

Abstract
More than 150 point mutations have now been identified in the ATP7A gene. Most of these mutations lead to the classic form of Menkes disease (MD), and a few lead to the milder occipital horn syndrome (OHS). To get a better understanding of molecular changes leading to classic MD and OHS, we took advantage of the unique finding of three patients with similar mutations but different phenotypes. Although all three patients had mutations located in the splice-donor site of intron 6, only two of the patients had the MD phenotype; the third had the OHS phenotype. Fibroblast cultures from the three patients were analyzed by reverse transcriptase (RT)–PCR to try to find an explanation of the different phenotypes. In all three patients, exon 6 was deleted in the majority of the ATP7A transcripts. However, by RT-PCR amplification with an exon 6–specific primer, we were able to amplify exon 6–containing mRNA products from all three patients, even though they were in low abundance. Sequencing of these products indicated that only the patient with OHS had correctly spliced exon 6–containing transcripts. We used two different methods of quantitative RT-PCR analysis and found that the level of correctly spliced mRNA in this patient was 2%–5% of the level found in unaffected individuals. These findings indicate that the presence of barely detectable amounts of correctly spliced ATP7A transcript is sufficient to permit the development of the milder OHS phenotype, as opposed to classic MD.
Introduction
Menkes disease (MD; MIM 309400) is a multisystemic lethal disorder of copper metabolism inherited as an X-linked recessive trait. The phenotypic features of MD are dominated by severe neurological degeneration and connective-tissue abnormalities (for review, see DiDonato and Sarkar 1997; Kodama et al. 1999). The disease is caused by mutations in the ATP7A gene, which encodes a Cu-transporting P-type ATPase involved in copper efflux from cells and in intracellular transport of copper to copper-requiring proteins (for review, see Pena et al. 1999). The ATP7A protein is localized to the trans-Golgi membrane (Yamaguchi et al. 1996) but translocates to the plasma membrane in response to increased copper concentrations (Petris et al. 1996). To a large extent, the clinical features of MD can be attributed to malfunction of one or more copper-requiring enzymes, such as lysyl oxidase, cytochrome c oxidase, and dopamine β-hydroxylase, caused by the deficiency of ATP7A (Kodama and Murata 1999).
In addition to the severe classic form of MD leading to death in early childhood, milder forms are observed in ∼5%–10% of the patients. Occipital horn syndrome (OHS) is the mildest allelic form of MD (Proud et al. 1996), and we are aware of 35–40 affected individuals in the world. The neurological symptoms of OHS patients are milder than those found in patients suffering from MD, leading to a clinical picture mainly characterized by connective-tissue manifestations.
To date, a total of 162 different mutations affecting ATP7A has been identified in 192 unrelated MD patients with the classic severe form or with one of the atypical phenotypes (reviewed in Tümer et al. 1999). Of these mutations, 7 were chromosome abnormalities and 35 were partial gene deletions (unpublished data). The total number of point mutations identified within ATP7A is 120, and ∼30% of these are splice-site mutations (Das et al. 1994, 1995; Kaler et al. 1994, 1995, 1996; Tümer et al. 1994, 1996, 1997; Levinson et al. 1996; Ronce et al. 1997; Qi and Byers 1998; Z. Tümer, L. B. Møller, N. Horn, C. Lund, unpublished data).
Until now, seven point mutations have been reported in patients who were diagnosed with OHS (Kaler et al. 1994; Das et al. 1995; Levinson et al. 1996; Ronce et al. 1997; Qi and Byers 1998); in five of these patients, the mutations were at the consensus splice-site sequences affecting the normal RNA splicing (Kaler et al. 1994; Das et al. 1995; Qi and Byers 1998). The milder phenotype of these patients could be explained by the presence of lower amount of normal transcripts or the presence of partially functional proteins.
To obtain a better understanding of the molecular defects leading to one form of MD or another, we made a comparative study of patients with similar ATP7A mutations but different clinical outcomes. Such a study requires identification of several mutations in a large patient cohort. The possibility of finding similar mutations leading to different phenotypes is not very high, taking into account the rarity of MD and especially that of OHS. However, we were able to make such a comparison, because we found one person with OHS and two with MD, all with mutations affecting the same splice-donor site of ATP7A.
The ATP7A-specific mRNAs produced in fibroblast cultures from the three patients were characterized and quantified by reveres transcriptase (RT)–PCR analysis to search for an explanation of the very different phenotypes. Only the cells from the patient with OHS were found to contain detectable amounts of correctly spliced ATP7A mRNA, the amount of which was determined by two different quantitative RT-PCR methods. One method involved the use of homologous competitor fragments (Siebert and Larrick 1992), whereas the other involved the use of a LightCycler, a real-time fluorescence detection system (Wittwer et al. 1997). We found, in agreement with previous studies, that the milder phenotype of the OHS patient could be correlated with the presence of normal ATP7A mRNA. However, to our surprise, the amount was only 2%–5% of the level found in unaffected individuals.
Materials and Methods
Patients
The clinical symptoms of patient MD1 suggested MD, and he was referred to the John F. Kennedy Institute for biochemical confirmation of the initial diagnosis when he was 6 mo old. In the child's neonatal period, hypoglycemia and repeated episodes of hypothermia were present. At the age of 8 wk, he was hospitalized because of feeding difficulties that were soon accompanied by therapy-resistant seizures. At 10 wk of age, his hair started to fall out. It was replaced by hair with an abnormal texture, raising suspicion of MD. Serum copper and ceruloplasmin levels were very low. Over the next few months, patient MD1 developed clinical features typical of MD, including subdural hematomas, a high arched palate, and wormian bones in the lambdoid suture of the occipital region. Bladder diverticulae were diagnosed at age 1.5 years. Copper histidine therapy was initiated when he was 8 mo old and continued until his death at age 21 mo. Copper incorporation studies confirmed the diagnosis of MD. Later, the patient's mother had an affected fetus diagnosed prenatally.
