)+(1-θ)γ=θ+γ-2θγ>θ, when θ<0.5 and γ>0.
Figure 2.

Hypercomplex recombination fraction between a disease locus and a diallelic marker locus. The recombination fraction is modeled in the hypercomplex number system, with a “real” component for the true probability of recombination (θ) between the loci and with two “imaginary” components for misclassification of recombination status resulting from genotyping errors at the disease locus (εi) and the marker locus (γj). All three components are orthogonal. The observed frequency of recombination is given by  , which is equal to the formula for adding three recombination fractions (θ, ε, and γ) under the assumption of no interference.
, which is equal to the formula for adding three recombination fractions (θ, ε, and γ) under the assumption of no interference.
The vector in hypercomplex map-distance space corresponding to Θ would be X(Θ)=x(θ)+x(ε)i+x(γ)j, with 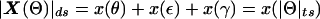 . Here, x( ) represents a mapping function such as the Haldane map function (Haldane 1919), and “ds” denotes “distance summing” (for details, see the companion article by Göring and Terwilliger 2000a). In what is often referred to as a “taxicab geometry” (Krause 1975), the ds metric in our hypercomplex three-space is defined as
. Here, x( ) represents a mapping function such as the Haldane map function (Haldane 1919), and “ds” denotes “distance summing” (for details, see the companion article by Göring and Terwilliger 2000a). In what is often referred to as a “taxicab geometry” (Krause 1975), the ds metric in our hypercomplex three-space is defined as 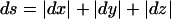 (i.e., the distance between two points is the sum of the lengths of the three orthogonal vectors rather than the Euclidean distance (for an explanation of taxicab geometry, see the companion article by Göring and Terwilliger [2000a]). The set of all points,
(i.e., the distance between two points is the sum of the lengths of the three orthogonal vectors rather than the Euclidean distance (for an explanation of taxicab geometry, see the companion article by Göring and Terwilliger [2000a]). The set of all points, 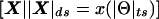 , equidistant from a marker locus, is represented by a taxicab sphere, which looks like the surface of a cube in Euclidean geometry. Since both ε⩾0 and γ⩾0, the set is restricted to be the surface of one quadrant of a taxicab sphere, as is shown in figure 3.
, equidistant from a marker locus, is represented by a taxicab sphere, which looks like the surface of a cube in Euclidean geometry. Since both ε⩾0 and γ⩾0, the set is restricted to be the surface of one quadrant of a taxicab sphere, as is shown in figure 3.
Figure 3.

Presentation of hypercomplex recombination fraction in hypercomplex map-distance space. The set of all points,  , equidistant from a marker locus, is represented by a taxicab sphere (with the marker locus at its center). In Euclidean geometry, this sphere looks like the surface of a cube. Since ε⩾0 and γ⩾0, the set is restricted to one quadrant of the surface of a taxicab sphere.
, equidistant from a marker locus, is represented by a taxicab sphere (with the marker locus at its center). In Euclidean geometry, this sphere looks like the surface of a cube. Since ε⩾0 and γ⩾0, the set is restricted to one quadrant of the surface of a taxicab sphere.
Multiple Two-Point and Multipoint Linkage Analysis with Hypercomplex Recombination Fractions
In joint analysis of multiple marker loci versus a trait locus, each marker locus, m, will have a unique and mutually orthogonal misclassification parameter, γm. The trait-locus misclassification parameter, ε, is fixed to be identical for all marker loci, since ε is a parameter of the trait locus alone. Because it is difficult to visualize the error vectors in multidimensional space, for the purposes of the following discussion, all figures are drawn so that the γm component of the hypercomplex recombination fraction for each marker locus is shown as a vector pointing downward from the real line—that is, the chromosome—whereas the ε component for the trait locus is shown as a vector pointing upward from the real line. In actuality, however, all such vectors are mutually orthogonal, and apparent recombination fractions between adjacent marker loci can be much more inflated than these simplified figures might imply. Furthermore, the hypercomplex recombination-fraction vector, Θ, will be drawn as a diagonal line connecting the ends of vectors εi and γj, to emphasize that Θ refers to the correlation between inferred trait-locus genotypes and observed marker-locus genotypes, even though its magnitude is defined in a non-Euclidean, taxicab metric space.
Multiple two-point analysis (Morton 1988; Morton and Andrews 1989; Shields et al. 1991) of a disease locus (D) against a set of marker loci (M1, M2, and M3) can then be visualized as shown in figure 4A. In the illustrated example,  , even though the true genetic distance between D and M2 (θD2) is smaller than that between D and M3 (θD3). This apparent discrepancy is the result of an excess of errors in the observed genotypes of M2, relative to M3, in this example. When one allows for marker- and disease-locus errors in this manner, the only restriction imposed on the recombination-fraction estimates in multiple two-point analysis is that the magnitude of ΘDm must be at least as large as the magnitude of θDm+εi, since the γm component must be non-negative because it is a probability. This constraint is more relaxed than that which is imposed when one does not allow for marker-locus genotyping errors.
, even though the true genetic distance between D and M2 (θD2) is smaller than that between D and M3 (θD3). This apparent discrepancy is the result of an excess of errors in the observed genotypes of M2, relative to M3, in this example. When one allows for marker- and disease-locus errors in this manner, the only restriction imposed on the recombination-fraction estimates in multiple two-point analysis is that the magnitude of ΘDm must be at least as large as the magnitude of θDm+εi, since the γm component must be non-negative because it is a probability. This constraint is more relaxed than that which is imposed when one does not allow for marker-locus genotyping errors.
Figure 4.

