

Abstract
The autosomal dominant spinocerebellar ataxias (SCAs) are a group of late-onset, neurodegenerative disorders for which 10 loci have been mapped (SCA1, SCA2, SCA4–SCA8, SCA10, MJD, and DRPLA). The mutant proteins have shown an expanded polyglutamine tract in SCA1, SCA2, MJD/SCA3, SCA6, SCA7, and DRPLA; a glycine-to-arginine substitution was found in SCA6 as well. Recently, an untranslated (CTG)n expansion on chromosome 13q was described as being the cause of SCA8. We have now (1) assessed the repeat size in a group of patients with ataxia and a large number of controls, (2) examined the intergenerational transmission of the repeat, and (3) estimated the instability of repeat size in the sperm of one patient and two healthy controls. Normal SCA8 chromosomes showed an apparently trimodal distribution, with classes of small (15–21 CTGs), intermediate (22–37 CTGs), and large (40–91 CTGs) alleles; large alleles accounted for only0.7% of all normal-size alleles. No expanded alleles (⩾100 CTGs) were found in controls. Expansion of the CTG tract was found in five families with ataxia; expanded alleles (all paternally transmitted) were characterized mostly by repeat-size contraction. There was a high germinal instability of both expanded and normal alleles: in one patient, the expanded allele (152 CTGs) had mostly contraction in size (often into the normal range); in the sperm of two normal controls, contractions were also more frequent, but occasional expansions into the upper limit of the normal size range were also seen. In conclusion, our results show (1) no overlapping between control (15–91) and pathogenic (100–152) alleles and (2) a high instability in spermatogenesis (both for expanded and normal alleles), suggesting a high mutational rate at the SCA8 locus.
Introduction
The spinocerebellar ataxias (SCAs) are a heterogeneous group of usually late-onset, neurodegenerative disorders, characterized by gait, limb, and speech incoordination, in addition to other variable signs. Ten loci for autosomal dominant SCAs have already been mapped (Zoghbi et al. 1988; Gispert et al. 1993; Takiyama et al. 1993; Gardner et al. 1994; Koide et al. 1994; Nagafuchi et al. 1994; Ranum et al. 1994; Benomar et al. 1995; Zhuchenko et al. 1997; Koob et al. 1999; Zu et al. 1999). The mutant proteins show an expanded polyglutamine tract in SCA1, SCA2, Machado-Joseph disease (MJD/SCA3), SCA6, SCA7, and DRPLA (Orr et al. 1993; Kawaguchi et al. 1994; Imbert et al. 1996; Pulst et al. 1996; Sanpei et al. 1996; David et al. 1997; Zhuchenko et al. 1997), whereas a glycine-to-arginine substitution has also been shown in SCA6 (Yue et al. 1997). Recently, a new ataxia gene, named SCA8 (MIM 603680), was found on chromosome 13q; the mutation consisted of the expansion of an untranslated composite, (CTA)n(CTG)n (Koob et al. 1999). This mutation was identified in a very large pedigree with seven generations. The range of ages at onset of symptoms was 18–65 years (mean 39 years). The initial clinical symptoms included dysarthria, mild aspiration, and gait instability. On neurological examination, spastic and ataxic dysarthria, nystagmus, limb and gait ataxia, limb spasticity, and diminished vibration perception were found. The disease progressed slowly over time; severely affected family members were nonambulatory by the fourth to fifth decades. The repeat length of pathogenic alleles was 110–130 combined CTA-CTG repeats, whereas alleles in a control population of 1,200 chromosomes had 16–92 repeat units. Like the (CTG)n expansion in myotonic dystrophy (DM [Ashizawa et al. 1994]), the repeat length of mutant alleles tended to contract with paternal transmission (−86 to +7) and to expand further with maternal transmission (−11 to +600 [Koob et al. 1999]). An interesting feature observed in DM and not reported in any of the SCAs is that the intergenerational contraction of the CTG repeat may accompany apparent anticipation (Ashizawa et al. 1994). Maternal bias toward expansion is not a feature seen in other dominant SCAs (Chung et al. 1993; Maciel et al. 1995; Imbert et al. 1996; David et al. 1997; Zhuchenko et al. 1997). The results of Koob et al. (1999) indicate that this repeat is not translated into a polyglutamine tract, since no open-reading frames were found to extend through the CAG repeat. In SCA8, the expanded CTG repeat lies in the 3′-UTR of a transcribed RNA and has a size, orientation, and general location similar to those of the 3′ untranslated CTG expansion that causes DM (Koob et al. 1999).
