

Abstract
Classical Ehlers-Danlos syndrome (EDS) is characterized by skin hyperelasticity, joint hypermobility, increased tendency to bruise, and abnormal scarring. Mutations in type V collagen, a regulator of type I collagen fibrillogenesis, have been shown to underlie this type of EDS. However, to date, mutations have been found in only a limited number of patients, which suggests genetic heterogeneity. In this article, we report two unrelated patients with typical features of classical EDS, including excessive skin fragility, in whom we found an identical arginine→cysteine substitution in type I collagen, localized at position 134 of the α1(I) collagen chain. The arginine residue is highly conserved and localized in the X position of the Gly-X-Y triplet. As a consequence, intermolecular disulfide bridges are formed, resulting in type I collagen aggregates, which are retained in the cells. Whereas substitutions of glycine residues in type I collagen invariably result in osteogenesis imperfecta, substitutions of nonglycine residues in type I collagen have not yet been associated with a human disease. In contrast, arginine→cysteine substitutions in type II collagen have been identified in a variety of chondrodysplasias. Our findings show that mutations in other fibrillar collagens can be causally involved in classical EDS and point to genetic heterogeneity of this disorder.
The Ehlers-Danlos syndrome (EDS) comprises a heterogeneous group of disorders affecting skin, ligaments and joints, and blood vessels (Steinmann et al. 1993). The most recent classification recognizes six subtypes, most of which have been linked with genetic defects affecting one of the fibrillar collagens (Beighton et al. 1998). Mutations in type III collagen have been identified in the vascular type of EDS (EDS IV [MIM 130050]) (Kuivaniemi et al. 1997). Structural mutations affecting the procollagen I N-proteinase cleavage site were found in the rare arthrochalasis variants of EDS (EDS VII A and B [MIM 130060]) (Byers et al. 1997), whereas defects of the enzymes lysylhydroxylase and procollagen I N-proteinase are implicated in the recessive occuloscoliotic (EDS VI [MIM 225400]) (Kuivaniemi et al. 1997) and dermatosparaxis (EDS VII C [MIM 225410]) (Colige et al. 1999) variants, respectively.
Insights into the molecular basis of the common, classical types of EDS (EDS I and II [MIM 130000 and MIM 130010]) have only recently become available. These EDS subtypes are characterized mainly by hyperelasticity of the skin, hyperlaxity of the joints, increased tendency to bruise, and abnormal scarring. Transgenic mouse experiments (Andrikopoulos et al. 1995) provided the first evidence for the causal involvement of type V collagen in this disorder. So far, however, ⩽10 mutations in the COL5A1 and COL5A2 genes have been reported in patients with classical EDS (Nicholls et al. 1996; Wenstrup et al. 1996; De Paepe et al. 1997; Burrows et al. 1998; Michalickova et al. 1998; Richards et al. 1998). This raises the question of genetic heterogeneity in this EDS variant. In this study, we found an identical arginine→cysteine substitution in the α1(I) collagen chain in two unrelated patients with classical EDS. These findings provide the first evidence for the causal involvement of another fibrillar collagen in this disorder.
Patient 1, a 5-year-old girl and the only child of a healthy nonconsanguineous couple, was born at >37 wk of gestation, after premature rupture of membranes. She had a history of easy bruising and scarring after minimal trauma and presented a soft, velvety, and hyperextensible skin. In addition, she had atrophic paper scars on the face, elbows, knees, and shins; ecchymoses on the lower legs; and generalized joint hyperlaxity. Her facial appearance, which included redundant skin folds on the eyelids and very soft earlobes, was reminiscent of classical EDS. Her sclerae were white, and X-ray examination indicated that she had no signs of osteoporosis.