Multiple two-point and multipoint linkage analysis with hypercomplex recombination fractions. Multiple two-point linkage analysis (A) and multipoint linkage analysis (B) are shown. Each locus has a unique error parameter. Although these parameters are mutually orthogonal in reality, they are drawn so that they are pointing above or below the real line—that is, the chromosome—for simplicity. Intermarker recombination fractions, which would also be overestimated, are not indicated, to avoid confusion. In this example, 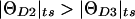 (i.e., more recombinants are observed between D and M2, even though M2 is actually closer to D than M3, as a result of the error vector being larger for M2 than for M3).
(i.e., more recombinants are observed between D and M2, even though M2 is actually closer to D than M3, as a result of the error vector being larger for M2 than for M3).
The frequency of the apparent recombination between observed marker-locus genotypes at two marker loci (e.g., M1 and M2) can be computed in an analogous manner, with 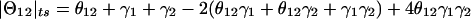 and
and 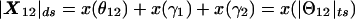 . Therefore, conditional on a well-known genetic marker-locus map, the observed marker-marker recombination frequencies can contribute information about the marker-locus-specific error rates. (For simplicity of presentation, the marker-marker recombination probabilities were omitted in fig. 4.)
. Therefore, conditional on a well-known genetic marker-locus map, the observed marker-marker recombination frequencies can contribute information about the marker-locus-specific error rates. (For simplicity of presentation, the marker-marker recombination probabilities were omitted in fig. 4.)
Multipoint analysis (Lathrop et al. 1984; Lander and Green 1987) of a disease locus against several marker loci can be performed with the use of the probability model shown in figure 4B. An example of how one would compute the likelihood in the presence of these misclassification parameters is given in figure 5. For simplicity, we only consider errors at D and M2 (errors in the other loci can be accommodated by means of direct analogy). In the meiosis used as an example, an apparent recombination was observed in interval M1-D, no recombination was observed in interval D-M2, and recombination was observed between M2 and M3. Since the genotypes at both D and M2 are potentially erroneous, there are four (=22) possible explanations for the observed meiotic outcome, in terms of recombination and misclassification. These four possibilities and their likelihoods are indicated in figure 5. Their sum would give the overall likelihood for this observed meiosis, allowing for errors at both D and M2. This procedure could be extended to allow for errors at all loci jointly (for four loci, as shown here, there would be 24=16 possible explanations for an observed meiotic outcome). All of the multilocus recombination information could then be used jointly for estimation of the locus-specific error rates.
Figure 5.

Example of multipoint likelihood computation with misclassification vectors. For simplicity, genotype errors resulting in misclassification of the meiotic-recombination status are allowed to occur only at the disease locus D and the marker locus M2; however, error components could be included for the other marker loci, by means of direct analogy. There are 22=4 possibilities (in terms of true recombination and misclassification) for the joint underlying true recombination status that could explain the observed meiotic outcome. The sum of the likelihoods of all four possibilities gives the likelihood of this meiotic observation.
In practice, the error rates at the individual marker and trait loci are typically not of primary interest, since the main goal is to estimate the chromosomal location of the disease-predisposing gene. The magnitudes of the marker-locus-specific values of γm are useful, however, for identification of marker loci that either have high rates of genotyping errors (those marker loci could subsequently be either rechecked or censored from the analysis) or are mapped to the wrong chromosomal location (since such errors in the marker-locus map are highly confounded with these error vectors; see Göring and Terwilliger [2000b]).
Asymmetry in Misclassification of Recombination Status
It was assumed above that the two types of misclassification of recombination status have equal probability—that is, γ=P(Nobs|Rtrue)=P(Robs|Ntrue). In a companion article (Göring and Terwilliger 2000a), a similar assumption was made for meiotic misclassification resulting from errors at the trait locus. This assumption is reasonable, as long as either the locus with the errors is diallelic or the parental genotypes are assumed to be known with accuracy, in the absence of any systematic types of bias (see Ott 1977). For diallelic loci, only a single parental genotype (D/+ or 1/2) is informative for linkage, which leads to the symmetry that any possible error in the genotyping of offspring would convert a recombinant to a nonrecombinant—and vice versa—with equal probability. (Genotyping errors are independent of the true recombination status, since such errors occur at each locus independently.) When an error occurs in the genotyping of a parent, this either results in censoring of the truly informative meioses from the analysis or leads to inclusion of truly uninformative meioses, which are expected to appear as recombinants 50% of the time—irrespective of whether recombination occurred in reality. Neither effect would lead to asymmetry in the two types of misclassification. If the parental genotype is known with accuracy, then the same argument also holds for a marker locus with more than two alleles, since then only the alleles (two at most) observed in the typed parent could have been transmitted to the offspring, with all other alleles leading to a Mendelian inconsistency. When parents are not genotyped, however, the symmetry between the two types of misclassification no longer holds for a multiallelic marker locus. In that situation, errors in the genotyping of offspring would influence the likelihood of each possible parental genotype, such that a larger proportion of all possible genotyping errors would lead to misclassification of a true nonrecombinant rather than of a true recombinant.
Let us allow for separate probabilities for the two types of misclassification in recombination status, by defining η=P(Robs|Ntrue) and ν=P(Nobs|Rtrue). The frequency of an observed recombination event (when it is assumed, for the moment, that the disease-locus genotype is known with certainty) would then be 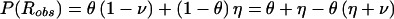 . Reparameterization, by generalization of the previously introduced misclassification parameter to γ=(η+ν)/2 and by definition of τ=(η-ν)/2, leads to
. Reparameterization, by generalization of the previously introduced misclassification parameter to γ=(η+ν)/2 and by definition of τ=(η-ν)/2, leads to 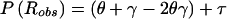 . When misclassification symmetry holds (i.e., η=ν), then γ=η=ν and τ=0, and this equation reduces to the relationship obtained above—namely, that
. When misclassification symmetry holds (i.e., η=ν), then γ=η=ν and τ=0, and this equation reduces to the relationship obtained above—namely, that 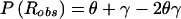 .