In an attempt to improve our knowledge about the molecular basis of this recently found dynamic mutation, we have (1) assessed the size of the (CTG)n at the SCA8 gene, in a large group of patients with ataxia of unknown cause and in a control population (1,818 chromosomes); (2) looked at meiotic instability of this repeat in families; and (3) examined the instability during the spermatogenesis of one patient with SCA8 and two normal controls.
Subjects and Methods
Subjects
We ascertained 73 unrelated individuals with SCA, from 21 families with dominant inheritance, 9 kindreds with apparently recessive transmission, and 43 isolated cases, all previously excluded for SCA1, SCA2, MJD/SCA3, SCA6, SCA7, and DRPLA (Silveira et al. 1996, 1998). A large control population (950 blood spots conserved on filter paper) was obtained from the Portuguese national screening for PKU, performed at IGM. We also obtained sperm samples from one patient with SCA8 and from two control donors.
Methods
Peripheral blood was collected from patients and their relatives; written informed consent was obtained from each individual. Genomic DNA was isolated from peripheral blood leukocytes by standard techniques (Sambrook et al. 1989).
Molecular analysis of the (CTG)n was performed by PCR amplification using the published primer sequences SCA8-F4 and SCA8-R4 (Koob et al. 1999). PCR was carried out with 1 μM of each primer; 200 μM of dNTPs; 1.0 mM MgCl2; 10 mM Tris, pH 9.0; 50 mM KCl; 1 U of Taq polymerase; and 2% formamide, in a final volume of 25 μl. Samples were processed through 35 cycles (at 95°C for 45 s, 54°C for 75 s, and 72°C for 75 s). PCR products were analyzed on 6% polyacrylamide gels. Allele sizes were determined by comparison of migration relative to an M13 sequencing ladder. Both the CTA and the CTG repeats on the SCA8 gene are polymorphic, and PCR assay determines the combined size of the two repeats. Low-copy PCR of sperm DNA was performed after sperm dilution to ∼1–10 cells by reaction.
Haplotype analysis was performed by use of four polymorphic markers, within 2 cM of the SCA8 locus: cen-D13S1309-D13S1268-D13S275-D13S1296-tel, according to the Center for Medical Genetics and Fondation Jean Dausset CEPH databases. Because the families were too small for linkage analysis, the relative positioning of the SCA8 locus could not be established; thus, for haplotype construction, the (CTG)n was arbitrarily considered to be telomeric to the markers. The numbering of marker alleles, according to their sizes, was based on data from Généthon.
Statistical Analysis
Allele frequencies at the SCA8 locus were estimated by the gene-count method, and heterozygosity (H) was calculated as 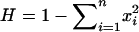 , where xi is the estimated frequency of the ith allele at the locus.
, where xi is the estimated frequency of the ith allele at the locus.
Results
Genotyping
The analysis of the SCA8 combined (CTA)n(CTG)n showed that seven patients, from five unrelated families, presented an expanded allele (table 1, figs. 1 and 2): three patients (from families PT and VC) had one allele with 132–152 repeats—in the expanded range, as previously described—whereas four other patients (from families CH, LE, and BR) presented an expanded allele with 100–106 repeat units, slightly below that pathogenic range (Koob et al. 1999). In addition, six unaffected subjects (aged 12–77 years) carried one allele with 128–170 CTGs.
Table 1.
Genetic and Clinical Characteristics of Patients[Note]
| Family and Patient | Sex/Age(years) | Expanded(CTG)n | Age atOnset(years) | Dysarthria | Gait Ataxia | Epilepsy | MemoryImpairment | MentalRetardation |
| PT: | ||||||||
| II:2 | M/39 | 152 | 28 | ++ | ++ | − | ++ | − |
| CH: | ||||||||
| III:1 | F/12 | 105 | 0 | +++ | +++ | ++ | NA | ++ |
| LE: | ||||||||
| III:1 | M/43 | 106 | 25 | + | ++ | − | − | − |
| III:3 | F/36 | 100 | 20 | ++ | ++ | − | − | − |
| VC: | ||||||||
| II:1 | M/46 | 132 | 26 | ++ | ++ | − | + | − |
| III:1 | M/20 | 132 | 20 | + | − | − | − | − |
| BR | F/42 | 104 | 31 | + | + | − | − | − |
Note.— M = male; F = female; − = absent; + = mild; ++ = moderate; +++ = severe; NA = data not available.
Figure 1.

Pedigrees and genotyping from four families with SCA8. Circles and squares with black dots indicate asymptomatic carriers of expanded alleles. Haplotypes of four microsatellite markers spanning 2 cM within the SCA8-locus region are shown. The haplotype that segregates with the expansion is boxed, and inferred haplotypes are bracketed. The (CTG)n (position unknown) was arbitrarily positioned as being telomeric to all markers. The fifth family (BR) is not shown, since no family genotyping was available.
Figure 2.