Patient 2 was a 7-year-old boy, the only affected child in a sibship of four children from healthy, nonconsanguineous parents. He was born at >37 wk of gestation and showed hypotonia in the first months of life. An operation was performed for strabismus. When clinically examined at age 5 years, he had typical features of classical EDS, including a soft and doughy skin texture, moderate skin hyperextensibility, and joint hyperlaxity. In addition, he had a pronounced tendency for splitting of the skin, easy bruising, and impaired wound healing. He also presented an unusual tenderness of the skin and soft tissues, evident when he was touched. He had a pectus excavatum and flat feet. His sclerae were white, and radiographic examination showed no signs of osteoporosis.
Ultrastructural studies performed on a skin biopsy from patient 2 showed, on transverse section, a distinct variability in the size of the collagen fibrils, with their diameter in the range of 70–250 nm (fig. 1A). Some of the fibrils were thicker and showed an irregular outline. Longitudinal sections showed the presence of granulofilamentous material along the fibrils (fig. 1B). In some foci, collagen fibrils had an unraveled aspect and ran in a disorganized pattern. Mild dilatation of the rough endoplasmic reticulum was seen.
Figure 1 .

Ultrastructural findings in the skin of patient 2. A, On the transverse section (28,400×), the collagen fibrils show a distinct variability in diameter; some fibrils are larger and irregular in outline (arrowhead). B, On the longitudinal section (28,400×), the deposition of granulofilamentous material along the collagen fibrils is visible (arrowhead). In some foci, collagen fibrils have an unraveled and disorganized aspect (arrow).
A dermal fibroblast culture was established from both patients, and collagen and procollagen were studied by SDS-PAGE after metabolic labeling with 14C-proline. Analysis of the collagen and procollagen molecules secreted into the medium showed a normal pattern for type I and type III collagen compared with control cell lines. In patient 1, analysis of the cell-layer collagens showed the presence of an additional band corresponding to dimeric collagen α-chains (fig. 2), which migrated higher up in the gel. In patient 2, a similar collagen profile was seen, although the additional band representing the collagen dimers was of lesser intensity. The dimers disappeared after reduction with β-mercaptoethanol, which suggests the presence of disulfide-bounded collagen α-chains. Two-dimensional cyanogen bromide peptide mapping showed that this additional band was composed of α1(I) collagen chains (data not shown). The thermal stability of the collagen dimers was decreased by 1°C compared with the normal α1(I) collagen chains.
Figure 2 .
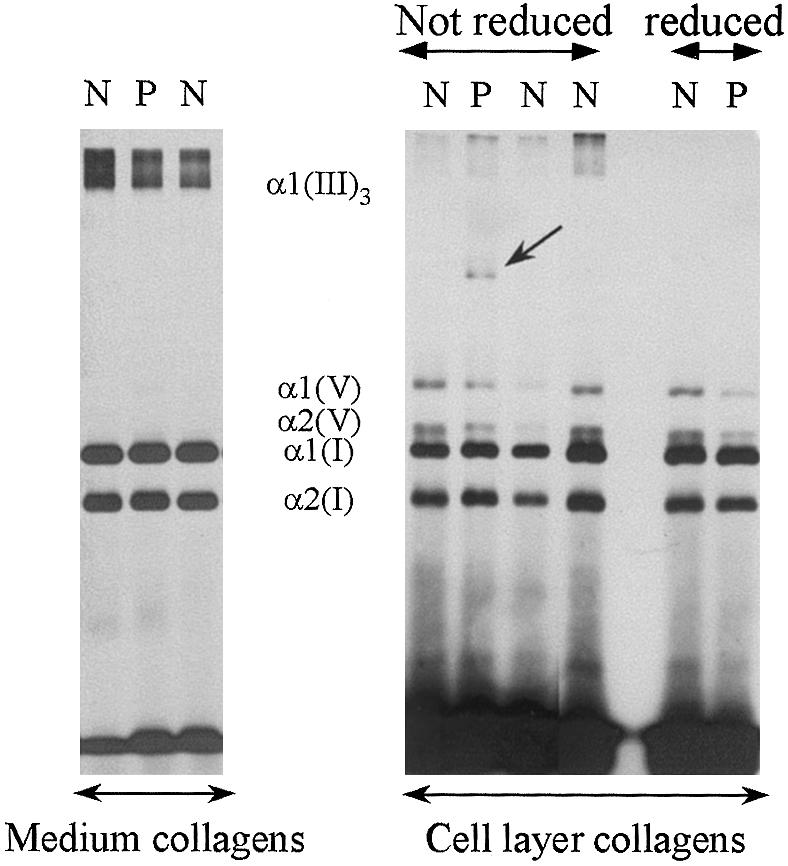
SDS-PAGE of radiolabeled, pepsin-digested collagens from the medium (left) and the cell layer (right), produced by the fibroblasts of patient 1. The pattern of the medium collagens is comparable to that of controls. In the cell layer, a population of dimeric collagen α-chains is seen (arrow) that disappears after reduction. P = patient 1; N = control samples.