.
Figure 6 summarizes the probability model that allows for errors in both the assigned disease-locus genotypes and the observed marker-locus genotypes. τ can be modeled as an additional error component in a four-dimensional hypercomplex recombination fraction, denoted as Θ=θ+εi+γj+τk. As shown in figure 7, the two marker-locus vectors, γj and τk, arise from the same marker locus, whereas the disease-locus error vector, εi, arises orthogonally from the disease locus. The previously defined ts and ds modes are not directly applicable, and we need to define a new metric (referred to here as the “τs” mode) for the frequency of apparent recombination between assigned genotypes at the disease loci and the marker loci. The magnitude of the observed recombination fraction would be 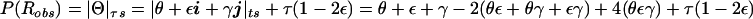 (see fig. 6). If either τ=0 or ε=0.5, then the τs mode is exactly identical to the familiar ts mode and, thus, represents a generalization of the ts mode to allow for asymmetry in the two types of misclassification in recombination status.
(see fig. 6). If either τ=0 or ε=0.5, then the τs mode is exactly identical to the familiar ts mode and, thus, represents a generalization of the ts mode to allow for asymmetry in the two types of misclassification in recombination status.
Figure 6.

Probability model allowing for misclassification of the recombination status as a result of errors at both the disease locus and a multiallelic marker locus. The real-valued probability of recombination is given by 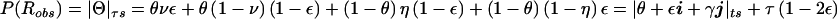 , which is obtained by calculating the sum of the probabilities of the relevant paths and by substituting
, which is obtained by calculating the sum of the probabilities of the relevant paths and by substituting  and
and 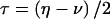 .
.
Figure 7.

Hypercomplex recombination fraction between a disease locus and a multiallelic marker locus. The probabilities of the two types of misclassification in recombination status (mistaking either a true nonrecombinant for a recombinant or a true recombinant for a nonrecombinant) are sometimes unequal, for reasons outlined in the text. Two separate imaginary components (γ and τ) are therefore added to the hypercomplex recombination fraction, with both imaginary vectors originating from the position of the marker locus. The recombination fraction is then defined as Θ=θ+εi+γj+τk. The real-valued probability of recombination is given by 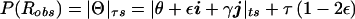 (see fig. 6 for derivation).
(see fig. 6 for derivation).
For the case of random errors in the genotyping of multiallelic marker loci, τ>0 since η>ν, which can lead to recombination-fraction estimates >0.5 under the null hypothesis of no linkage, since 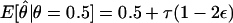 . As shown below, this can cause problems with the null-hypothesis distribution of the LOD score. However, there is also the possibility of systematic genotyping errors—that is, τ<0 when ν>η—which may have the opposite effect. An example where this might apply would be if, for some technical reason, there was a propensity to misread heterozygotes as homozygotes (see Lindqvist et al. [1996]), which may occur with diallelic as well as with multiallelic marker loci. This apparent increase in marker-locus homozygosity among sets of affected sibs might cause the illusion that such affected sib pairs received these marker-locus alleles identical by descent (IBD) from their parents. When ascertainment is primarily of affected relatives, this may give rise to false-positive findings of linkage. If there were random ascertainment with respect to phenotype or if equal numbers of affected and unaffected siblings were included in the analysis, then such systematic biases would be less likely. Affecteds-only linkage analysis will therefore tend to magnify the effects of such systematic errors. Another potential source of systematic bias in favor of nonrecombinants arises when genotyping is done with knowledge of the phenotypes, such that investigators might resolve ambiguous genotypes in such a way as to minimize recombinants in the data set. The proposed model intrinsically allows for this possibility as well, since it allows τ to take negative values. Other effects of such ascertainment biases, which are related to errors in parameters of the marker loci and their map, are discussed elsewhere (Göring and Terwilliger 2000b; Terwilliger and Göring, in press).
. As shown below, this can cause problems with the null-hypothesis distribution of the LOD score. However, there is also the possibility of systematic genotyping errors—that is, τ<0 when ν>η—which may have the opposite effect. An example where this might apply would be if, for some technical reason, there was a propensity to misread heterozygotes as homozygotes (see Lindqvist et al. [1996]), which may occur with diallelic as well as with multiallelic marker loci. This apparent increase in marker-locus homozygosity among sets of affected sibs might cause the illusion that such affected sib pairs received these marker-locus alleles identical by descent (IBD) from their parents. When ascertainment is primarily of affected relatives, this may give rise to false-positive findings of linkage. If there were random ascertainment with respect to phenotype or if equal numbers of affected and unaffected siblings were included in the analysis, then such systematic biases would be less likely. Affecteds-only linkage analysis will therefore tend to magnify the effects of such systematic errors. Another potential source of systematic bias in favor of nonrecombinants arises when genotyping is done with knowledge of the phenotypes, such that investigators might resolve ambiguous genotypes in such a way as to minimize recombinants in the data set. The proposed model intrinsically allows for this possibility as well, since it allows τ to take negative values. Other effects of such ascertainment biases, which are related to errors in parameters of the marker loci and their map, are discussed elsewhere (Göring and Terwilliger 2000b; Terwilliger and Göring, in press).