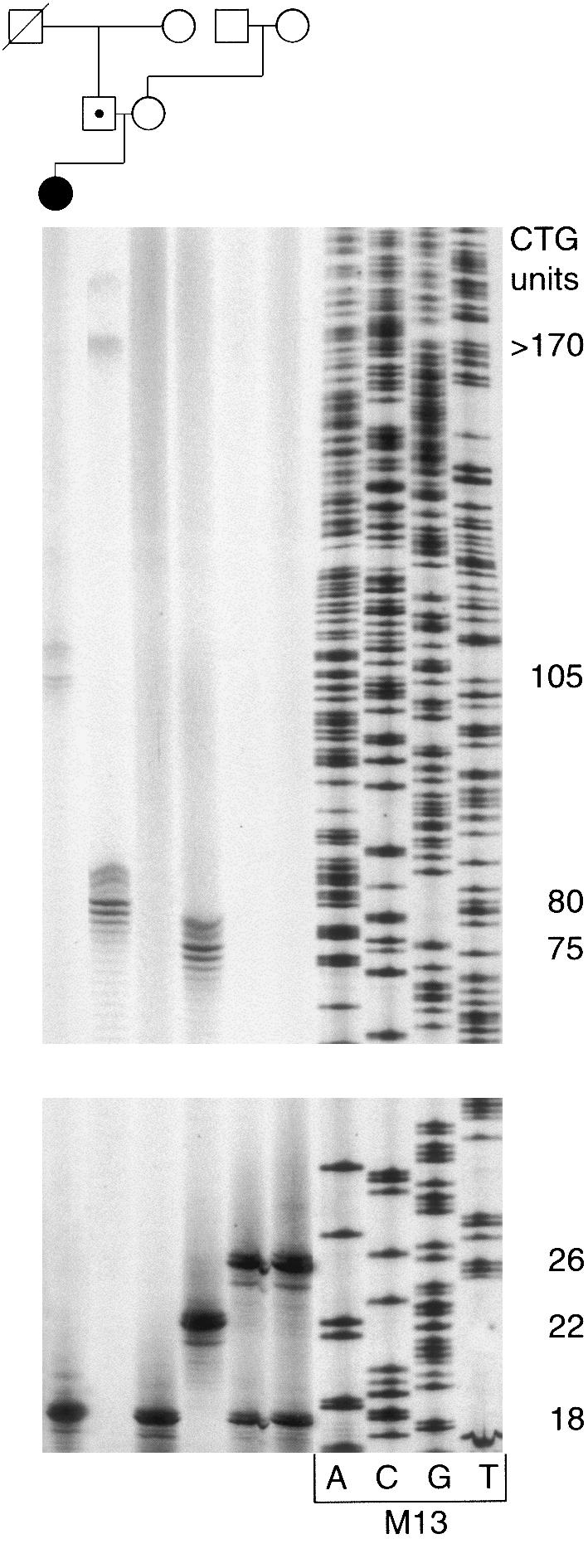
Pedigree and PCR products of family CH. Genomic DNA was amplified by use of primers SCA8-F4 and SCA8-R4. PCR products were analyzed on 6% polyacrylamide gels. Allele sizes were determined by comparison of migration relative to an M13 sequencing ladder. Normal alleles had sizes varying from 15 to 91 units, and expanded alleles varied from 100 to 170.
Clinical Features
Only two families (VC and LE) had more than one patient described. Age at onset of first symptoms was 20–31 years, except for one patient, in whom symptoms were present since the first year of life (table 1). The initial clinical symptoms included dysarthria and diplopia. On neurological examination, limb and gait ataxia were present in all patients; memory impairment was present in two patients (with repeat sizes of 132 and 152).
The young girl with congenital ataxia had one allele with 105 CTGs; she had had severe cerebellar signs since her first year of life, walked (with aid) only at age 5 years, had myoclonic epilepsy since age 3 years, and was mentally retarded. Cental nervous system malformations were excluded by magnetic resonance imaging. A skin and muscle biopsy revealed a normal pathology and showed a normal function of the mitochondrial respiratory chain; other metabolic causes of ataxia were also excluded: amino- and organic-acid chromatography and β-hexosaminidase and β-galactosidase activity were all normal. No MERRF mutations were found. Study of α-fetoprotein, immunoglobulins, and chromosome breakage excluded ataxia telangiectasia; this patient had also been excluded for Friedreich ataxia (in addition to DRPLA and the other dominant ataxias, as mentioned). This is thus the first report of a case of congenital ataxia that may be due to a mutation for a dominant SCA. Congenital forms of DM with distinct clinical features, when compared with adult-onset cases, are well known (Tsilfidis et al. 1992).