Patient MD2 was referred to the John F. Kennedy Institute at age 2 years for biochemical confirmation of the clinical diagnosis. At birth, a right-sided cephalohematoma was observed; the child had neonatal bilirubinemia that did not require therapy. Hypothermia was noted in the neonatal period and at age 1 year. By age 1 year, he had developed seizures and had symptoms of MD including hair changes, connective-tissue abnormalities comprising bilateral inguinal hernias, and typical bone changes such as pectus excavatum. Serum copper and ceruloplasmin values were low. He did not receive any copper therapy, and he died at age 33 mo. Copper-incorporation studies indicated MD. His family's history did not indicate that any members had suffered from MD. Subsequent DNA analysis showed that his mother did not carry the disease mutation.
Patient OS1 is presently aged 24 years. His clinical picture was typical of OHS, including narrow thorax, joint deformities, right-sided genital hernia, bladder diverticulae, vascular abnormalities, and chronic diarrhea. The occipital horns that name the disease were diagnosed when the patient was 18 years of age; the horns had a length of about 5 cm. The patient's skin was dry, loose, and hypopigmented and his hair was coarse, but other changes typical of OHS were not obvious. Recent complications were aneurysms of abdominal vessels, hepatic artery, and splenic artery, which were treated surgically. The patient was psychomotorically retarded, with psychotic traits (manic-depressive behavior). He was able to walk without support at age 3 years, and he started talking at age 3.5 years. Serum copper and ceruloplasmin levels were significantly below normal (<10 μmol/liter and 0.15 g/liter, respectively). Copper-incorporation studies showed abnormal accumulation and retention, confirming that patient OS1 suffered from a variant of MD. A brother had died at age 8 years; he had had similar connective-tissue disturbances and coarse hair but was more severely retarded than patient OS1 (Mentzel et al. 1999).
Samples
Skin samples were collected from the three patients for diagnostic investigation. Similar samples from unaffected male patients were used as controls. The fibroblast cells were cultured in a 1:1 mixture of RPMI 1640 with 20 mM N-2-hydroxyethylpiperazine-N′-2-ethane sulphonic acid buffer and nutrient mixture F-10 Ham's media, supplemented with 7.5% Amnio Max (Life Technologies) C100 supplement, 4% fetal calf serum, penicillin, and streptomycin.
PCR Amplifications
PCR reactions were generally performed in a reaction mixture containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, and 0.01% gelatin, with addition of primers, deoxynucleotides (dNTPs), and Taq polymerase (PE Biosystems), as indicated. Table 1 describes the DNA oligonucleotides used as primers in PCR reactions.
Table 1.
DNA Primers Used in PCR Reactions
| Primer Name | Sequence | Comment |
| MNK-1 | 5′-GCTCTAGAGAAACGTTGAGAGGAGCAAT-3′ | In exon 4 (forward direction); contains XbaI restriction site |
| MNK-2 | 5′-CCCAAGCTTTCACAGTGGCTCCAAATCCAAG-3′ | In exon 6 (reverse direction); contains HindIII restriction site |
| MNK-3 | 5′-CAAAAAGCAGCCCAAGTACCTC-3′ | In exon 4 (forward direction) |
| MNK-4 | 5′-GGTGGTTGCCAGCACAATCAGTACGTCC-3′ | In exon 10 (reverse direction) |
| MNK-5 | 5′-CAGAAGGGTCACAGCAAAGG-3′ | In exon 4 (forward direction) |
| MNK-6 | 5′-AGACAATCCTGGAAGAATCTGGCGC-3′ | In exon 9 (reverse direction) |
| MNK-7 | 5′-GTGATAGAAAATGCTGAT-3′ | In exon 6 (forward direction) |
| 18S-1 | 5′-GGGGTACCATGCATGTCTAAGTACGCAC-3′ | Contains KpnI restriction site |
| 18S-2 | 5′-CCCAAGCTTGGACACTCAGCTAAGAGCATC-3′ | Contains HindIII restriction site |
| GAPDH-1 | 5′-CCCAAGCTTTCCATGACAACTTTGGTATCGTGG-3′ | Contains HindIII restriction site |
| GAPDH-2 | 5′-CCGGAATTCGTCGCTGTTGAAGTCAGAGGAGAC-3′ | Contains EcoRI restriction site |
Detection of Mutations in ATP7A
Genomic DNA was isolated from cultured skin fibroblasts by the NaCl extraction method (Grimberg et al. 1989). Mutation screening was performed either by single-strand conformational polymorphism as described previously (Tümer et al. 1997) or by dideoxy fingerprinting modified after Sarkar et al. (1992). In brief, dideoxy fingerprinting was performed by PCR amplification of each exon with the previously described primers (Tümer et al. 1997). The PCR amplification of genomic DNA was performed in a total volume of 25 μl containing 100 nM of each primer, 20 μM dNTP, and 0.25 U Taq polymerase. A set of termination products was generated by performing a limited sequencing of the amplified exon with a single dideoxy terminator, 32P-labeled primer, and ThermoSequenase (United States Biochemical). The termination products were separated on a 6% nondenaturing mutation detection enhancement gel (FMC Bioproducts). Exons showing an abnormal band pattern were sequenced directly without further purification.