Effects of Marker-Locus Genotyping Errors on Linkage Analysis
In this section, we focus on the effects of marker-locus genotyping errors on linkage analysis. It has been shown that linkage analysis is impacted not only when it is done under the alternative hypothesis of linkage (H1), which is well known (Smith 1937; Terwilliger et al. 1990; Buetow 1991; Shields et al. 1991; Lincoln and Lander 1992; Goldstein et al. 1997), but also under the null hypothesis of no linkage (H0) between the disease locus and the marker loci. Under H0, if τ>0 (i.e., there are random genotyping errors), then the LOD score is expected to be maximized at 0 more than 50% of the time, and positive LOD scores are reduced toward 0, making the behavior of the LOD score conservative relative to theoretical expectations (see Nordheim 1984; Tai and Chen 1989). If τ<0 (i.e., there are systematic genotyping errors), then the LOD score behaves anticonservatively, with a propensity for false-positive findings. Counterintuitively, the larger the sample size, the greater are the effects of marker-locus genotyping errors, because one has more “power” to detect the error-induced deviation from the expected 50% frequency of observed recombinations.
To examine, in more detail, the effects of marker-locus genotyping errors on linkage analysis, let us—for reasons of simplicity—focus on the situation in which recombinant and nonrecombinant meioses can be counted. In this situation, there is a one-to-one correspondence between the LOD score
 |
and the statistic 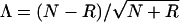 , where N and R represent the number of nonrecombinant and recombinant meioses, respectively, that were observed in a given data set. Λ is used here, rather than the LOD score, since Λ has a simpler algebraic representation. To single out the effects of marker-locus genotyping errors, genotype-assignment errors at the trait locus have been ignored throughout this section (i.e., ε=0); however, the results can be generalized to include those errors as well. Under H0 (θ=0.5), when an absence of genotyping errors of any kind is assumed, Λ∼N(0,1). The mean and the variance of Λ are given as
, where N and R represent the number of nonrecombinant and recombinant meioses, respectively, that were observed in a given data set. Λ is used here, rather than the LOD score, since Λ has a simpler algebraic representation. To single out the effects of marker-locus genotyping errors, genotype-assignment errors at the trait locus have been ignored throughout this section (i.e., ε=0); however, the results can be generalized to include those errors as well. Under H0 (θ=0.5), when an absence of genotyping errors of any kind is assumed, Λ∼N(0,1). The mean and the variance of Λ are given as 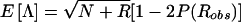 and Var
and Var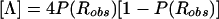 , as derived in the Appendix. By substituting the value that P(Robs) takes for selected parameter values (sample size N+R, θ, γ, and τ), the mean and the variance of Λ can be derived as a function of these parameters. The power under the alternative hypothesis (or the P value under the null hypothesis) can be computed as
, as derived in the Appendix. By substituting the value that P(Robs) takes for selected parameter values (sample size N+R, θ, γ, and τ), the mean and the variance of Λ can be derived as a function of these parameters. The power under the alternative hypothesis (or the P value under the null hypothesis) can be computed as
 |
for a chosen cutoff value c, where Φ( ) is the cumulative distribution function for a standard normal random variable (see Appendix).
Let us focus on the effects of marker-locus genotyping errors under the null hypothesis of no linkage (θ=0.5), since this will be shown to lead to a simple approach for estimation of error rates from the results of a genome scan. When the two types of misclassifications have equal probability, τ=0 and P(Robs)=0.5+γ-2(0.5)γ=0.5, which is independent of γ and τ. Thus, P(Robs)=P(Rtrue)=0.5, E[Λ]=0 and Var[Λ]=1, and the statistic behaves appropriately under H0. However, when the symmetry arguments no longer apply, τ≠0 and 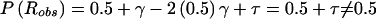 . Thus, P(Robs)≠P(Rtrue),
. Thus, P(Robs)≠P(Rtrue),  , Var[Λ]=1-4τ2<1, and the LOD-score statistic no longer behaves properly under H0. (If one wanted to allow for trait-locus errors as well, one would need to replace τ with τ(1-2ε) throughout.) Notice that γ does not appear in these expressions. Therefore, it is only through τ that marker-locus genotyping errors affect linkage analysis under H0.
, Var[Λ]=1-4τ2<1, and the LOD-score statistic no longer behaves properly under H0. (If one wanted to allow for trait-locus errors as well, one would need to replace τ with τ(1-2ε) throughout.) Notice that γ does not appear in these expressions. Therefore, it is only through τ that marker-locus genotyping errors affect linkage analysis under H0.
Figure 8A shows the point mass at 0, computed as
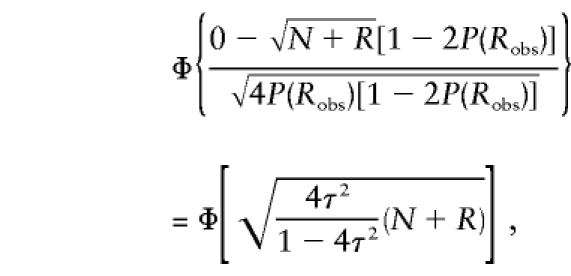 |
as a function of τ for different sample sizes. For nonsystematic genotyping errors on multiallelic marker loci, τ⩾0. As τ increases in magnitude, so does the point mass at 0, which is expected to be 0.5 in the absence of genotype errors. The larger the sample size, N+R, the more pronounced the effect: For τ=0.01 and a sample size of 500 meioses—a fairly small data set for the mapping of complex traits—the point mass at 0 is >0.6; however, in a very large study with a sample size of 5,000 meioses, the point mass at 0 is >0.9! Figure 8B shows the P values corresponding to a LOD score of 1—that is, 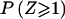 —where Z is the LOD score, again as a function of τ for different sample sizes. The larger the value of τ, the more conservative is the behavior of the LOD score in comparison with its theoretical distribution. As mentioned above, the effect is more pronounced in large sample sizes. Although a LOD score of 1 asymptotically has a theoretical P value of ∼.016 in the absence of marker-locus genotyping errors, it is shown here that the P value would be <.005 when τ=0.01 in a sample size of 500 meioses.