Instability of SCA8 Alleles
Analysis of the SCA8 locus in families (fig. 1) showed instability on transmission of expanded alleles, as well as of a large normal allele (family CH, individuals I:2 and II:1).
In family PT, the proband had an expansion of 152 repeat units, whereas his two asymptomatic sibs (aged 38 and 31 years) had expansions of 130 and 160 repeat units; furthermore, one of his children (aged 12 years) presented an allele with 130 units on the paternal chromosome. In family CH, a girl with congenital ataxia had inherited an expanded allele with 105 repeats from her 42-year-old unaffected father (fig. 2), who, in turn, had inherited an expanded and a large normal allele from his unaffected parents. Given the large instability of both expanded and large normal alleles, we cannot be completely sure that he received the expansion from his deceased father (which then underwent a contraction from 170 to 105 repeats when transmitted to his own daughter) and the large normal allele (80) from his mother (75 CTGs). The alternative, that the mother’s 75 CTGs allele expanded to 170 repeats, is very unlikely (because this would imply an expansion from 80 to 105 repeats on transmission of the other allele to his daughter). In fact, there is a very strong trend for contraction of paternal expanded alleles; further expansion has been seen in just two cases, both with an increase of nine repeats or less (Koob et al. 1999). In family LE, the proband inherited an allele with 100 CTGs, whereas her affected brother inherited an allele with 106 repeats from their 77-year-old, unaffected father, who had 170 repeat units. This is the largest expansion found so far in an unaffected subject; an unaffected individual with 140 repeats was described by Koob et al. (1999). In family VC, patient II:1 had an expanded allele with 132 CTGs that contracted to 128 repeats in the unaffected child (aged 14 years) but was stably transmitted to the affected one. In family BR, no relatives could be genotyped.
In summary, expanded alleles were unstable in five of six known transmissions (in the second generation of family PT, the mutation may have been inherited from either side, because the father died and the mother was unavailable for genotyping); all known transmissions were of paternal origin. This instability was characterized by a contraction of the CTG repeat tract in 4–70 units (average change of −38).
DNA analysis of the (CTG)n at the SCA8 locus always showed multiple bands for the expanded alleles, thus indicating mitotic instability in peripheral blood leukocytes (fig. 2).
Control Population
Analysis of the SCA8 repeat in the control population was possible for 1,818 chromosomes: normal alleles ranged in size from 15 to 91 CTG repeat units. SCA8 is a highly polymorphic locus (fig. 3 and table 2), with a heterozygosity of 87.9%. The distribution of normal alleles appears to comprise (1) a class of small alleles, within a repeat interval of 15–21 (18.9%); (2) a class of intermediate-sized alleles, with 22–37 repeats (80.4%); and (3) a rather heterogeneous class of larger alleles, with 40–91 repeat units (only 0.7%). The allele with 24 CTGs was the most common, contributing 18% of all normal alleles; it was found in the homozygous state in only 2% of the population.
Figure 3.

Distribution of CTG repeat size of alleles at the SCA8 locus in a normal population of Portuguese origin (n=1,818).
Table 2.
CTG Repeat Size in Normal Alleles at the SCA8 Locus in a Control Population (1,818 chromosomes) of Portuguese Origin
| RepeatSize | na | Frequency(%) | Mean (± SD) | Mode |
| 15–21 | 344 | 18.9 | 18 (±.93) | 18 |
| 22–37 | 1,462 | 80.4 | 25 (±2.14) | 24 |
| 40–91 | 12 | .7 | 56 (±18.83) | 48 |
No. of chromosomes studied from Guthrie blood spots of presumably unrelated individuals.
Sperm-Cell Analysis
Analysis of the SCA8 repeat, by low-copy PCR, in sperm from the proband of family PT (152/28 CTGs) showed multiple bands, with sizes of 19–∼240 repeat units (fig. 4A). The most frequent allele in the normal range had 24 CTGs (contrasting with the 28 CTGs allele found in leukocytes), which also suggested mosaicism for the normal SCA8 allele.
Figure 4.

Low-copy PCR of sperm cells. DNA was amplified with primers SCA8-F4 and SCA8-R4 and analyzed on 6% polyacrylamide gels. Allele sizes were determined by comparison of migration relative to a M13 sequencing ladder. A, Patient with alleles of 152 and 28 repeats in peripheral blood; alleles with ⩽37 CTGs were assumed to have derived from the 28-repeat allele. B, Healthy control, homozygote for a 25-repeat allele on blood leukocytes; similar results (not shown) were obtained with another healthy control with 25/23 CTGs in peripheral leukocytes.