Mutation analysis of the COL1A1 cDNA sequence was performed, and in half the clones a C→T transition (nucleotide 934→exon 14) was identified, which resulted in the substitution of arginine (CGC) for cysteine (TGC) in position 134 of the procollagen α1(I) chain (fig. 3). Analysis of the sequencing of the genomic DNA confirmed the mutation in both patients and showed it to be absent in the parents. Because the α1(I)-R134C mutation was not detectable by restriction digestion, genomic DNA from 50 control individuals was investigated by SSCP analysis for the COL1A1–exon 14 area and surrounding sequences, but no abnormal migration shift was seen. The mutation was not found in 30 unrelated patients with classical EDS in whom a COL5A1 and COL5A2 mutation screening was negative. Mutation analysis of the COL3A1, COL5A1, and COL5A2 cDNA was normal in both patients.
Figure 3 .

Sequencing data from patient 1 representing the normal (upper panel) and the mutant (lower panel) COL1A1 allele. A C→T transition changes the codon for arginine (CGC), in position 134 of the proα1(I) collagen chain, to the codon for cysteine (TGC).
The mutation identified in both EDS patients was a substitution of a basic arginine in the X position of the Gly-X-Y triplet of the type I collagen α1 chain by an uncharged polar cysteine, an amino acid residue, which is not normally present in the triple helical domain of type I collagen. The most common type of mutation in type I collagen is the substitution of a glycine residue by a bulkier amino acid, which has invariably been associated with osteogenesis imperfecta (OI) (Kuivaniemi et al. 1997). The presence of a glycine residue in every third position of the collagen α-chain is essential for correct folding of the helical domain of fibrillar collagen molecules. Glycine→cysteine substitutions in the α1(I) collagen chain are easily detected on biochemical analysis by the presence of an additional band migrating in the upper part of the gel, which consists of disulfide-bounded α1(I) collagen chains.
In this study, however, the cysteine substitution involves an arginine residue in the X position of the Gly-X-Y triplet. As such, the cysteine residue projects on the outside of the triple helix, enabling the formation of intermolecular disulfide bridges between two adjacent collagen molecules. This results in the formation of molecular aggregates that cannot be secreted efficiently. This may explain the dilatation of the endoplasmic reticulum and the fact that the extra band representing the dimeric type I collagen α-chains is detectable only in the cell-layer samples and not in the medium samples of the patient’s fibroblasts.
Mutant type I collagen molecules, which are not involved in the intermolecular aggregates but harbor one or two unbounded sulfhydryl groups, may cause local intracellular perturbations and, once secreted, may interfere with normal physiological interactions of the extracellular matrix components, disturbing normal collagen fibril assembly. Glycine→cysteine substitutions in the α1(I) or α2(I) collagen chain, which cause OI, act mainly at the level of molecular folding and fibril formation, whereas the substitution of a nonglycine residue by a cysteine most probably affects the molecular packing of the collagen fibrils and fibers through the presence of unbounded sulfhydryl groups.