—where Z is the LOD score, again as a function of τ for different sample sizes. The larger the value of τ, the more conservative is the behavior of the LOD score in comparison with its theoretical distribution. As mentioned above, the effect is more pronounced in large sample sizes. Although a LOD score of 1 asymptotically has a theoretical P value of ∼.016 in the absence of marker-locus genotyping errors, it is shown here that the P value would be <.005 when τ=0.01 in a sample size of 500 meioses.
Figure 8.

Effects of marker-locus genotyping errors in the absence of linkage. A, point mass at 0; B,P value corresponding to a LOD score of 1 in the presence of misclassification, as a function of τ and the sample size (given as the number of countable meioses).
When marker-locus genotyping errors are systematic, it may be the case that τ<0. This has the opposite effect, leading to a propensity for false-positive findings, as also shown in figure 8. When τ=-0.01 in 500 meioses, the point mass at 0 is decreased to <0.4 , and the P value of a LOD score of 1 is inflated by more than threefold from its theoretical expectation. As previously mentioned, the impact is greater for larger sample sizes—that is, the point mass at 0 is <0.1 and the P value of a LOD score of 1 is inflated by more than 15-fold when N+R=5,000. The reason why the effect is stronger for larger sample sizes can be seen in the expressions of the mean and variance of Λ. Only the mean, as the “directional component” of the statistic, depends on the sample size, whereas the variance does not. In summary, for a multiallelic marker locus, there will tend to be >50% recombinants observed in the presence of marker-locus genotyping errors, with the assumption of the absence of a systematic bias, and the null-hypothesis behavior of the LOD-score statistic will thus be over-conservative, with increased point mass at 0 and fewer positive LOD scores of any magnitude than would be expected in the absence of genotyping errors. If there were systematic biases and if τ<0, then the situation might be more critical, since there would be a propensity for false-positive findings.
Of course, marker-locus genotyping errors also affect linkage analysis under the alternative hypothesis of linkage (θ<0.5 [Smith 1937; Terwilliger et al. 1990; Buetow 1991; Shields et al. 1991; Lincoln and Lander 1992; Goldstein et al. 1997]). The frequency of apparent recombination is then given by 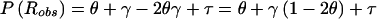 , which is a function of both γ and τ, in contrast with the situation under H0, where P(Robs) depends only on τ, as shown above. For this reason, marker-locus genotyping errors will have an effect on power even if the two types of misclassification have equal probability, as is typically the case for diallelic marker loci. Note that the impact of γ depends on the true recombination fraction, whereas that of τ does not. Table 1 gives the power for LOD-score thresholds of 1, 2, and 3, for a sample size of 500 countable meioses, for several different combinations of γ and τ.
, which is a function of both γ and τ, in contrast with the situation under H0, where P(Robs) depends only on τ, as shown above. For this reason, marker-locus genotyping errors will have an effect on power even if the two types of misclassification have equal probability, as is typically the case for diallelic marker loci. Note that the impact of γ depends on the true recombination fraction, whereas that of τ does not. Table 1 gives the power for LOD-score thresholds of 1, 2, and 3, for a sample size of 500 countable meioses, for several different combinations of γ and τ.
Table 1.
Effect of Nonsystematic Marker-Locus Genotyping Errors on the Power of a Linkage Study[Note]
|
Power for LOD Score at |
|||
| γ | τ = 0 | τ = γ/2 | τ = γ |
| Threshold 1: | |||
| 0 | .815 | .815 | .815 |
| .01 | .798 | .729 | .649 |
| .02 | .781 | .627 | .449 |
| .03 | .762 | .515 | .262 |
| .04 | .743 | .402 | .126 |
| .05 | .722 | .297 | .049 |
| Threshold 2: | |||
| 0 | .500 | .500 | .500 |
| .01 | .476 | .387 | .305 |
| .02 | .451 | .284 | .154 |
| .03 | .427 | .196 | .063 |
| .04 | .404 | .127 | .021 |
| .05 | .380 | .077 | .006 |
| Threshold 3: | |||
| 0 | .246 | .246 | .246 |
| .01 | .227 | .165 | .116 |
| .02 | .209 | .104 | .044 |
| .03 | .192 | .062 | .014 |
| .04 | .176 | .034 | .003 |
| .05 | .160 | .017 | .001 |
Note.—The recombination fraction was chosen such that the power for a LOD-score threshold of 2, with the use of 500 countable meioses, is 0.5 in the absence of any marker-locus genotyping errors. Only random genotyping errors are considered here—that is, τ⩾0 (by definition, 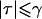 ).
).
Estimation of τ
As previously indicated, the distribution of the LOD score under the null hypothesis is affected by genotyping errors, when the errors lead to an asymmetry of the misclassification rates (i.e., τ⩽0). Although two-point linkage analysis typically does not allow for (or benefit from) separation of an observed recombination-fraction estimate into its various components (i.e., the true recombination fraction and the various misclassification parameters), estimates of τ may be obtained from the LOD scores obtained in a genome scan. Furthermore, the marker-locus-specific error rate can be estimated from the null-hypothesis distribution of the LOD-score statistic, via simulation.
For marker loci with multiple alleles, random genotyping errors can cause the null-hypothesis expectation of the recombination-fraction estimate to be raised from 0.5 to 0.5+τ. If one does not constrain 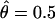 (i.e.,
(i.e., 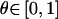 ), then one can compute the average value of this recombination-fraction estimate over all M marker loci analyzed in two-point analysis in a genome scan. A crude estimator of τ would be given as
), then one can compute the average value of this recombination-fraction estimate over all M marker loci analyzed in two-point analysis in a genome scan. A crude estimator of τ would be given as 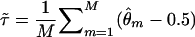 . An analogous estimator of τ would be provided by the genomewide average value of
. An analogous estimator of τ would be provided by the genomewide average value of 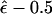 , in “complex” multipoint analysis (Göring and Terwilliger 2000a) performed under the assumptions that γ=0 and τ=0. Note that these are estimates of the genomewide average value of τ, which may be estimated to be 0, even in the presence of substantial locus-specific rates of error.