Thus, to investigate whether germinal mosaicism was also present in normal individuals, we analyzed sperm cells from two control donors, with 25/25 and 23/25 CTGs in leukocytes: again, multiple bands were found, with allele sizes of 14–97 and 19–68 repeats, respectively. Most alleles were within the range of 19–37, with rare alleles <15 or >37 CTG repeats. The results implied the occurrence of frequent contractions and of occasional expansions over the normal range of 15–91 CTGs (fig. 4B).
Single-sperm-cell PCR (with haplotype analysis, to identify the parental origin of mutated alleles) would have been the only manner to assess the instability rate of both normal and expanded alleles. Nevertheless, in an attempt to estimate the change in size, during meiosis, for the expanded allele in the proband of family PT, sperm alleles with >37 repeats (0.7% in our controls) were considered to arise from a contraction of the 152-CTG allele. Under this assumption, an analysis of 227 sperm cells presumably carrying the mutant chromosome showed the most common allele to have 97 CTGs (152 repeats in leukocytes; fig. 5A). Variation in size of the expanded repeat in sperm cells (as compared with its size in peripheral leukocytes) ranged from −112 to ∼+88 units (range, 40–240; fig. 5B). Altogether, there were 98.7% changes in repeat size (95% CI, 97.2–100); contractions occurred in 87.7% (95% CI, 83.4–92.0), whereas expansions accounted for only 11% of all sperm cells (95% CI, 7.1–14.9).
Figure 5.

A, Frequency of variation in repeat size of a 152-repeat SCA8 allele in sperm cells of a heterozygous patient with 152/28 CTGs; the arrow indicates the length of the donor’s allele in peripheral blood leukocytes (alleles with ⩽37 CTGs are not shown, since they were assumed to be derived from the 28-repeat allele). B, Distribution of repeat-size changes during meiosis (determined by subtracting the size of repeats in leukocytes to that in sperm).
Of interest, only 31.3% (95% CI, 25.3–37.3) of the gametes had a repeat size in the range expected to give a clinical phenotype (⩾100 repeats, in our affected subjects): the chance of this male transmitting a mutated allele would be significantly less than the expected 50% (χ21=30.48; P<.0001).
Discussion
Normal and Expanded Ranges
Our results support the previous findings that implicate a (CTG)n expansion on chromosome 13q21 in SCA8, a new form of SCA (Koob et al. 1999); however, the possibility that the (CTG)n is only in linkage disequilibrium with the SCA8 locus still cannot be completely excluded.
All affected subjects showed an expanded allele between 100 and 152 repeats (110–130 in the previous report). Alleles in our large control population ranged from 15 to 91 repeats (16–92 in the previous report). Taking both studies, there is thus no overlap between normal alleles in controls (>3,000 chromosomes altogether) and expanded alleles found in patients with SCA8.
The allele distribution in controls (three classes) closely resembles that at the DM locus (Zerylnick et al. 1995). Alleles <21 repeats may not show instability, whereas those with 22–37 CTGs (the most frequent) may be somewhat more unstable; this is in fact suggested by the degree of germinal mosaicism found in two healthy controls with 25/23 and 25/25 CTGs, respectively (fig. 4B). Large alleles, with 40–91 repeats, are very rare in the control population, but may be particularly unstable, and thus undergo occasional expansion up to full mutation; this is also suggested by the expansion of five units found on transmission of a large normal allele with 75 repeats (figs. 1 and 2). A similar pattern has been found for other trinucleotide-repeat diseases (Imbert et al. 1993; Zhang et al. 1994) and for ERDA1 (Deka et al. 1999), a trinucleotide-repeat locus to which no association with a pathological phenotype has yet been shown, in which different classes of repeat sizes present different mutation rates, according to age, sex, and repeat size of the transmitting parent. Additional data (more SCA8 and control families with haplotype analysis) are needed to support these findings.
The SCA8 mutation may be fairly common; it was found in five of all our families that were previously excluded for SCA1-2, MJD, SCA6-7, and DRPLA. This may be related to a pool of large normal alleles (40–91 CTGs) found to have a frequency of .7% in the general population.
Mitotic Instability
Mitotic instability is a feature of trinucleotide-repeat diseases, including DM (Jansen et al. 1994; Cancel et al. 1995; Chong et al. 1995). The finding of multiple bands for each expanded SCA8 allele in peripheral blood leukocytes and the presence of mosaicism between sperm and blood cells (both for normal and expanded alleles) indicate a significant mitotic instability at the SCA8 locus. In DM, the heterogeneity in length of expanded CTG repeats in blood cells is also known to progress with time (Martorell et al. 1998). Prospective studies are now needed to confirm whether this is also the case with SCA8.