The fact that we identified this substitution in two unrelated patients and found it to be absent in the unaffected parents and 50 control individuals supports a causal role for this mutation in classical EDS. Moreover, the arginine residue involved is a highly conserved amino acid in most of the fibrillar collagens and is therefore probably important for normal fibrillar collagen assembly.
So far, the substitution of a nonglycine residue in type I collagen has not been associated with a clinical phenotype in humans. However, in type II collagen, the substitution of an arginine by a cysteine residue is associated with a variety of chondrodysplasia phenotypes, including spondyloepiphyseal dysplasia and Stickler-like syndromes (Chan et al. 1993; Williams et al. 1993; Ballo et al. 1998).
Mutations in the COL1A1 and COL1A2 gene that abolish normal processing of the procollagen I N-proteinase cleavage site have been shown to cause the rare arthrochalasis variant of EDS. A certain degree of clinical overlap between this form of EDS and OI exists, since arthrochalasis patients commonly present osteoporosis, whereas patients with OI may show joint laxity and some degree of skin hyperextensibility and abnormal scarring. The children with EDS whom we studied, however, lacked the major features of the arthrochalasis type of EDS and showed no signs of osteoporosis.
To date, only mutations in the COL5A1 and COL5A2 genes have been identified in patients with classical EDS. Type V collagen is a quantitatively minor component among the fibrillar collagens, and it is coexpressed with type I collagen in skin and tendon (Birk et al. 1990). Type V collagen regulates the diameter of type I collagen fibrils through its huge N-propeptide domain, which protrudes from the surface of the type I collagen fibrils, in which the type V molecules are buried (Birk et al. 1990). Since the α1(I)-R134C substitution results in the same clinical phenotype as that associated with type V mutations, it is possible that the mutation disturbs the regulatory function of type V collagen in normal collagen fibrillogenesis. Indeed, the ultrastructural abnormalities in the skin from patient 2 showed similar collagen fibril abnormalities, as seen in the EDS patients in whom a type V collagen mutation was identified.
The identification of a structural mutation in type I collagen in two unrelated patients with classical EDS provides formal proof of the genetic heterogeneity of this EDS subtype. Moreover, recent studies, including knock-out experiments, suggest that other connective-tissue components, such as decorin (Danielson et al. 1997), thrombospondin-2 (Kyriakides et al. 1998), lumican (Chakravarti et al. 1998), and tenascin-X (Burch et al. 1997), may be causally involved in classical EDS.
In conclusion, our findings show that the substitution of a nonglycine residue in type I collagen can cause classical EDS. Further study of the contribution of type I collagen defects in this EDS subtype is warranted.
Acknowledgments
We are grateful to K. Wettinck, M. Van Thielen, and N. Aslan for excellent technical assistance. This work is supported by the Fund for Scientific Research, Flanders, project G.0090.99N.
