

Summary
We describe a variance-components method for multipoint linkage analysis that allows joint consideration of a discrete trait and a correlated continuous biological marker (e.g., a disease precursor or associated risk factor) in pedigrees of arbitrary size and complexity. The continuous trait is assumed to be multivariate normally distributed within pedigrees, and the discrete trait is modeled by a threshold process acting on an underlying multivariate normal liability distribution. The liability is allowed to be correlated with the quantitative trait, and the liability and quantitative phenotype may each include covariate effects. Bivariate discrete-continuous observations will be common, but the method easily accommodates qualitative and quantitative phenotypes that are themselves multivariate. Formal likelihood-based tests are described for coincident linkage (i.e., linkage of the traits to distinct quantitative-trait loci [QTLs] that happen to be linked) and pleiotropy (i.e., the same QTL influences both discrete-trait status and the correlated continuous phenotype). The properties of the method are demonstrated by use of simulated data from Genetic Analysis Workshop 10. In a companion paper, the method is applied to data from the Collaborative Study on the Genetics of Alcoholism, in a bivariate linkage analysis of alcoholism diagnoses and P300 amplitude of event-related brain potentials.
Introduction
The availability of suites of correlated phenotypes for many common multifactorial traits can facilitate the study of these traits, by providing information beyond what is contained in the traits individually. In principle, statistical genetic analyses in which the correlations between the phenotypes are explicitly modeled can provide greater power than that provided by univariate analysis of the individual traits. Multitrait analysis can also improve the detection of quantitative-trait loci (QTLs) whose effects are too small to be found in single-trait analyses, and it can facilitate the investigation of genetic mechanisms such as pleiotropy and close linkage (Jiang and Zeng 1995; Mangin et al. 1998). Joint genetic linkage analysis of multiple correlated traits has been shown to improve the power to detect, localize, and estimate the effect of genes jointly influencing a complex disease (Amos et al. 1990; Amos and Laing 1993; Schork 1993; Jiang and Zeng 1995; Korol et al. 1995; Weller et al. 1996; Almasy et al. 1997; Blangero et al. 1997; De Andrade et al. 1997; Wijsman and Amos 1997; Mangin et al. 1998).
Multitrait analysis is not a panacea, however. In general, joint analysis of multiple traits increases the number of model parameters that must be estimated, and the additional df increase the critical value of the test statistic required to achieve a given level of statistical significance. These factors can offset the potential gains from joint consideration of the correlated characters, with the result that multitrait analysis may actually be less powerful than single-trait analysis, even with traits that are highly correlated (Mangin et al. 1998). If the correlations between traits are not appropriately handled—for example, by true multivariate analysis or by means of orthogonal canonical variables—then some correction for multiple nonindependent tests must be applied to control the rate of type I error (Ducrocq and Besbes 1993; Weller et al. 1996).
A frequently encountered situation in which the potential benefits of joint multitrait linkage analysis are likely to be realized is that of a discrete disease trait and a correlated quantitative character. Many multifactorial diseases—such as diabetes, glaucoma, hypertension, schizophrenia, and alcoholism—are conventionally studied as qualitative traits but are also associated with correlated quantitative precursors, physiological risk factors, or other biological markers. Although the quantitative character may not be used explicitly to define the qualitative disease state, both kinds of information are useful and mutually supportive (Moldin 1994; Ott 1995). A quantitative risk factor may lend a degree of classificatory flexibility to the clinical diagnosis of the disease, whereas a definitive clinical presentation on the basis of established criteria can be valuable in elucidation of physiological relationships between the disease state and related quantitative measures.
Considerable precedent exists for the joint analysis of qualitative and quantitative traits in statistical genetics, particularly in the areas of segregation analysis and parametric linkage analysis (e.g., see Morton and MacLean 1974; Elston et al. 1975; Lalouel et al. 1985; Bonney et al. 1988; Borecki et al. 1990; Moldin et al. 1990; Blangero et al. 1992; Ott 1995). In the present report we describe a variance-components method for joint multipoint linkage analysis of correlated discrete and continuous traits in pedigrees of arbitrary size and complexity, and we illustrate the properties of the method by using simulated pedigree data from Genetic Analysis Workshop 10 (Goldin et al. 1997). In contrast to fully parametric penetrance-based linkage methods for which a mode of inheritance must be specified, the variance-components approach requires fewer parameters to be estimated (Lander and Schork 1994; Blangero 1995; Tiwari and Elston 1997).