Incomplete Penetrance
Several unaffected individuals were found to have a (CTG)n in the expanded range; two are very young (12 and 14 years) and two are still in their 30s; however, another unaffected subject (family LE), with a repeat size of 170 units, was 77 years old. Of note is also the fact that the 42-year-old, unaffected father of the girl with congenital ataxia (family CH) had one allele in the upper normal range (80 repeats) and a large expanded allele (170 repeats).
If the expanded (CTG)n is indeed the cause of ataxia in these families, possible explanations for incomplete penetrance include (1) somatic mosaicism with the presence of significantly different repeat sizes between affected tissues and peripheral blood, as is often seen in DM (Jansen et al. 1994); (2) a repeat length that is now different from that at the time of birth because of a progression of somatic mosaicism, as occurs in DM (Martorell et al. 1998); (3) operation of modifying factors (interruptions of the CTG tract, other polymorphisms, or imprinting); or (4) the fact that larger expansions may not be pathogenic, as suggested by Ranum et al. (1999) for CTG tracts >250 units; we now have some evidence that this threshold may be as low as 170 CTGs. The possibility still remains, however, that these older carriers with large expansions are not completely asymptomatic but may have other undetected phenotypes—for example, some form of psychiatric disorder, as described by Kennedy et al. (1999).
Intergenerational Instability
The expanded CTG tract showed instability during meiotic transmission (fig. 1), as is seen in other trinucleotide-repeat diseases (Tsilfidis et al. 1992; Chung et al. 1993; Duyao et al. 1993; Maciel et al. 1995; David et al. 1997). We could verify the origin of the expanded allele in six cases, all of which were paternal transmissions; thus, we could not confirm the maternal bias toward transmission of SCA8 reported by Koob et al. (1999). On the other hand, the expanded repeat showed a reduction in size in all instances but one (where its size remained the same); this is also in agreement with previous results, which showed a trend for contraction during paternal transmissions (Koob et al. 1999).
If there is indeed a threshold above which large expansions are not pathogenic, we must thus expect an additional bias for paternal transmission of SCA8, with contractions into the pathogenic range. We must consider, however, the alternative hypothesis that genomic imprinting may be in operation in this instance, leaving paternal offspring unprotected.
Intergenerational instability of the CTG repeat in the normal range, like the one documented in family CH (individuals I:2 and II:1; fig. 2), was also described at the DM locus (Dow et al. 1997; Meiner et al. 1998). Recently, meiotic instability of normal alleles was reported for the CAG repeat at the SCA7 locus (Giunti et al. 1999).
Sperm Studies
Our results from germinal mosaicism (in low-copy genome PCR of sperm cells) in a patient with 152/28 CTGs (fig. 4A) confirmed the trend for contraction on paternal transmission of expanded repeats seen in pedigrees. But we also detected a significant degree of germinal mosaicism of normal alleles, both from that patient and from the two controls (fig. 4B). This indicates that the SCA8 locus is very unstable, undergoing both frequent contractions and expansions; although contractions were more frequent, some expansions over the normal range (>91 CTGs) were observed in sperm cells of our normal controls (fig. 4B). These results enable us to predict the frequent occurrence of de novo mutations.
Implications for Genetic Counselling
All of these findings, if confirmed, will have strong implications on genetic counseling. The risk for the general population may not be negligible, whereas the risk for the offspring of affected fathers will probably be significantly less than 50%. Counseling will have to include data on penetrance, expansion and contraction rates, and parental origin. It would be reasonable that it include as well empiric information on allele-size change in the sperm of any given affected male. Any counselling based on information about the (CTG)n, however, should be postponed until confirmation of the present results and until the role of the expanded (CTG)n is no longer disputed (as in Durr et al. 1999).
Haplotype Studies
Analysis of markers spanning 2 cM within the SCA8-locus region showed the existence of distinct haplotypes associated with the expansion in each of the five families. These results further support the hypothesis of a high mutation rate at this locus. Haplotype analysis in sperm cells also needs to be performed, to assess accurately the parental origin of each allele and to quantify its mutation rate. Further studies are also needed to look for cis-acting factors, such as the polymorphic (CTA)n preceding the (CTG)n, interruptions in the CTG tract, or other polymorphisms that may have a role in stabilizing the expansion and reducing penetrance.
Acknowledgments
We thank Dr. M. Koob for providing us with positive controls for the SCA8 expansion. We are also particularly indebted to Dr. Vaz Osório (IGM), for supplying the blood spots on filter paper from the IGM national screening for PKU. C. Cunha was of great help with her secretarial assistance. We also thank the staff of the Neurobiology unit at IBMC for their technical support and all patients and their families for their cooperation. This work was supported by research grants PRAXIS/PSAU/C/SAU/084/96, PRAXIS/PSAU/C/SAU/13226/98, and Financiamento Plurianual de Unidades de I&DE, all from F.C.T. (Ministry of Science and Technology, Portugal). I.A., L.G., P.M., and C.S. were the recipients of scholarships from the PRAXIS Program, F.C.T., Portugal.