Electronic-Database Information
Accession numbers and URL for data in this article are as follows:
References
- Andrikopoulos K, Liu X, Keene DR, Jaenisch R, Ramirez F (1995) Targeted mutation in the COL5A2 gene reveals a regulatory role for type V collagen during matrix assembly. Nat Genet 9:31–36 [DOI] [PubMed] [Google Scholar]
- Ballo R, Beighton PH, Ramesar R (1998) Stickler-like syndrome due to a dominant negative mutation in the COL2A1 gene. Am J Med Genet 80:6–11 [DOI] [PubMed] [Google Scholar]
- Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup R (1998) Ehlers-Danlos syndromes: revised nosology, Villefranche 1997. Am J Med Genet 77:31–37 [DOI] [PubMed] [Google Scholar]
- Birk DE, Fitch JM, Babiarz JP, Doane KJ, Linsenmayer TF (1990) Collagen fibrillogenesis in vitro: interaction of types I and V collagen regulates fibril diameter. J Cell Sci 95:649–657 [DOI] [PubMed] [Google Scholar]
- Burch GH, Gong Y, Liu W, Dettmann RW, Curry CJ, Smith L, Miller WL, et al (1997) Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet 17:104–108 [DOI] [PubMed] [Google Scholar]
- Burrows NP, Nicholls AC, Richards AJ, Luccarini C, Harrison JB, Yates JR, Pope FM (1998) A point mutation in an intronic branch site results in aberrant splicing of COL5A1 and in Ehlers-Danlos syndrome type II in two British families. Am J Hum Genet 63:390–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers PH, Duvic M, Atkinson M, Robinow M, Smith LT, Krane SM, Greally MT, et al (1997) Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am J Med Genet 72:94–105 [DOI] [PubMed] [Google Scholar]
- Chakravarti S, Magnuson T, Lass JH, Jepsen KJ, LaMantia C, Carroll H (1998) Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J Cell Biol 141:1277–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D, Taylor TKF, Cole WG (1993) Characterization of an arginine 789 to cysteine substitution in alpha1(II) collagen chains of a patient with spondyloepiphyseal dysplasia. J Biol Chem 268:15238–15245 [PubMed] [Google Scholar]
- Colige A, Sieron AL, Li S-W, Schwarze U, Petty E, Wertelecki W, Wilcox W, et al (1999) Human Ehlers-Danlos Syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet 65:308–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV (1997) Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol 136:729–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paepe A, Nuytinck L, Hausser I, Anton-Lamprecht I, Naeyaert JM (1997) Mutations in the COL5A1 gene are causal in the Ehlers-Danlos syndromes I and II. Am J Hum Genet 60:547–554 [PMC free article] [PubMed] [Google Scholar]
- Kuivaniemi H, Tromp G, Prockop DJ (1997) Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum Mutat 9:300–315 [DOI] [PubMed] [Google Scholar]
- Kyriakides TR, Zhu YH, Smith LT, Bain SD, Yang Z, Lin MT, Danielson KG, et al (1998) Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, and increased vascular density, and a bleeding diathesis. J Cell Biol 140:419–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalickova K, Susic M, Willing MC, Wenstrup RJ, Cole WG (1998) Mutations of the alpha2(V) chain of type V collagen impair matrix assembly and produce Ehlers-Danlos syndrome type I. Hum Mol Genet 7:249–255 [DOI] [PubMed] [Google Scholar]
- Nicholls AC, Oliver JE, McCarron S, Harrison JB, Greenspan DS, Pope FM (1996) An exon skipping mutation of a type V collagen gene (COL5A1) in Ehlers-Danlos syndrome. J Med Genet 33:940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AJ, Martin S, Nicholls AC, Harrison JB, Pope FM, Burrows NP (1998) A single base mutation in COL5A2 causes Ehlers-Danlos syndrome type II. J Med Genet 35:846–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann B, Royce PM, Superti-Furga A (1993) The Ehlers-Danlos syndrome. In: Royce PM, Steinmann B (eds) Connective tissue and its heritable disorders. Wiley-Liss, New York, pp 351–407 [Google Scholar]
- Wenstrup RJ, Langland GT, Willing MC, D’Souza VN, Cole WG (1996) A splice-junction mutation in the region of COL5A1 that codes for the carboxyl propeptide of pro alpha 1(V) chains results in the gravis form of the Ehlers-Danlos syndrome (type I). Hum Mol Genet 5:1733–1736 [DOI] [PubMed] [Google Scholar]
- Williams CJ, Considine EL, Knowlton RG, Reginato A, Neumann G, Harrison D, Buxton P, et al (1993) Spondyloepiphyseal dysplasia and precocious osteoarthritis in a family with an Arg75→Cys mutation in the procollagen type II gene (COL2A1). Hum Genet 92:499–505 [DOI] [PubMed] [Google Scholar]