We also describe likelihood-based tests for pleiotropy—that is, whether the same QTL influences both the discrete-trait state and the quantitative phenotype—and for close or coincident linkage—that is, whether there is independent linkage of the traits to distinct, nonpleiotropic genes that happen to be linked (Jiang and Zeng 1995; Mangin et al. 1998). In a companion paper (Williams et al. 1999 [in this issue]) we apply the qualitative-quantitative–trait linkage method to bivariate data on alcoholism and P300 amplitude of event-related brain potentials from the Collaborative Study on the Genetics of Alcoholism (Begleiter et al. 1995).
Multivariate Analysis of Quantitative and Qualitative Traits
Variance-components linkage analysis of a collection of discrete and continuous traits can be approached as a form of bivariate (i.e., two-trait) analysis in which one variable represents the (possibly multivariate) discrete-trait state and the other variable represents a (possibly multivariate) correlated quantitative character. Following previous usage (Lange and Boehnke 1983; Boehnke et al. 1986; Lange 1997), we use the terms “univariate,” “bivariate,” and “multivariate” to refer to the number of phenotypes analyzed, although analysis of a single phenotype in a pedigree containing more than one individual can involve multivariate distributions in the strict statistical sense.
The theoretical foundation for polygenic multivariate quantitative-trait variance-components analysis was described by Lange and Boehnke (1983) and Boehnke et al. (1986). Extensions of the variance-components approach to multipoint linkage analysis were later introduced by Goldgar (1990), Schork (1993), Amos (1994), and Almasy and Blangero (1998). In this section we review briefly the theoretical framework for multipoint variance-components linkage analysis of multivariate phenotypes, and then we show how the method can be modified to accommodate a mixture of qualitative and quantitative data. For simplicity, the bivariate problem of a discrete trait and an associated continuous biological marker is used for illustration, but the model is easily extended to include additional variables of either type. For the special case of univariate discrete or continuous data, the multivariate qualitative-quantitative model reduces to the expected univariate variance-components model.
Bivariate Quantitative-Trait Linkage Analysis
Let x=(x1,…,xm)′ and y=(y1,…,yn)′ be the pedigree-trait vectors for two quantitative phenotypes X and Y, measured on m and n individuals, respectively. For a univariate variance-components linkage analysis, we assume that x and y are each normally distributed with means μX=(μx1,μx2,…,μxm) and μY=(μy1,μy2,…,μyn) and variance-covariance matrices ΣX and ΣY. The log likelihoods (ln L) of the phenotypes X and Y considered individually are then given by
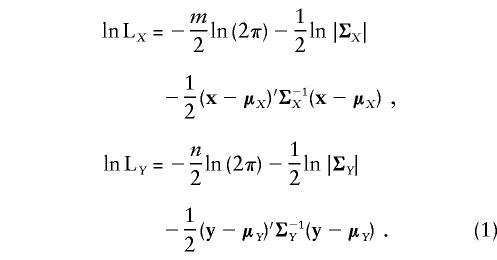 |
We note in passing that there are theoretical reasons for assuming within-pedigree multivariate normality (Lange 1978), as well as many indications that the assumption is robust to distributional violations (Beaty et al. 1985; Searle et al. 1992; Amos et al. 1996; Iturria et al., in press). The assumption of multivariate normality can be examined empirically (Gnanadesikan 1977; Hopper and Mathews 1982; Beaty et al. 1985, 1987), and univariate or multivariate normality of the phenotypes can be induced by data transformations (Andrews et al. 1971; Boehnke and Lange 1984; Clifford et al. 1984; Boehnke et al. 1986). Alternatively, one can either implement robust estimators of standard errors for the model parameters (Beaty et al. 1985, 1987; Beaty and Liang 1987) or reformulate the pedigree likelihood in terms of the multivariate t distribution (Lange et al. 1989). Provided that the phenotypic distribution is not patently discontinuous (e.g., because it contains extreme outliers) or characterized by extreme kurtosis, the assumption of multivariate normality of the pedigree phenotypic vector has been found to be remarkably robust to reasonable distributional violations (Iturria et al., in press; J. Blangero, unpublished data).