, in “complex” multipoint analysis (Göring and Terwilliger 2000a) performed under the assumptions that γ=0 and τ=0. Note that these are estimates of the genomewide average value of τ, which may be estimated to be 0, even in the presence of substantial locus-specific rates of error.
More information about τ can be obtained from the full distribution of either 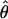 or the LOD score (or, equivalently, from the statistic Λ), for all marker loci, regardless of whether θ is constrained to ⩽0.5. If one considers the results of a sparse genome scan with two-point linkage analysis, one would expect ∼1.6% of the marker loci to show a LOD score >1 (see above), whereas the number could be significantly lower in the presence of genotyping errors (systematic bias may lead to the opposite effect). One could estimate the genomewide average value of τ by fitting the distribution of observed LOD scores to the form of the distribution described above, as a function of τ. If significantly >50% of the marker loci show a maximum LOD score of 0 and if there is a significant contraction of the distribution toward 0, then a high rate of error in genotyping may be indicated. This method also assumes that τ has the same value for all marker loci in the genome, and, thus, it provides only a crude genomewide estimate of the error rates. In this case, one could compute the test statistic
or the LOD score (or, equivalently, from the statistic Λ), for all marker loci, regardless of whether θ is constrained to ⩽0.5. If one considers the results of a sparse genome scan with two-point linkage analysis, one would expect ∼1.6% of the marker loci to show a LOD score >1 (see above), whereas the number could be significantly lower in the presence of genotyping errors (systematic bias may lead to the opposite effect). One could estimate the genomewide average value of τ by fitting the distribution of observed LOD scores to the form of the distribution described above, as a function of τ. If significantly >50% of the marker loci show a maximum LOD score of 0 and if there is a significant contraction of the distribution toward 0, then a high rate of error in genotyping may be indicated. This method also assumes that τ has the same value for all marker loci in the genome, and, thus, it provides only a crude genomewide estimate of the error rates. In this case, one could compute the test statistic 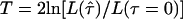 , where the likelihood under either hypothesis is computed from the density function of the statistic under H0 as a function of τ (see the Appendix).
, where the likelihood under either hypothesis is computed from the density function of the statistic under H0 as a function of τ (see the Appendix).
An even better approach would be to estimate the locus-specific error rates by means of simulation. If one simulates error-free genotypes for a fully informative marker locus in the data set, independent of the observed genotyping data, then one can then perform linkage analysis between the simulated marker locus and each of the genotyped marker loci spanning the genome. With a large number of replicates, one can estimate the distribution of the LOD scores and the value of τ independently for each genotyped marker locus, with use of either of the approaches described above. This provides a locus-specific test of the asymmetry in the genotyping error rate. This will not allow estimation of γ, however, since the null-hypothesis distribution is only a function of τ (the overall genotyping error rate must be at least as large as τ). The simulation-based approach will not detect systematic genotyping errors in the analysis of affected individuals only, since the asymmetry in misclassification of recombination seen in such an analysis is a result of ascertainment bias (i.e., in affecteds-only analysis, one ascertains that the affected individuals are similar with respect to disease-locus genotypes, so that, when marker-locus genotypes are similar, an asymmetry in misclassification results solely from the ascertainment on trait-locus genotypes), which cannot be estimated by such simulation procedures. This proposed method only estimates the asymmetry in recombination-status misclassification resulting from random genotyping errors.
A contraction of the positive part of the distribution toward 0 may also occur when the mode of inheritance is assumed to be very weak (Göring and Terwilliger 2000c), as a result of the reduction in the variance of the LOD-score statistic, compared with its theoretical expectation. Asymptotically, this problem may dissipate; however, in sample sizes that are as large as 1,000 sib pairs, an excessively weak penetrance ratio (as formerly advocated by Terwilliger and Ott 1994 [see section 25.3]) leads to a significant contraction of the null distribution (although not necessarily to an increase in the size of the point mass at 0), since the effective number of equivalent meioses can be rendered quite small, potentially causing a large deviation from the asymptotic distributions.
Discussion
Errors in genotyping of the marker loci are not infrequent. It is therefore important to understand the consequences of high error rates in marker-locus genotypes and to have a model in which these errors can be handled mathematically, to minimize their consequences. In the present study, marker-locus genotyping errors are handled by adding additional “imaginary” components to the recombination fraction. The results of the present study, together with findings from a companion article (Göring and Terwilliger 2000a), provide the basis for a logical framework by which multipoint likelihoods can be computed, allowing for errors in the genotype assignments at trait and marker loci jointly, with use of all the available data to estimate these error rates in a locus-specific manner. A summary of linkage analysis in the presence of genotyping errors at both disease locus and marker loci, under the described model, is shown in figure 9. The method is applicable to both “model-based” and “model-free” analyses, as described elsewhere (Göring and Terwilliger 2000c). Through the application of this technique, the earlier admonition that “one should not do multipoint analysis with a complex trait because of the increased propensity for false-negative results when there are model misspecifications” (Terwilliger and Ott 1994 [see p. 220]) need not be followed, since we have demonstrated an equivalence between two-point LOD scores without allowing for misclassification errors and multipoint LOD scores in the presence of misclassification errors at the trait locus (Göring and Terwilliger 2000a), a result that can be trivially extended to marker-locus genotyping errors as well (proof is available on request).
Figure 9.

Overview of linkage analysis in the presence of genotyping errors at trait and marker loci, under the outlined model for higher-dimensional recombination fractions.