Preparation of RNA and cDNA
Total RNA or mRNA was isolated from 104–106 cells of cultured skin fibroblasts with the RNeasy Mini Kit (QIAgen) or MicroPoly(A) pure mRNA Isolation Kit (Ambion), respectively. Total RNA was eluted in 50 μl, whereas mRNA was eluted in 20 μl. Single-stranded cDNA was synthesized by reverse transcription of 7.5 μl RNA in a total volume of 20 μl with Superscript II RNase H− Reverse Transcriptase (GibcoBRL) and a mixture of random hexamer primers (Pharmacia). The RNA used was isolated from the following cell lines: B9729997H/B9836567H (control cells); D9523179H (MD1 cells); D9626782H (OS1 cells); and 119310634H (MD2 cells).
Characterization of ATP7A mRNA by RT-PCR
The cDNA amplifications were performed as nested PCR reactions, except for the quantitative determinations. The PCR amplification was done in a total volume of 50 μl containing 100 nM of each primer, 100 μM dNTP, and 2.5 U Taq polymerase. Two microliters of cDNA was used as template in the first PCR reaction, whereas 2 μl of the first PCR reaction was used as template in the second and final PCR reaction. The PCR primers used to analyze the skipping of exon 6 were MNK-3 and MNK-4 in the first PCR reaction, then MNK-5 and MNK-6 in the second (table 1). The PCR primers used to specifically amplify exon 6–containing products were MNK-4 and MNK-7 in the first PCR reaction, then MNK-6 and MNK-7 in the second. PCR amplifications were performed for 40 cycles (94°C for 30 s, 60°C for 1 min, and 72°C for 1 min) followed by 7 min at 72°C. The amplified products were separated on a 3% Metaphor agarose gel (FMC Bioproducts) and visualized by ethidium bromide staining.
Sequence Analysis of RT-PCR Products
The products were separated on a 1% agarose gel, and the fragments were excised from the gel and eluted in 50 μl of water at room temperature. Direct sequencing of the eluted fragment was performed with ThermoSequenase (United States Biochemical) and a [32P]-labeled primer.
Quantitative RT-PCR Analysis by Homologous Competitor DNA
The relative amounts of exon 6–containing ATP7A mRNAs in normal control cells and in cells from patient OS1 were determined by competitive RT-PCR (Siebert and Larrick 1992). As a competitor, we used a homologous plasmid, pMNKΔ, which contained a segment of ATP7A cDNA with a small internal deletion, that had been engineered in vitro. The amount of ATP7A mRNA was normalized to the amount of 18S rRNA in the same sample, which was measured in parallel with the homologous competitor plasmid, p18SΔ. The PCR reactions for determination of ATP7A transcript and 18S rRNA were performed in separate tubes with the primer pairs MNK-1 and MNK-2, and 18S-1 and 18S-2, respectively. In both cases, fourfold serial dilutions of the competitor DNA were added to PCR reactions containing equal amounts of target cDNA (2 μl of ATP7A cDNA or 1 μl of a 100-fold dilution of 18S cDNA). Because of the greater abundance of 18S rRNA compared with ATP7A mRNA, much higher concentrations of competitor DNA had to be used in the 18S reactions. The PCR amplifications were performed in a total volume of 20 μl containing 500 nM of each primer, 80 μM dNTP (for 18S) or 18 μM dNTP (for ATP7A), 2.5 U Taq polymerase, and 0.01 μCi of α-[32P]-dCTP. PCR amplifications were performed for 60 cycles (94°C for 30 s, 60°C for 1 min, and 72°C for 1 min) followed by 7 min at 72°C. The large number of cycles was required to get sufficient signals for quantitation, as the amount of exon 6–containing transcript in patient OS1 was very low. The amplified products were separated on a 3% agarose gel. The bands were visualized by staining with ethidium bromide, and the amount of radioactivity in each band was determined by a PhospoImager (Molecular Dynamics).
Construction of Plasmids Encoding Homologous Competitor Fragments
The ATP7A competitor plasmid, pMNKΔ, contained a segment of ATP7A cDNA (1439–1821, numbered according to Tümer et al. 1995) with a 30-bp deletion (1552–1581), which was introduced by gene SOEing (Horton 1993). This 353-bp–long cDNA fragment was amplified by use of the MNK-1 and MNK-2 primers (table 1), digested with HindIII and XbaI, and ligated into a pDP19 vector.
For construction of the 18S rRNA competitor plasmid, p18SΔ, a 735-bp PCR fragment (149–883, numbered according to GenBank accession number M10098) was amplified from cDNA derived from 18S rRNA by use of the 18S-1 and 18S-2 primers (table 1). The PCR product was cloned into pDP19 by use of KpnI and HindIII restriction enzymes, and a deletion of 133 bp was subsequently introduced by excision of an XbaI restriction fragment (263–395). The resulting plasmid thus contained a 602-bp 18S cDNA competitor fragment.
Quantitative RT-PCR Analysis with a Real-Time Fluorescence Detection System
Real-time quantitative PCR analysis was performed with a LightCycler Instrument with the DNA Master SYBR Green I mix according to the manufacturer's protocol (Roche Molecular Biochemicals). SYBR Green I is a double-strand–specific dye, and its fluorescence is greatly enhanced by its binding to double-stranded DNA. Thus, the amount of amplified PCR product can be estimated during each cycle of amplification by its fluorescence (Wittwer et al. 1997). PCR amplifications were performed on twofold serial dilutions of cDNA reverse transcribed from OS1 or control mRNA samples. The primers MNK-1 and MNK-2 were used for determination of the amount of ATP7A mRNA. The relative amounts of glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA in the same RNA samples were determined in parallel in separate tubes with the primers GAPDH-1 and GAPDH-2 (table 1), which give rise to a cDNA fragment of 376 bp (corresponding to 3978–5447 in the genomic sequence with removal of intron; GenBank accession number J04038). The PCR amplifications were performed in a total volume of 20 μl containing 2 μl of LightCycler DNA Master SYBR Green I, 4 mM MgCl2, 10 μg bovine serum albumin, 500 nM of each primer, and 5 μl of cDNA. The amplification parameters used were as follows: preheating at 95°C for 30 s, followed by 50 cycles of amplification (95°C for 2 s, 60°C for 7 s, 72°C for 35 s, and 79°C for 0 s, single-acquisition mode), followed by extension (72°C for 30 s), terminated by melting (95°C for 0 s, 65°C for 15 s, and 95°C for 0 s, continuous-acquisition mode). In the last segment in the melting file, the slope was 0.1°C/s; in all other cases, the slope was 20°C/s. The fluorescence settings for gains F1, F2, and F3 were 5, 15, and 30, respectively. The obtained data were analyzed by the second derivative maximum method.