The vector means in equation (1) are typically modeled as functions of covariate information on the pedigree members; that is, μX=1mαX+WXβX, μY=1nαY+WYβY, where 1m is a vector of m 1's and, for each trait X and Y, α is the grand mean; W is the design matrix whose m or n rows contain l covariates such as age, sex, and environmental factors for each pedigree member; and β is the l×1 vector of regression coefficients. For linkage analysis, the covariance matrices can be modeled as
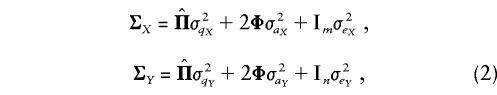 |
where 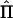 is a matrix whose elements
is a matrix whose elements 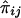 are the estimated proportion of genes shared identical by descent (IBD), at the QTL, by individuals i and j; σ2q is the additive genetic variance due to the major locus; Φ is the kinship matrix for the pedigree; σ2a is the variance due to residual additive genetic effects (i.e., polygenes and, potentially, other major genes); I is the identity matrix; and σ2e is the variance due to random, individual-specific environmental effects (Lange et al. 1976; Amos 1994; Almasy and Blangero 1998).
are the estimated proportion of genes shared identical by descent (IBD), at the QTL, by individuals i and j; σ2q is the additive genetic variance due to the major locus; Φ is the kinship matrix for the pedigree; σ2a is the variance due to residual additive genetic effects (i.e., polygenes and, potentially, other major genes); I is the identity matrix; and σ2e is the variance due to random, individual-specific environmental effects (Lange et al. 1976; Amos 1994; Almasy and Blangero 1998).
The matrix 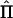 in equation (2) is an estimate of the true IBD-sharing matrix Π (whose elements are necessarily 0, 1/2, or 1) and is determined, by use of a regression-based approach, on the basis of (a) the estimated IBD matrices at a set of markers and (b) the distances of the markers from the QTL (Fulker et al. 1995; Almasy and Blangero 1998). The covariance matrices in equation (2) model the resemblance between relatives that is due solely to the effect of a major gene on a background of residual additive genetic effects and individual-specific environmental variation, but additional genetic and environmental effects and interactions are easily incorporated (Hopper and Mathews 1982; Blangero 1993; Williams and Blangero 1999).
in equation (2) is an estimate of the true IBD-sharing matrix Π (whose elements are necessarily 0, 1/2, or 1) and is determined, by use of a regression-based approach, on the basis of (a) the estimated IBD matrices at a set of markers and (b) the distances of the markers from the QTL (Fulker et al. 1995; Almasy and Blangero 1998). The covariance matrices in equation (2) model the resemblance between relatives that is due solely to the effect of a major gene on a background of residual additive genetic effects and individual-specific environmental variation, but additional genetic and environmental effects and interactions are easily incorporated (Hopper and Mathews 1982; Blangero 1993; Williams and Blangero 1999).
If phenotypes X and Y are correlated, univariate analysis of the individual phenotypes disregards the additional information implicit in the correlational structure. A bivariate analysis in which this phenotypic correlation is explicitly modeled will exploit more of the information content of the data and will improve the power of statistical tests for hypotheses of interest.
To implement a bivariate variance-components analysis, assume that the composite phenotype Z has the pedigree-trait vector 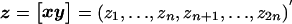 , which follows a 2n-variate normal distribution with mean μZ and covariance matrix ΣZ. The log likelihood of the bivariate data z is then
, which follows a 2n-variate normal distribution with mean μZ and covariance matrix ΣZ. The log likelihood of the bivariate data z is then
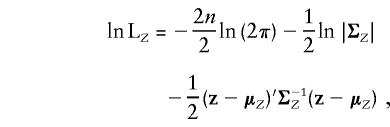 |
where
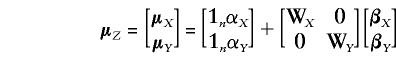 |
and α, W, and β have their previous definitions and interpretation. Separate design matrices and vectors of regression coefficients are retained for each trait, to emphasize that different sets of covariates could be used for the discrete and the continuous data; for example, smoking or alcohol consumption might be a significant covariate for one of the traits but not for the other.