Since genotyping errors can affect multipoint LOD scores more dramatically than they can affect two-point LOD scores (as a result of the apparent deviation from linearity of the map at both disease locus and marker loci), traditional multipoint LOD scores may be lower than traditional two-point LOD scores in the presence of linkage, despite the use of more “information” in the multipoint analysis. More information will only lead to a more powerful test, if this additional information is accurate. Errors in the genotypes of marker loci, much like errors in the marker-locus map, can lead to false-negative multipoint LOD scores, unless one allows for such errors by use of a scheme such as the one proposed in the present study. It is therefore not necessarily the case that one most likely has a false-positive finding when use of traditional multipoint statistics are less significant traditional two-point statistics. In fact, such findings may be expected in the presence of errors. Furthermore, if >50% of the genome has a maximum LOD score (or any other one-sided linkage test statistic) that is 0, then a high rate of genotyping errors may be indicated, and careful scrutiny of the data—as well as skepticism of the conclusions—may be warranted.
An alternative option for handling errors in genotyping of the marker loci would be to model them directly, through penetrances, rather than indirectly, through the resulting misclassifications in recombination status (Ott 1985; Terwilliger et al. 1990; Lincoln and Lander 1992). Rather than using a one-to-one correspondence between the readings of marker-locus genotypes (i.e., the marker phenotypes) and the true marker-locus genotypes, one could specify penetrance functions similar to those used for disease loci. Another option would be to model errors in genotyping of the marker locus through mutation rates (Ott 1985; Terwilliger et al. 1990). The described model does not deal with other forms of marker-locus errors that are known to lead to spurious results in linkage and linkage-disequilibrium tests, such as incorrect specification of the marker-locus allele frequencies (Ott 1992), incorrect specification of marker-marker linkage disequilibria, and incorrect specification of the marker-locus order and intermarker genetic distances. These difficulties can be dealt with by treating the “offending” parameters as nuisance parameters, by use of profile likelihoods in the computation of the LOD score (Göring and Terwilliger 2000b). Similar treatment is advised for the marker-locus-specific error vectors as well.
Acknowledgments
A Hitchings-Elion Fellowship from the Burroughs-Wellcome Fund (to J.D.T.) is gratefully acknowledged, as is grant HG00008 from the National Institute of Health (to Jürg Ott, thesis advisor of H.H.H.G.).
Appendix: Derivation of Statistical Distribution of Λ in Terms of θ, γ, and τ
In the absence of linkage,  , assuming lack of any genotyping errors. Bearing in mind that the total number of meioses, N+R, is a constant, the mean and the variance of Λ are given by
, assuming lack of any genotyping errors. Bearing in mind that the total number of meioses, N+R, is a constant, the mean and the variance of Λ are given by
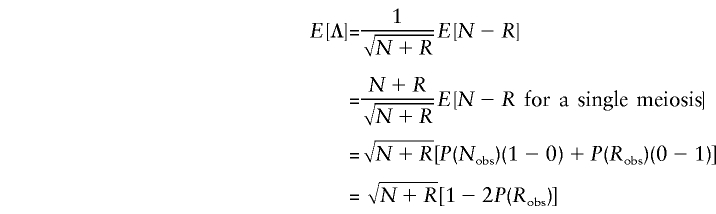 |
and
 |
Since 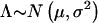 , where
, where  and σ2=Var[Λ]=4P(Robs)[1-2P(Robs)],
and σ2=Var[Λ]=4P(Robs)[1-2P(Robs)], 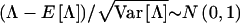 , and the power (or the P value) for a given critical value c can be computed as
, and the power (or the P value) for a given critical value c can be computed as
 |
where Φ( ) is the cumulative-distribution function of a N(0, 1) random variable.
By substituting the value of P(Robs) for different parameter values (N+R, θ, ε, γ, and τ), the mean and variance of Λ as well as the power or the P value can be determined for different situations. For example, under the null hypothesis of no linkage (θ=0.5), when no genotyping errors exist at either the disease locus or the marker locus (ε=γ=0), P(Robs)=P(Rtrue)=0.5, so that E[Λ]=0 and Var[Λ]=1, and Λ is distributed as an N(0, 1) random variable (for any sample size that is sufficiently large for the asymptotic normal approximation to hold). The P value for a cutoff point of c=3.72 (the equivalent of a LOD score of 3) would then be 1-Φ(3.72)=0.0001, and the point mass at 0 would be Φ(0)=0.5, and the statistic would behave as expected (see Nordheim 1984; Tai and Chen 1989). (The point mass at 0 is given by Φ(0)=0.5, since the test is conducted in a one-sided manner—that is, θ⩽0.5. Any negative value of Λ, however, corresponds to a value of 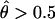 , which is not consistent with the alternative hypothesis of linkage. All negative values of Λ are therefore customarily truncated to 0—that is, the maximum LOD score is 0 at recombination fraction 0.5.)
, which is not consistent with the alternative hypothesis of linkage. All negative values of Λ are therefore customarily truncated to 0—that is, the maximum LOD score is 0 at recombination fraction 0.5.)