Results
Identification of Genomic Mutations in the ATP7A Gene
The three patients investigated in this study were chosen because they had very different clinical phenotypes in spite of the fact that they all had a mutation at the splice-donor site of exon 6 (IVS6,DS). Two of the patients (MD1 and MD2) had the classic severe form of MD, whereas the third patient (OS1) had the milder OHS form.
The ATP7A mutations of the patients were identified with genomic DNA isolated from skin fibroblasts. In patient MD1, an abnormal band pattern of exon 6 was detected with single-strand conformational polymorphism, and, in patients MD2 and OS1, a mutation was predicted with dideoxy fingerprinting. To identify the mutations, exon 6, in each patient, was amplified with intron-specific primers and sequenced on both strands with the same primers. The normal sequence of splice-donor site of exon 6 is TTgtaagtaaga (Dierick et al. 1995; Tümer et al. 1995). In patient OS1, one of two 4-bp tandem repeats (taag) was deleted at the splice-donor site, whereas patients MD1 and MD2 contained base pair substitutions at positions +1 and +5, respectively (table 2).
Table 2.
Molecular Findings and Phenotype Related to Three Patients with IVS6,DS Mutations
| Individual | Nucleotide Sequence at IVS6,DSa | Effect on mRNA | Cryptic Splice-Site Sequencea,b | Phenotype |
| Control | GTTgtaagtaagat | Normal mRNA | Normal | |
| OS1 | GTTgtaagtaagat→ GTTgtaagat | Skipping of exon 6, leading to frameshift | OHS | |
| Normal mRNA | ||||
| MD1 | GTTgtaagtaagat→GTTataagtaagat | Skipping of exon 6, leading to frameshift | MD | |
| Skipping of exons 6 and 7, leading to frameshift | ||||
| Cryptic splice site, leading to frameshift | GTTATAAgtaagat (exon 6 enlarged with 4 bp) | |||
| MD2 | GTTgtaagtaagat→GTTgtaaataagat | Skipping of exon 6, leading to frameshift | MD | |
| Skipping of exons 6 and 7, leading to frameshift | ||||
| Cryptic splice site, leading to frameshift | GTTGTAAATAAGATTTTTTGTGTGATTAATAAAACTTCCAGAAAAAAAACTTGgtaaggt (exon 6 enlarged with 50 bp) |
Lowercase letters indicate sequence in introns, whereas uppercase letters indicate sequence in exons.
Boldface letters indicate the sequence included in exon 6 caused by the new exon/intron boundaries.
Exon Skipping in the Three Patients
The predicted effect of a mutation at the IVS6 donor site is the skipping of exon 6. To determine whether exon 6 was skipped in all three patients, a cDNA segment from exon 4 to exon 9 was amplified by RT-PCR (fig. 1). Amplification of the control mRNA from an unaffected individual resulted in a single product of normal size (1,300 bp), but the results obtained from the three patients diverged from the normal. The OS1 patient had two transcripts, a major product of 1,150 bp, and a minor product of 1,300 bp. Patient MD1 revealed three transcripts: a major product of 1,150 bp and two minor products of 1,300 bp and 1,000 bp, respectively. Patient MD2 had a 1,150-bp major product and a 1,000-bp minor product, and a smear was observed at 1,300 bp instead of a distinct band. Sequencing of the 1,150-bp major product of each patient revealed skipping of exon 6, since exons 5 and 7 were precisely spliced. The 1,000-bp band observed in both of the patients with MD was a consequence of concomitant skipping of exons 6 and 7. Skipping of exon 6 alone (164 bp) or in combination with exon 7 (164 + 162 bp) leads to a shift in reading frame, thus resulting in a premature termination of translation. Attempts to sequence the 1,300-bp bands present in patients OS1 and MD1 were not successful because of contamination with the major bands lacking exon 6.
Figure 1.

Analyses of exon skipping in patients OS1, MD1, and MD2. The region from exon 4 to exon 9 in the ATP7A cDNA sequence was PCR-amplified from cDNA obtained from the three patients (see Materials and Methods). cDNA from an unaffected person was used as a control (C). The PCR products were separated on a 3% Metaphor gel and visualized by ethidium bromide staining. Lane M, φX174DNA-HaeIII digest.
Characterization of Exon 6–Containing ATP7A Transcripts
It was crucial to determine whether the normal or a cryptic splice site was activated in the apparently normal transcript that gave rise to the 1,300-bp PCR product in patients OS1 and MD1. Furthermore, it should be clarified whether a normal transcript could have been present in the 1,300-bp region of the gel, where a smear was observed instead of a distinct band in patient MD2. To investigate these issues, we performed RT-PCR with primers located in exons 6 and 9, which exclusively would allow amplification of exon 6–containing cDNAs, even though they were in low abundance (fig. 2).
Figure 2.