The covariance matrix for Z has the partitioned structure
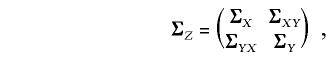 |
where ΣX and ΣY are as in equation (2), and the matrix ΣXY=ΣYX of cross-covariances is given by
Note that the cross-covariance matrix ΣXY is defined only when m=n—that is, when both phenotypes X and Y have been measured for each pedigree member. This restriction can be relaxed, however, by appropriate evaluation of the pedigree likelihood; this is discussed further below.
The natural bounds on the cross-covariances in equation (3) are 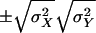 . The parameterization in terms of cross-covariances is computationally inconvenient, however (Boehnke et al. 1986), and a parameterization in terms of correlations is achieved by writing σ2XY=σXσYρXY, where ρXY is the correlation between traits X and Y. The covariance matrix for Z can then be expressed compactly, as
. The parameterization in terms of cross-covariances is computationally inconvenient, however (Boehnke et al. 1986), and a parameterization in terms of correlations is achieved by writing σ2XY=σXσYρXY, where ρXY is the correlation between traits X and Y. The covariance matrix for Z can then be expressed compactly, as 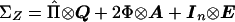 , where matrices Q, A, and E are the QTL, polygenic, and environmental variance components, respectively, and ⊗ is the Kronecker-product operator (Searle 1971). In general, when k traits in a pedigree having n individuals are being considered, matrices
, where matrices Q, A, and E are the QTL, polygenic, and environmental variance components, respectively, and ⊗ is the Kronecker-product operator (Searle 1971). In general, when k traits in a pedigree having n individuals are being considered, matrices 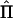 , Φ, and I have dimension n×n; matrices Q, A, and E are k×k; and ΣZ is nk×nk. In a univariate analysis Q, A, and E reduce to their scalar equivalents σ2q, σ2a, and σ2e. In a bivariate analysis Q, A, and E are 2×2 matrices of the form
, Φ, and I have dimension n×n; matrices Q, A, and E are k×k; and ΣZ is nk×nk. In a univariate analysis Q, A, and E reduce to their scalar equivalents σ2q, σ2a, and σ2e. In a bivariate analysis Q, A, and E are 2×2 matrices of the form
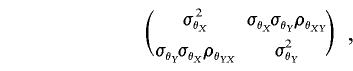 |
where ρθXY is the correlation, between X and Y, that is due to effect θ, and θ is q, a, or e; thus, ρaXY is the correlation between the additive genetic components of the two traits, ρeXY is the correlation between the individual trait environmental factors, and ρqXY is the correlation between the major-gene effects.
Joint Analysis of Qualitative and Quantitative Data
When all phenotypic data are of a single type—that is, are either quantitative or qualitative in nature—the likelihood of the complete pedigree data can be specified without any special partitioning of the variables. When some phenotypes are continuous and others are discrete, however, it becomes convenient to partition the total likelihood into factors descriptive of each type of data and to develop each factor accordingly.
To modify the bivariate quantitative approach outlined above for the situation involving mixed discrete and continuous data, associate one of the pedigree phenotype vectors (x, say) with the continuous phenotype and the other (y) with an underlying, continuous liability value on the basis of which the discrete trait is determined by a threshold process (Wright 1934a, 1934b; Crittenden 1961; Falconer 1965, 1989; Mendell and Elston 1974; for alternative approaches to polychotomous traits, see Morton et al. 1970). The joint likelihood of observing a particular configuration of continuous phenotype values and discrete-trait statuses within a pedigree can be factored as L(x,y)=L(x)L(y|x), where L(x) is the likelihood of observing the continuous data on the pedigree members and L(y|x) is the conditional likelihood of observing liabilities consistent with the affection statuses of the pedigree members, given their values for the continuous phenotype. No distributional assumptions are implied by this factorization, although multivariate normality of the joint distribution for x and y must be invoked so that the separate factors can themselves be modeled as multivariate normal.