References
- Brzustowicz LM, Mérette C, Xie X, Townsend L, Gilliam TC, Ott J (1993) Molecular and statistical approaches to the detection and correction of errors in genotype databases. Am J Hum Genet 53:1137–1145 [PMC free article] [PubMed]
- Buetow KH (1991) Influence of aberrant observations on high-resolution linkage analysis outcomes. Am J Hum Genet 49:985–994 [PMC free article] [PubMed]
- Daw EW, Thompson EA, Wijsman EM (1998) Bias in multipoint linkage analysis arising from map misspecification. Am J Hum Genet Suppl 63:A17 [DOI] [PubMed] [Google Scholar]
- de la Chapelle A, Wright FA (1998) Linkage disequilibrium mapping in isolated populations: the example of Finland revisited. Proc Natl Acad Sci USA 95:12416–12423 [DOI] [PMC free article] [PubMed]
- Ehm MG, Kimmel M, Cottingham RW (1996) Error detection for genetic data using likelihood methods. Am J Hum Genet 58:225–234 [PMC free article] [PubMed]
- Goldstein DR, Zhao H, Speed TP (1997) The effects of genotyping errors and interference on estimation of genetic distance. Hum Hered 47:86–100 [DOI] [PubMed]
- Göring HHH, Terwilliger JD (2000a) Linkage analysis in the presence of errors I. complex-valued recombination fractions and complex phenotypes. Am J Hum Genet 66:1095–1106 (in this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2000b) Linkage analysis in the presence of errors III. marker loci and their map as nuisance parameters. Am J Hum Genet (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2000c) Linkage analysis in the presence of errors IV. joint pseudomarker analysis of linkage and/or linkage disequilibrium on a mixture of pedigrees and singletons when the mode of inheritance cannot be accurately specified. Am J Hum Genet (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göring HHH, Terwilliger JD, Ott J. (1997) A likelihood-based approach to extended haplotype analysis of shared segments using a Markov branching process. Am J Hum Genet 61:1614 [Google Scholar]
- Haldane JBS (1919) The combination of linkage values and the calculation of distances between the loci of linked factors. J Genet 8:299–309 [Google Scholar]
- Holmans P, Craddock N (1997) Efficient strategies for genome-scanning using maximum-likelihood affected-sib-pair analysis. Am J Hum Genet 60:657–666 [PMC free article] [PubMed]
- Jaynes ET (1996) Probability theory: the logic of science. http://bayes.wustl.edu/etj/prob.html
- Krause EF (1975) Taxicab geometry—an adventure in non-Euclidean geometry. Addison-Wesley, Menlo Park, NJ [Google Scholar]
- Lander ES, Green P (1987) Construction of multilocus genetic linkage maps in humans. Proc Natl Acad Sci USA 84:2363–2367 [DOI] [PMC free article] [PubMed]
- Lathrop GM, Hooper AB, Huntsman JW, Ward RH (1983) Evaluating pedigree data. I. The estimation of pedigree error in the presence of marker mistyping. Am J Hum Genet 35:241–262 [PMC free article] [PubMed]
- Lathrop, GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446 [DOI] [PMC free article] [PubMed]
- Lincoln SE, Lander ES (1992) Systematic detection of errors in genetic linkage data. Genomics 14:604–610 [DOI] [PubMed]
- Lindqvist AK, Magnusson PK, Balciuniene J, Wadelius C, Lindholm E, Alarcón-Riquelme ME, Gyllensten UB (1996) Chromosome-specific panels of tri- and tetra-nucleotide microsatellite markers for multiplex fluorescent detection and automated genotyping: evaluation of their utility in pathology and forensics. Genome Res 6:1170–1176 [DOI] [PubMed]
- Morton NE (1988) Multipoint mapping and the emperor's clothes. Ann Hum Genet 52:309–318 [DOI] [PubMed]
- Morton NE, Andrews V (1989) MAP, an expert system for multiple pairwise linkage analysis. Ann Hum Genet 53:263–269 [DOI] [PubMed]
- Nordheim EV (1984) On the performance of a likelihood ratio test for genetic linkage. Biometrics 40:785–790 [Google Scholar]
- Ott, J (1977) Linkage analysis with misclassification at one locus. Clin Genet 12:119–124 [DOI] [PubMed]
- ——— (1985) Analysis of human genetic linkage, 1st ed. Baltimore, Johns Hopkins University Press [Google Scholar]
- ——— (1992) Strategies for characterizing highly polymorphic markers in human gene mapping. Am J Hum Genet 51:283–290 [PMC free article] [PubMed]
- Shields DC, Collins A, Buetow KH, Morton NE (1991) Error filtration, interference, and the human linkage map. Proc Natl Acad Sci USA 88:6501–6505 [DOI] [PMC free article] [PubMed]
- Smith HF (1937) Test of significance for Mendelian ratios when classification is uncertain. Ann Eugen 8:94–95 [Google Scholar]
- Tai JJ, Chen CL (1989) Asymptotic distribution of the LOD score for familial data. Proc Natl Sci Counc Repub China B 13:38–41 [PubMed]
- Terwilliger JD. On the resolution and feasibility of genome scanning approaches to unraveling the genetic components of multifactorial phenotypes. In: Rao DC, Province MA (eds) Genetic dissection of complex phenotypes: challenges for the new millennium. Academic Press, San Diego (in press) [Google Scholar]
- Terwilliger JD, Göring HHH. A review of gene mapping in the 20th and 21st centuries: statistical methods, data analysis, and experimental design. Hum Biol 72:63–132 [PubMed] [Google Scholar]
- Terwilliger JD, Ott J (1994) Handbook of human genetic linkage. The Johns Hopkins University Press, Baltimore [Google Scholar]
- Terwilliger JD, Shannon WD, Lathrop GM, Nolan JP, Goldin LR, Chase GA, Weeks DE (1997) True and false positive peaks in genomewide scans: applications of length-biased sampling to linkage mapping. Am J Hum Genet 61:430–438 [DOI] [PMC free article] [PubMed]
- Terwilliger JD, Weeks DE, Ott J (1990) Laboratory errors in the reading of marker alleles cause massive reductions in LOD score and lead to gross overestimation of the recombination fraction. Am J Hum Genet Suppl 47:A201 [Google Scholar]
- Walley P (1991) Statistical reasoning with imprecise probabilities. Chapman and Hall, London [Google Scholar]