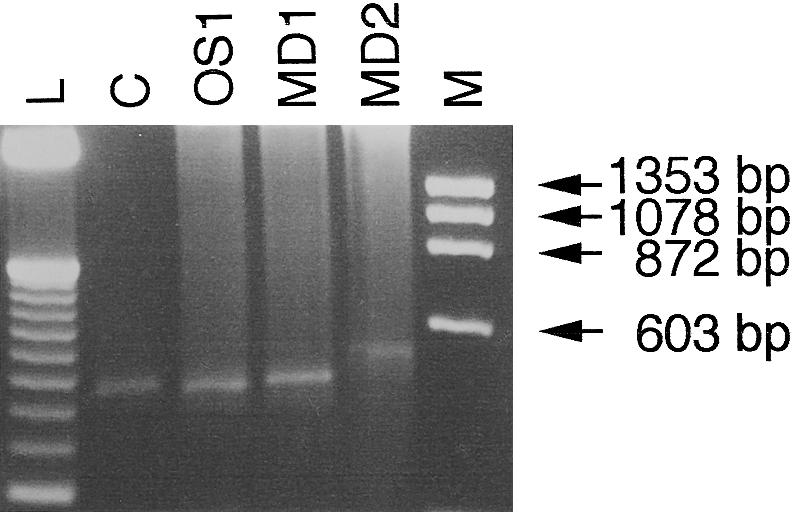
Amplification of exon 6–containing transcripts from patients OS1, MD1, and MD2. Exon 6–containing cDNAs from three patients were amplified by the use of primers located in exons 6 and 9 (see Materials and Methods). cDNA obtained from an unaffected person was used as a control (C). The PCR products were separated on a 3% Metaphor gel and visualized by ethidium bromide staining. Lane M, φX174DNA-HaeIII digest; lane L, 50-bp DNA ladder (Gibco BRL).
The size of the RT-PCR product of the patients OS1 and MD1 and of the control individual was ∼450 bp. However, in the sample from patient MD2, a larger band of ∼500 bp was observed. Sequencing of the RT-PCR product from patient OS1 revealed a correctly spliced exon 6–containing product, as was the case with the control individual. The RT-PCR product from patient MD1 represented an alternatively spliced transcript that used a cryptic donor site, leading to an extension of exon 6 with four nucleotides. Sequencing of the 500-bp fragment from patient MD2 showed activation of another cryptic splice site, resulting in enlargement of exon 6 by 50 nucleotides. Use of the cryptic splice sites in both cases causes a frameshift, resulting in premature termination of the reading frame. Furthermore, examination of the sequences showed that both transcripts were spliced at the first available cryptic splice site downstream of the mutations (table 2).
In summary, these results showed that the OHS patient could be clearly distinguished from the MD patients by the presence of some amount of correctly spliced ATP7A mRNA. The amount of normal transcript in the OHS patient was determined by two different quantitative RT-PCR–based methods, as described in the following two sections.
Quantitation of ATP7A mRNA by Competitive RT-PCR
The amount of correctly spliced ATP7A mRNA in cells from the patient with OHS was determined by competitive RT-PCR, in which reverse transcription of the mRNA is followed by competitive PCR on the generated cDNA. In competitive PCR, a DNA fragment containing the same primer template sequences as the target competes for primer binding and amplification. The amount of target DNA in a PCR reaction can thus be estimated from the amount of PCR product formed from target DNA relative to the amount of PCR product generated from a known amount of homologous competitor DNA (Bouaboula et al. 1992; Nedelman et al. 1992; Siebert and Larrick 1992).
As competitor to ATP7A cDNA we used a plasmid, pMNKΔ, containing an ATP7A cDNA fragment from exon 4 to exon 6 with an internal deletion of 30 bp. With the PCR primers MNK-1 and MNK-2, amplification of the ATP7A target and competitor DNA resulted in PCR products of 383 bp or 353 bp, respectively (fig. 3). In addition to these two products, a slowly migrating third product appeared. This band apparently represented heteroduplexes formed between target and competitor PCR products, because it was resolved into two bands of 383 and 353 bp when excised and electrophoresed on a denaturing polyacrylamide gel (data not shown). Formation of heteroduplexes frequently occurs when the competitor DNA is homologous to the target (Henco and Heibey 1990).
Figure 3.

Determination of the amount of ATP7A transcript in patient OS1 by competitive RT-PCR. Total RNA isolated from patient OS1 and control fibroblasts, respectively, was used for the synthesis of cDNA. By use of primers MNK-1 and MNK-2, equal amounts of cDNA were amplified in the presence of increasing amounts of homologous ATP7A competitor DNA, pMNKΔ (from left to right). Quantitation of the reaction products was accomplished by labeling with α-[32P]-dCTP included in the PCR reactions. A, Ethidium bromide–stained agarose gel with separated ATP7A RT-PCR products from control fibroblast. “T” and “C” indicate the positions of the target and competitor PCR products, respectively, and HD represents a heteroduplex formed between these. B, Ethidium bromide–stained agarose gel with separated ATP7A RT-PCR products from patient OS1. C, Ratio of PCR products generated from target and competitor DNA, respectively, as a function of the amount of competitor plasmid DNA added to the PCR reactions. The amount of the two different products in the heteroduplex band was taken into account in the calculation of the product ratios. The ratio of target/competitor product has been plotted only for lanes in which both signals are above background—that is, lanes 2–7 in the OHS experiment and lanes 4–9 in the control experiment.
The amount of ATP7A mRNA was normalized to the amount of 18S rRNA in the same sample determined by competitive RT-PCR. We chose 18S rRNA as an internal standard because the abundance of actin mRNA, which is frequently used for this purpose, is known to be altered in people with MD (Hamalainen et al. 1996). As competitor in the 18S rRNA determinations, we used a plasmid, p18SΔ, containing an 18S cDNA fragment with an internal deletion of 133 bp. With the 18S-1 and 18S-2 primers (table 1), the target RNA gave rise to an RT-PCR product of 735 bp, whereas the competitor gave a product of only 602 bp (fig. 4A,B).