If the notation and assumptions of the previous section are used, the marginal likelihood of the continuous data x can be written as
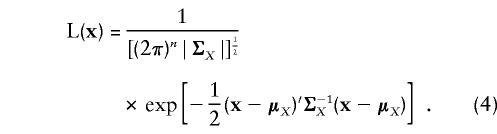 |
The likelihood of the discrete data for an individual i is given by the integral of the liability function over a range determined by the individual's trait status; thus,
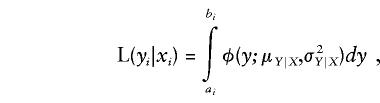 |
where φ(y;μY|X,σ2Y|X) denotes the univariate-normal probability-density function centered at μY|X with conditional variance σ2Y|X, and ai and bi are the threshold values on the liability distribution between which the individual will express the observed trait. For a dichotomous trait
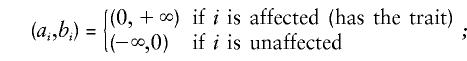 |
the model is completely general, however, and readily accommodates polychotomous data (Hasstedt 1993).
For a pedigree of n individuals, the conditional likelihood of the discrete data is given by the multiple integral
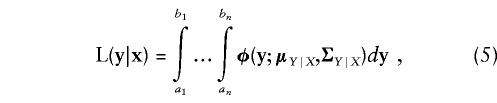 |
where φ(y;μY|X,ΣY|X) is the multivariate-normal density function having conditional mean μX|Y and covariance matrix ΣY|X. The mean vector and covariance matrix for the conditional liability distribution are determined by means of a standard result for conditioning a multivariate normal on a subset of its variables (Searle 1971; Tong 1990); thus, μY∣X=μY+ΣYXΣ-1X(x-μX) and ΣY∣X=ΣY-ΣYXΣ-1XΣXY, where μX, μY, ΣX, ΣY, and ΣYX=ΣXY have their previous meanings. The conditional mean liability μY|X is, in general, different for each individual, depending on any covariates that are introduced as fixed effects.
The likelihoods in equations (4) and (5) are finally multiplied to give the total joint likelihood of the discrete and continuous observations in a pedigree:
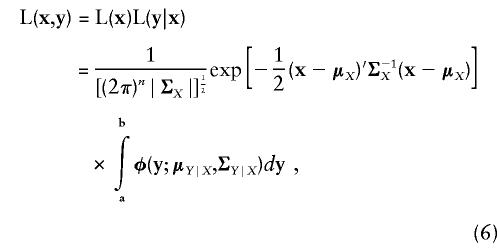 |
where vector limits of integration a,b are used as a notational convenience to represent the multiple integral in equation (5). For a collection of independent (i.e., unrelated) pedigrees, the total likelihood is simply the product of the individual pedigree likelihoods. Equation (6) fully specifies the joint likelihood of a bivariate mixture of discrete- and continuous-trait data. Note that the regression on covariates and the estimation of variance components are not explicitly separated in equation (6), consistent with our practice of maximizing the likelihood jointly with respect to mean and random effects. This is not essential, however, and the regression of trait values on covariates could be performed independently of the estimation of the variance components, with relatively little effect on the final estimates for either set of parameters.
Likelihood Evaluation
The expression for the joint likelihood L(x,y) in equation (6) must, in general, be evaluated numerically, and locating the allowable configuration of parameters that maximizes the likelihood can become computationally intensive. The results reported below were obtained by means of a likelihood-estimation algorithm described by Hasstedt (1993). The algorithm is a generalization of an iterated conditional univariate-integration strategy for evaluation of the multivariate-normal distribution (Pearson 1903; Aitken 1934; Mendell and Elston 1974; Rice et al. 1979; Van Eerdewegh 1982) and can accommodate mixtures of major-locus effects, quantitative data, polychotomous traits, and multivariate phenotypes. The algorithm is computationally extremely fast and, in general, yields good approximations even for large pedigrees. The primary disadvantages of the algorithm are the lack of a specifiable error bound on the resulting approximation and the potential for systematic bias in the estimated likelihood (J. T. Williams, unpublished data). When applied to the likelihood in equation (6), the technique of iterated conditioning and univariate-probability calculation has several useful consequences: the total likelihood is estimated without explicit separation of the marginal and conditional factors, obviating the task of determining μY|X and ΣY|X; explicit matrix inversion is not required; and the working assumption that both the discrete and continuous phenotypes have been measured for each pedigree member can be relaxed, minimizing the information loss that is due to incomplete observations.