Electronic-Database Information
The accession number and URLs for data in this article are as follows:
- Center for Medical Genetics database, http://www.marshmed.org/genetics
- Fondation Jean Dausset CEPH database, http://www.cephb.fr [Google Scholar]
- Généthon, http://www.genethon.fr
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nim.nih.gov/omim (for SCA8 [MIM 603680])
References
- Ashizawa T, Anvret M, Baiget M, Barceló JM, Brunner H, Cobo AM, Dallapiccola B, et al (1994) Characteristics of intergenerational contractions of the CTG repeat in myotonic dystrophy. Am J Hum Genet 54:414–423 [PMC free article] [PubMed]
- Benomar A, Krols L, Stevanin G, Cancel G, LeGuern E, David G, Ouhabi H, et al (1995) The gene for autosomal dominant cerebellar ataxia with pigmentary macular dystrophy maps to chromosome 3p12-p21.1. Nat Genet 10:84–88 [DOI] [PubMed]
- Cancel G, Abbas N, Stevanin G, Dürr A, Chneiweiss H, Néri C, Duyckaerts C, et al (1995) Marked phenotypic heterogeneity associated with expansion of a CAG repeat sequence at the spinocerebellar ataxia 3/Machado-Joseph disease locus. Am J Hum Genet 57:809–816 [PMC free article] [PubMed]
- Chong SS, McCall AE, Cota J, Subramony SH, Orr HT, Hughes MR, Zoghbi HY (1995) Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet 10:344–350 [DOI] [PubMed]
- Chung M, Ranum LPW, Duvick LA, Servadio A, Zoghbi HY, Orr HT (1993) Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type 1. Nat Genet 5:254–258 [DOI] [PubMed]
- David G, Abbas N, Stevanin G, Dürr A, Yvert G, Cancel G, Weber C, et al (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17:65–70 [DOI] [PubMed]
- Deka R, Guangyun S, Wiest J, Smelser D, Chunhua S, Zhong Y, Chakraborty R (1999) Patterns of instability of expanded CAG repeats at the ERDA1 locus in general populations. Am J Hum Genet 65:192–198 [DOI] [PMC free article] [PubMed]
- Dow DJ, Rubinsztein DC, Yates JRW, Barton DE, Ferguson-Smith MA (1997) Instability of normal (CTG)n alleles in the DM kinase gene. J Med Genet 34:871–873 [DOI] [PMC free article] [PubMed]
- Durr A, Stevanin G, Herman A, Lebre A-S, Frontali M, Brice A (1999) Screening of the SCA8 expansion. Am J Hum Genet Suppl 65:A247 [Google Scholar]
- Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, et al (1993) Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet 4:387–392 [DOI] [PubMed]
- Gardner K, Alderson K, Galster B, Kaplan C, Leppert M, Ptácek L (1994) Autosomal dominant spinocerebellar ataxia: clinical description of a distinct hereditary ataxia and genetic localization to chromosome 16 (SCA4) in a Utah kindred. Neurology (Suppl 2) 44:A361 [PMC free article] [PubMed] [Google Scholar]
- Gispert S, Twells R, Orozco G, Brice A, Weber J, Heredero L, Scheufler K, et al (1993) Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1. Nat Genet 4:295–299 [DOI] [PubMed]
- Giunti P, Stevanin G, Worth PF, David G, Brice A, Wood NW (1999) Molecular and clinical study of 18 families with ADCA type II: evidence for genetic heterogeneity and de novo mutation. Am J Hum Genet 64:1594–603 [DOI] [PMC free article] [PubMed]
- Imbert G, Kretz C, Johnson K, Mandel J-L (1993) Origin of the expansion mutation in Myotonic Dystrophy. Nat Genet 4:72–76 [DOI] [PubMed]
- Imbert G, Saudau F, Yvert G, Devys D, Trottier Y, Garnier J-M, Weber C, et al (1996) Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 14:285–291 [DOI] [PubMed]
- Jansen G, Willems P, Coerwinkel M, Nillesen W, Smeets H, Vits L, Höwler C, et al (1994) Gonosomal mosaicism in myotonic dystrophy patients: involvement of mitotic events in (CTG)n repeat variation and selection against extreme expansion in sperm. Am J Hum Genet 54:575–585 [PMC free article] [PubMed]
- Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, et al (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8:221–228 [DOI] [PubMed]
- Kennedy JL, Neves-Pereira ML, Paterson AD, Yamamoto E, Parikh SV, Macciardi F, Petronis A, et al (1999) Trinucleotide repeat for SCA8 on 13q21: super expansion in psychosis individuals unaffected by ataxia. Am J Hum Genet Suppl 65:A278 [Google Scholar]
- Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, Takahashi H, et al (1994) Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat Genet 6:9–13 [DOI] [PubMed]
- Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LPW (1999) An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 21:379–384 [DOI] [PubMed]
- Maciel P, Gaspar C, DeStefano AL, Silveira I, Coutinho P, Radvany J, Dawson DM, et al (1995) Correlation between CAG repeat length and clinical features in Machado-Joseph disease. Am J Hum Genet 57:54–61 [PMC free article] [PubMed]
- Martorell L, Monckton DG, Gamez J, Johnson KJ, Gich I, de Munain AL, Baiget M (1998) Progression of somatic CTG repeat length heterogeneity in the blood cells of myotonic dystrophy patients. Hum Mol Genet 7:307–12 [DOI] [PubMed]
- Meiner A, Thamm B, Strenge S, Froster U (1998) Instability in the normal CTG repeat range at the myotonic dystrophy locus. J Med Genet 35:791 [DOI] [PMC free article] [PubMed]
- Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E, Bundo M, Takeda T, et al (1994) Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nat Genet 6:14–18 [DOI] [PubMed]
- Orr HT, Chung M, Banfi S, Kwiatkowski TJ, Jr., Servadio A, Beaudet AL, McCall AE, et al (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4:221–226 [DOI] [PubMed]
- Pulst S-M, Nechiporuk A, Nechiporuk T, Gispert S, Chen X-N, Lopes-Cendes I, Pearlman S, et al (1996) Moderate expansion of normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 14:269–275 [DOI] [PubMed]
- Ranum LPW, Moseley ML, Leppert MF, van den Engh G, La Spada AR, Koob MD, Day JW (1999) Massive CTG expansions and deletions may reduce penetrance of spinocerebelllar ataxia type 8. Am J Hum Genet Suppl 65:A466 [Google Scholar]
- Ranum LPW, Schut LJ, Lundgren JK, Orr HT, Livingston DM (1994) Spinocerebellar ataxia type 5 in a family descended from the grandparents of President Lincoln maps to chromosome 11. Nat Genet 8:280–284 [DOI] [PubMed]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, et al (1996) Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 14:277–283 [DOI] [PubMed]
- Silveira I, Coutinho P, Maciel P, Gaspar C, Hayes S, Dias A, Guimarães J, et al (1998) Analysis of SCA1, DRPLA, MJD, SCA2, and SCA6 CAG repeats in 48 Portuguese ataxia families. Am J Med Genet 81:134–138 [DOI] [PubMed]
- Silveira I, Lopes-Cendes I, Kish S, Maciel P, Gaspar C, Coutinho P, Botez MI, et al (1996) Frequency of spinocerebellar ataxia type 1, dentatorubropallidoluysian atrophy, and Machado-Joseph disease mutations in a large group of spinocerebellar ataxia patients. Neurology 46:214–218 [DOI] [PubMed]
- Takiyama Y, Nishizawa M, Tanaka H, Kawashima S, Sakamoto H, Karube Y, Shimazaki H, et al (1993) The gene for Machado-Joseph disease maps to human chromosome 14q. Nat Genet 4:300–304 [DOI] [PubMed]
- Tsilfidis C, MacKenzie AE, Mettler G, Barceló J, Korneluk RG (1992) Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat Genet 1:192–195 [DOI] [PubMed]
- Yue Q, Jen JC, Nelsen SF, Baloh RW (1997) Progressive ataxia due to a missense mutation in a calcium-channel gene. Am J Hum Genet 61:1078–1087 [DOI] [PMC free article] [PubMed]
- Zerylnick C, Torroni A, Sherman SL, Warren ST (1995) Normal Variation at the myotonic dystrophy locus in global human populations. Am J Hum Genet 56:123–130 [PMC free article] [PubMed]
- Zhang L, Leeflang EP, Yu J, Arnheim N (1994) Studying human mutations by sperm typing: instability of CAG trinucleotide repeats in the human androgen receptor gene. Nat Genet 7:531–535 [DOI] [PubMed]
- Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, et al (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the a1A-voltage-dependent calcium channel. Nat Genet 15:62–69 [DOI] [PubMed]
- Zoghbi HY, Pollack MS, Lyons LA, Ferrell RE, Daiger SP, Beaudet AL (1988) Spinocerebellar ataxia: variable age of onset and linkage to human leukocyte antigen in a large kindred. Ann Neurol 23:580–584 [DOI] [PubMed]
- Zu L, Figueroa KP, Grewal R, Pulst S (1999) Mapping of a new autosomal dominant spinocerebellar ataxia to chromosome 22. Am J Hum Genet 64:594–599 [DOI] [PMC free article] [PubMed]