Figure 4.

Determination of the amount of 18S rRNA by competitive RT-PCR. Total RNA isolated from OS1 and control fibroblasts, respectively, was used for the synthesis of cDNA. By use of primers 18S-1 and 18S-2, equal amounts of cDNA were amplified in the presence of increasing amounts of homologous 18S competitor DNA, p18SΔ (from left to right). Reaction products were quantitated as in figure 3. A, Ethidium bromide–stained agarose gel with separated 18S RT-PCR products from control fibroblast. T and C indicate the positions of target and competitor PCR products, respectively. B, Ethidium bromide–stained agarose gel with separated 18S RT-PCR products from patient OS1. C, Ratio of PCR products generated from target and competitor DNA, respectively, as a function of the amount of competitor plasmid DNA added to the PCR reactions. The ratio of target/competitor product has been plotted only for lanes in which both signals are above background—that is, lanes 4–7 in either experiment.
As shown in figure 3C, the amount of competitor DNA required to give equal formation of competitor and target PCR products was ∼60-fold higher for the control RNA sample compared with the RNA sample from the OHS patient. However, the determination of 18S rRNA (fig. 4C) indicated that the concentration of total RNA in the control sample was slightly higher (∼20%). Taking this into account, we estimate from this experiment that the amount of correctly spliced ATP7A mRNA in the patient with OHS is ∼2% of the level found in control cells. Two similar experiments with different samples of either total RNA or polyA-containing messenger RNA also yielded estimates in the range of 2%–4% of the normal level (data not shown).
Quantitation of ATP7A mRNA by Real-Time Monitoring of the Formation of Fluorescent RT-PCR Products
It was remarkable that the tiny amount of normal ATP7A mRNA in the patient with OHS was apparently sufficient to permit development of the milder phenotype as opposed to the more severe symptoms of classic MD. To validate this result, we measured the relative amount of exon 6–containing ATP7A transcript by another method, which involved real-time monitoring of the formation of fluorescent RT-PCR products in a LightCycler apparatus (see Materials and Methods). By this method, we determined the threshold number of PCR cycles required to give detectable exponential accumulation of PCR product for different amounts of input cDNA derived from OS1 or control RNA samples. These experiments were performed with mRNA samples because we observed an inhibition of the PCR reactions if samples of total RNA were used. Consequently, we could not use 18S rRNA for normalization, so instead we measured the amount of GAPDH mRNA in parallel amplification reactions.
As shown in figure 5A, the threshold cycle numbers for the OS1 sample were comparable to the values obtained for control samples that were 50-fold more dilute. However, the determinations of GAPDH mRNA (fig. 5B) indicated that the RNA sample from the control cells was 2.5-fold more concentrated than the OS1 sample. Thus, the relative amount of correctly spliced ATP7A mRNA in patient OS1 is estimated from this experiment to be ∼5% of the normal level. In another experiment using this methodology, an estimate of 2.5% was obtained (data not shown). Thus, cells from patient OS1 were found to contain 20- to 50-fold less correctly spliced ATP7A mRNA than did normal control cells, as determined both by competitive RT-PCR and by LightCycler amplifications.
Figure 5.

Quantitation of ATP7A mRNA by real-time monitoring of the formation of fluorescent RT-PCR products. cDNA was reverse transcribed from mRNA isolated from patient OS1's cells or from control cells, respectively. The amount of exon 6–containing ATP7A transcripts or GAPDH mRNA was estimated from twofold serial dilutions of the cDNAs. A, Plot of the threshold cycle number for the ATP7A amplification as a function of the dilution of the input cDNA sample. B, Plot of the threshold cycle number for the GAPDH amplification as a function of the dilution of the input cDNA sample.
Discussion
In this study, we have shown that patients with mutations in the same splice donor site of the ATP7A gene may have different clinical phenotypes—classic MD or milder OHS—depending on the presence of a normal transcript. In the patients with MD who we studied, normal ATP7A mRNA was not present at all, but in the patient with OHS, normally processed transcript could be detected, albeit in severely reduced amounts corresponding to 2%–5% of the normal level.
The consensus sequence for a 5′ splice-donor site is A58G78g100t100a/g96a71g84t47, where exon nucleotides are shown in uppercase and intron nucleotides in lowercase letters, and the numbers indicate the percentage occurrence of the different nucleotides (Ketterling et al. 1999). The wild-type sequence of the splice-donor site of exon 6 is TTgtaagt (Dierick et al. 1995; Tümer et al. 1995); that is, the junction matches the consensus sequence perfectly, except for the two nucleotides in the exon. It is noteworthy that the mutations in the two MD patients (TTataagt in patient MD1 and TTgtaaat in patient MD2) affected two of the most strongly conserved nucleotides in the splice site consensus sequence, whereas the splice site in patient OS1 (TTgtaaga) deviated from the wild-type sequence at the least conserved position (+6) in the intron. This may explain the use of cryptic splice sites in the patients with MD whom we studied, whereas some correctly spliced ATP7A transcripts could be detected in the patient with OHS.