Simulation Results
To illustrate the properties of the variance-components approach to joint linkage analysis of discrete and continuous traits, the method was applied to a subset of the simulated data from problem 2 of Genetic Analysis Workshop 10 (GAW10) (Goldin et al. 1997). The results show that joint consideration of a discrete trait and a correlated quantitative phenotype can improve the estimation of genetic parameters and increase the evidence for linkage of the traits to a major gene, compared with univariate analysis of the individual traits. Simulation results and power estimates for bivariate variance-components linkage analysis of strictly quantitative data have been reported by Almasy et al. (1997). Independent evaluation of a variance-components approach to linkage analysis of bivariate quantitative phenotypes is found in the work of Schork (1993), and specific applications of joint qualitative-quantitative linkage analysis to real data sets are found in the work of Williams et al. (1999 [in this issue]) and Czerwinski et al. (in press). Related investigation of the statistical performance for univariate variance-components analysis of discrete and continuous traits is available in the work of Duggirala et al. (1997), Williams et al. (1997), and Williams and Blangero (1999).
Data
The simulated data for GAW10 comprise two sets of family data for a common oligogenic disease (MacCluer et al. 1997). Each problem set has the same underlying additive genetic and environmental model, the same number of living individuals, the same number of sibships, and the same distribution of sibship sizes for living sibs (686 siblings distributed among 239 sibships, as 114 sib pairs, 68 sib triplets, 38 sib quartets, 12 sib quintets, and 7 sib sextets). In each data set, individuals <16 years of age are excluded. Phenotypic data are available for all living individuals, and genotypic data are available for all individuals, living or dead.
Problem 2A consists of 200 replications of 239 nuclear families, and each replication contains 1,164 individuals, 1,000 of whom are living. Nuclear families are randomly ascertained, subject to the constraint that there be at least two living offspring. Problem 2B consists of 200 replications of 23 extended pedigrees, and each replication has 1,497 individuals, 1,000 of whom are living. Pedigrees are randomly ascertained through a 40–60-year-old male or female having at least three living offspring and three full sibs. Pedigrees include the proband, the spouse of the proband, and all first-, second-, and third-degree relatives of the proband and of the spouse.
The genome for GAW10 comprises 10 chromosomes and is mapped by 367 markers spaced an average of 2.03 cM apart, with 24–50 markers per chromosome. The total genome length is 726 cM. The markers are highly polymorphic, having 4–15 alleles (mean 6.7) and a mean heterozygosity of .77. There is no disequilibrium among the markers.
Genetic Model
The generating genetic model used for the tests is diagrammed in figure 1. The model is a subset of the complete GAW10 generating model described by MacCluer et al. (1997) and is different from the genetic model investigated by Almasy et al. (1997). Quantitative traits Q4 and Q5 are influenced by major genes MG4, MG5, and MG6 but otherwise have no polygenic, nonrandom environmental (i.e., age and environmental factor) or sex-specific variance components. Major gene MG4 is located at 51.2 cM on chromosome 8, MG5 at 15.7 cM on chromosome 9, and MG6 at 13.7 cM on chromosome 10. Quantitative trait Q5 was dichotomized to simulate a discrete disease trait D5 having a population prevalence of KP=30% over all replications, by defining as “affected” all individuals exceeding a predetermined threshold value for the trait. The resulting genetic model demonstrates both pleiotropic action of MG4 and MG5 on phenotypes Q4 and D5 and the single-gene action of MG6 on Q4. The contribution of MG6 introduces a residual additive genetic component in univariate linkage analysis of Q4 and in bivariate linkage analysis of Q4 and D5.
Figure 1.

Genetic model for simulated data: random environmental effect (E), major genes with additive effects (MG4, MG5, and MG6), observable quantitative trait (Q4), unobservable quantitative trait (liability) (Q5), observable discrete disease trait derived from Q5 (D5), residual environmental correlation (.40) (ρe), and population prevalence of D5 (30%) (KP). Unlabeled numbers indicate the percentage of relative additive genetic and environmental variance components for each trait. (Adapted from MacCluer et al. [1997])
Parameter Estimation
Univariate and bivariate multipoint linkage analyses of Q4 and D5 were performed for all 200 replications for chromosomes 8 and 9 in the GAW10 nuclear-family and extended-pedigree data. Likelihoods were estimated by the algorithm described by Hasstedt (1993). The mean estimates of QTL effect size and residual additive heritability for each analysis are given in table 1, for MG4, and in table 2, for MG5. For the bivariate analyses, the mean estimates of the additive genetic and environmental correlations are also given. SDs representative of any single analysis are reported for all estimates. The expected parameter values are derived from the relative variances given in table 3 of a report by MacCluer et al. (1997).
Table 1.
Estimates of Parameters, at Location of Major Gene MG4 (Chromosome 8), in Univariate and Bivariate Multipoint Linkage Analyses of GAW10 Data, for 200 Replications
|
Mean ± SD Observed in |
|||
| Analysis and Parameter | Expected | Nuclear Families | Extended Pedigrees |
| Univariate Q4: | |||
| h2q | 28.0 | 28.1 ± 8.6 | 29.3 ± 8.9 |
| h2a | 27.0 | 26.4 ± 11.7 | 25.7 ± 11.5 |
| Univariate D5: | |||
| h2q | 14.0 | 16.8 ± 13.2 | 14.9 ± 10.6 |
| h2a | 23.0 | 24.5 ± 16.1 | 25.6 ± 12.7 |
| Bivariate Q4/D5: | |||
| Q4h2q | 28.0 | 27.8 ± 8.3 | 30.5 ± 7.0 |
| Q4h2a | 27.0 | 26.8 ± 11.3 | 24.4 ± 10.1 |
| D5h2q | 14.0 | 15.9 ± 10.4 | 15.7 ± 7.2 |
| D5h2a | 23.0 | 24.1 ± 13.4 | 23.8 ± 11.2 |
| ρa | 77.0 | 79.3 ± 32.8 | 78.5 ± 23.5 |
| ρe | 40.0 | 40.5 ± 8.1 | 39.6 ± 8.3 |
Table 2.
Estimates of Parameters, at Location of Major Gene MG5 (Chromosome 9), in Univariate and Bivariate Multipoint Linkage Analyses of GAW10 Data, for 200 Replications
|
Mean ± SD Observed in |
|||
| Analysis and Parameter | Expected | Nuclear Families | Extended Pedigrees |
| Univariate Q4: | |||
| h2q | 16.0 | 16.3 ± 8.5 | 17.9 ± 7.1 |
| h2a | 39.0 | 38.1 ± 10.6 | 36.8 ± 9.3 |
| Univariate D5: | |||
| h2q | 23.0 | 21.6 ± 12.8 | 23.8 ± 13.2 |
| h2a | 14.0 | 20.1 ± 16.0 | 17.2 ± 12.2 |
| Bivariate Q4/D5: | |||
| Q4h2q | 16.0 | 16.8 ± 8.0 | 18.0 ± 6.7 |
| Q4h2a | 39.0 | 37.6 ± 10.1 | 36.8 ± 8.8 |
| D5h2q | 23.0 | 19.7 ± 11.3 | 24.7 ± 9.5 |
| D5h2a | 14.0 | 21.2 ± 13.4 | 15.7 ± 9.7 |
| ρa | 84.7 | 86.5 ± 23.4 | 87.4 ± 15.2 |
| ρe | 40.0 | 40.3 ± 8.0 | 39.5 ± 8.0 |
Table 3.
Mean LOD Scores, P Values, and CVs at Location of Major Gene MG4 (Chromosome 8), in Univariate and Bivariate Analyses of GAW10 Data, for 200 Replications
|
Nuclear Families |
Extended Pedigrees |
|||||
| Analysis | LOD Score | P | CV | LOD Score | P | CV |
| Univariate D5 | .39 | .0888 | 1.21 | .59 | .0493 | 1.14 |
| Univariate Q4 | 2.36 | 4.86×10-4 | .55 | 5.05 | 7.10×10-7 | .47 |
| Bivariate Q4/D5a | 2.63 [2.15] | 8.23×10-4 | .51 | 5.62 [4.92] | 7.80×10-7 | .39 |
Values in square brackets are LOD[1] values.
The mean parameter estimates at the location of major gene MG4 on chromosome 8 are shown in table 1. Bivariate linkage analysis accurately recovers the generating parameters of the underlying genetic model. Estimates of the additive genetic and environmental correlation are in excellent agreement with expectation, although the SD for the estimate of the genetic correlation is large. The estimates of residual additive and QTL heritability from univariate and bivariate analysis are essentially unbiased. A similar pattern in parameter estimation is seen at the location of major gene MG5 on chromosome 9 (table 2).