To our knowledge, normal ATP7A transcripts have not been detected in any patients suffering from classic MD, although the presence of normal transcripts has been reported for three of five patients with OHS with splice-site mutations (Kaler et al. 1994; Das et al. 1995; Levinson et al. 1996). The amount of normal transcript was determined in only one of these cases; it was found to be 19% of the normal level by ribonuclease protection analysis (Kaler et al. 1994). Thus, we found it remarkable that cells from patient OS1 that we examined in the present work contained as little as 2%–5% of the normal amount of full-length mRNA. From the short clinical descriptions, it is not possible to correlate in any simple way the severity of the clinical symptoms of these two patients with OHS with their abundance of ATP7A mRNA. However, we do not think there is any contradiction between these results. Rather, they suggest that ATP7A mRNA concentrations up to 20% of the normal level may be sufficient to permit development of the mild OHS phenotype, whereas the MD phenotype obtains when functional mRNA is essentially absent. It remains to be shown how much normal transcript is needed to avoid disease symptoms entirely.
In two of the previously reported patients with OHS, normal ATP7A mRNA was not present (Kaler et al. 1994; Qi and Byers 1998). In one patient, the mutation was a single-base-pair substitution within the splice-site sequence of exon 11 (Ser833Gly). Besides two abnormal-sized transcripts, a normal-size transcript was present, but this transcript included the mutation (Kaler et al. 1994). This missense mutation was not within a region coding for a conserved motif, and its effect on the function of the protein was probably not severe. In the patient with OHS described by Qi and Byers (1998), a normal-size transcript was not present. The abnormal transcript was missing exon 10, which is alternatively spliced in normal individuals (Dierick et al. 1995). The protein product missing transmembrane domains 3 and 4 encoded by exon 10 was localized to the endoplasmic reticulum (Qi and Byers 1998) instead of the Golgi complex, where the normal ATP7A resides (Petris et al. 1996; Yamaguchi et al. 1996; Dierick et al. 1997; Francis et al. 1998). The milder phenotype of the patient suggests that at least some of the enzymes, other than lysyl oxidase and dopamine β-hydroxylase, could receive copper even though ATP7A is mislocalized. Thus, these two patients probably produced at least some partially functional ATP7A protein.
In contrast, none of the present patients contained any transcripts that are likely to encode partially functional protein, which might have complicated the interpretation of the results. All transcripts, including the major exon 6–deleted transcript, identified in the two patients suffering from classic MD could only encode severely truncated proteins, consistent with the severe phenotype. Except for the exon 6–deleted mRNA, the only transcript observed in the patient with OHS was the correctly spliced mRNA. Thus, we are confident that the mild phenotype of this patient was due to the presence of the small amount of normal ATP7A mRNA.
In several preliminary experiments, we performed RT-PCR amplifications with primers located in exons 4 and 9, but we were not always able to detect the exon 6–containing product in patient OS1 and MD1, and we were never able to detect it in patient MD2. This was probably because the large majority of ATP7A transcripts lacking exon 6 was preferentially amplified at the expense of the small fraction of transcripts containing exon 6. However, we found that this problem could be avoided by performing RT-PCR with primers located in exons 6 and 9, so that only exon 6–containing transcripts would be amplified. This strategy may be generally useful for identification and quantitation of very small amounts of specific mRNAs.
Acknowledgments
This research was supported by grants from the National Research Council, the Danish Health Insurance Foundation, Novo Nordisk Foundation, and the Foundation of 1870.
Electronic-Database Information
The accession number and URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for MD, MIM 309400) [PubMed]
References
- Bouaboula M, Legoux P, Pességué B, Delpech B, Dumont X, Piechaczyk M, Casellas P, et al (1992) Standardization of mRNA titration using a polymerase chain reaction method involving co-amplification with a multispecific internal control. J Biol Chem 267:21830–21838 [PubMed] [Google Scholar]
- Das S, Levinson B, Whitney S, Vulpe C, Packman S, Gitschier J (1994) Diverse mutations in patients with Menkes disease often lead to exon skipping. Am J Hum Genet 55:883–889 [PMC free article] [PubMed] [Google Scholar]
- Das S, Levinson B, Vulpe C, Whitney S, Gitschier J, Packman S (1995) Similar splicing mutations of the Menkes/mottled copper-transporting ATPase gene in occipital horn syndrome and the blotchy mouse. Am J Hum Genet 56:570–576 [PMC free article] [PubMed] [Google Scholar]
- DiDonato M, Sarkar B (1997) Copper transport and its alterations in Menkes and Wilson disease. Biochim Biophys Acta 1360:3–16 [DOI] [PubMed] [Google Scholar]
- Dierick HA, Adam AN, Escara-Wilke JF, Glover TW (1997) Immunocytochemical localization of the Menkes copper transport protein (ATP7A) to the trans-Golgi network. Hum Mol Genet 6:409–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierick HA, Ambrosini L, Spencer J, Glover TW, Mercer JFB (1995) Molecular structure of the Menkes disease gene (ATP7A). Genomics 28:462–469 [DOI] [PubMed] [Google Scholar]
- Francis MJ, Jones EE, Levy ER, Ponnambalam S, Chelly J, Monaco AP (1998) A Golgi localization signal identified in the Menkes recombinant protein. Hum Mol Genet 7:1245–1252 [DOI] [PubMed] [Google Scholar]
- Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A (1989) A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res 17:8390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamalainen ER, Kuiva Niemie H, Tromp G, Pihla Janimen T, Kimppainen R, Kivirikko KI (1996) Expression of mRNAs for lysyl oxidase and type III procollagen in cultured fibroblast from patients with Menkes and occipital horn syndrome as determined by quantitative polymerase chain reaction. Arch Biochem Biophys 328:101–106 [DOI] [PubMed] [Google Scholar]
- Henco K, Heibey M (1990) Quantitative PCR: the determination of template copy numbers by temperature gradient gel electrophoresis (TGGE). Nucleic Acids Res 18:6733–6734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM (1993) In vitro recombination and mutagenesis of DNA: SOEing together tailor-made genes. In: White BA (ed) PCR protocols: current methods and applications, pp 251–261 [DOI] [PubMed] [Google Scholar]
- Kaler SG, Buist NRM, Holmes CS, Goldstein DS, Miller RC, Gahl WA (1995) Early copper therapy in classic Menkes disease patients with a novel splicing mutation. Ann Neurol 38:921–928 [DOI] [PubMed] [Google Scholar]
- Kaler SG, Das S, Levinson B, Goldstein DS, Holmes CS, Patronas NJ, Packman S, et al (1996) Successful early copper therapy in Menkes disease associated with a mutant transcript containing a small in-frame deletion. Biochem Mol Med 57:37–46 [DOI] [PubMed] [Google Scholar]
- Kaler SG, Gallo LK, Proud VK, Percy AK, Mark Y, Segal NA, Goldstein DS, et al (1994) Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat Genet 8:195–202 [DOI] [PubMed] [Google Scholar]
- Ketterling RP, Drost JB, Scaringe WA, Liao D-Z, Liu J-Z, Kasper CK, Sommer SS (1999) Reported in vivo splice-site mutations in the factor IX gene: severity of splicing defects and a hypothesis for predicting deleterious splice donor mutations. Hum Mutat 13:221–231 [DOI] [PubMed] [Google Scholar]
- Kodama H, Murata Y (1999) Molecular genetics and pathophysiology of Menkes disease. Pediatr Int 41:430–435 [DOI] [PubMed] [Google Scholar]
- Kodama H, Murata Y, Kobayashi M (1999) Clinical manifestations and treatment of Menkes disease and its variants. Pediatr Int 41:423–429 [DOI] [PubMed] [Google Scholar]
- Levinson B, Conant R, Schnur R, Das S, Packman S, Gitschier J (1996) A repeated element in the regulatory region of the MNK gene and its deletion in a patient with occipital horn syndrome. Hum Mol Genet 5:1737–1742 [DOI] [PubMed] [Google Scholar]
- Mentzel HJ, Seidel J, Vogt S, Vogt L, Kaiser WA (1999) Vascular complications (splenic and hepatic artery aneurysms) in the occipital horn syndrome: report of a patient and review of the literature. Pediatr Radiol 29:19–22 [DOI] [PubMed] [Google Scholar]
- Nedelman J, Heagerty P, Lawrence F (1992) Quantitative PCR with internal controls. Comput Appl Biosci 8:65–70 [DOI] [PubMed] [Google Scholar]
- Pena MMO, Lee J, Thiele DJ (1999) A delicate balance: homeostatic control of copper uptake and distribution. J Nutr 129:1251–1260 [DOI] [PubMed] [Google Scholar]
- Petris MJ, Mercer JFB, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J (1996) Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane. a novel mechanism of regulated trafficking. EMBO J 15:6084–6095 [PMC free article] [PubMed] [Google Scholar]
- Proud VK, Mussell HG, Kaler SG, Young DW, Percy AK (1996) Distinctive Menkes disease variant with occipital horns: delineation of natural history and clinical phenotype. Am J Med Genet 65:44–51 [DOI] [PubMed] [Google Scholar]
- Qi M, Byers PH (1998) Constitutive skipping of alternatively spliced exon 10 in the ATP7A gene abolishes Golgi localization of the Menkes protein and produces the occipital horn syndrome. Hum Mol Genet 7:465–469 [DOI] [PubMed] [Google Scholar]
- Ronce N, Moizard MP, Robb L, Toutain A, Villard L, Moraie C (1997) A C2055T transition in exon 8 of the ATP7A gene is associated with exon skipping in an occipital horn syndrome family. Am J Hum Genet 61:233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar G, Yoon H-S, Sommer SS (1992) Dideoxy fingerprinting (ddF): a rapid and efficient screen for the presence of mutations. Genomics 13:441–443 [DOI] [PubMed] [Google Scholar]
- Siebert PD, Larrick W (1992) Competitive PCR. Nature 359:557–558 [DOI] [PubMed] [Google Scholar]
- Tümer Z, Horn N, Tønnesen T, Christodoulou J, Clarke JTR, Sarker B (1996) Early copper-histidine treatment for Menkes disease. Nat Genet 12:11–13 [DOI] [PubMed] [Google Scholar]
- Tümer Z, Lund C, Tolshave J, Vural B, Tønnesen T, Horn N (1997) Identification of point mutations in 41 unrelated patients affected with Menkes disease. Am J Hum Genet 60:63–71 [PMC free article] [PubMed] [Google Scholar]
- Tümer Z, Møller LB, Horn N (1999) Mutation spectrum of ATP7A, the gene defective in Menkes disease. In: Leone A, Mercer JFB (eds) Copper transport and its disorders: molecular and cellular aspects. Kluwer Academic/Plenum Publishers, New York, pp 83–95 [DOI] [PubMed] [Google Scholar]
- Tümer Z, Tønnesen T, Horn N (1994) Detection of genetic defects in Menkes disease by direct mutation analysis and its implications in carrier diagnosis. J Inherit Metab Dis 17:267–270 [DOI] [PubMed] [Google Scholar]
- Tümer Z, Vural B, Tønnesen T, Chelly J, Monaco AP, Horn N (1995) Characterization of the exon structure of the Menkes disease gene using Vectorette PCR. Genomics 26:437–442 [DOI] [PubMed] [Google Scholar]
- Wittwer CT, Ririe KM, Andrew RV, David DA, Gundry RA, Balis UJ (1997) The LightCycler: microvolume multisample fluorimeter with rapid temperature control. Biotechniques 22:176–181 [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Heiny ME, Suzuki M, Gitlin J (1996) Biochemical characterization and intracellular localization of the Menkes disease protein. Proc Natl Acad Sci USA 93:14030–14035 [DOI] [PMC free article] [PubMed] [Google Scholar]