Corresponding parameter estimates obtained in univariate and bivariate linkage analyses are not significantly different, but the estimates from the bivariate analysis are more precise as a result of exploitation of the correlation between the traits. The increase in precision is greatest for parameters related to D5 and is relatively minor for parameters related to continuous trait Q4, indicating that joint consideration of the discrete trait in the linkage analysis does not contribute greatly to parameter estimation for the quantitative trait, although the availability of the correlated quantitative trait leads to moderate improvement in the estimation of effects related to the discrete trait.
Type I Error Rate
The rate of type I error experienced in the joint multitrait linkage analysis can be estimated on the basis of the occurrence of false-positive signals observed in a series of independent tests of linkage to chromosomal regions that are unlinked to any of the major genes in the model shown in figure 1. A single location on each of chromosomes 1–7 from the GAW10 data was tested for linkage in all 200 replications of the nuclear-family data, constituting 1,400 independent tests of linkage. The following estimates 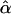 of the type I error rate corresponding to selected nominal values of α (in parentheses) were obtained: .0529±.0060 (.05), .0100±.0027 (.01), and .0007±.0007 (.001). Analysis of all 200 replications of the extended-pedigree data gave the following results: .0564±.0062 (.05), .0079±.0024 (.01), and .0007±.0007 (.001). These estimates are not significantly different from the nominal values; thus, there is no indication in these data that the type I error rate is inflated by joint linkage analyses of the two phenotypes.
of the type I error rate corresponding to selected nominal values of α (in parentheses) were obtained: .0529±.0060 (.05), .0100±.0027 (.01), and .0007±.0007 (.001). Analysis of all 200 replications of the extended-pedigree data gave the following results: .0564±.0062 (.05), .0079±.0024 (.01), and .0007±.0007 (.001). These estimates are not significantly different from the nominal values; thus, there is no indication in these data that the type I error rate is inflated by joint linkage analyses of the two phenotypes.
Mean LOD Scores
Mean LOD scores at the locations of major genes MG4 and MG5, together with their P values and coefficients of variation (CVs), are given in tables 3 and 4, respectively. The CV controls for the variation in the mean LOD score itself, with sampling unit and data type, and is a more appropriate measure of the variability of the LOD score between replications than is the standard error (Williams and Blangero 1999). The bivariate LOD score has 2 df and does not correspond directly with the familiar univariate LOD score; consequently, in tables 3 and 4, two scores are given for each bivariate analysis: the true bivariate LOD score having 2 df and the bivariate LOD score after adjustment to 1 df (denoted as “LOD[1]”). LOD[1] is determined as the 1-df LOD score required to give the same P value as is given by the true bivariate LOD score, and it can be compared directly with the univariate LOD scores.
Table 4.
Lod Scores, P Values, and CVs at the Location of Major Gene MG5 (Chromosome 9) in Univariate and Bivariate Analyses of GAW10 Data, for 200 Replications
|
Nuclear Families |
Extended Pedigrees |
|||||
| Analysis | LOD Score | P | CV | LOD Score | P | CV |
| Univariate D5 | .60 | .0488 | .96 | 1.44 | 5.03×10-3 | .89 |
| Univariate Q4 | .97 | .0174 | .85 | 2.15 | 8.23×10-4 | .70 |
| Bivariate Q4/D5a | 1.49 [1.09] | .0126 | .64 | 3.40 [2.87] | 1.37×10-4 | .53 |
Values in square brackets are LOD[1] values.
At the location of each major gene, comparison of the univariate LOD score for D5 versus the bivariate LOD score for Q4/D5 illustrates the effect of supplementing a focal qualitative trait (i.e., D5) with a correlated quantitative character (i.e., Q4). At MG4 on chromosome 8 (table 3) a nonsignificant LOD score (LOD=0.59, P=.0493) is obtained in univariate analysis of D5 in extended pedigrees. However, when the discrete trait is examined jointly with the correlated continuous character Q4 in a bivariate analysis, the LOD score increases to a statistically significant level (LOD[1]=4.92, P=7.80×10-7). Significant evidence for linkage to MG4 is not detected with either phenotype when nuclear families are used.
At the location of major gene MG5 on chromosome 9 (table 4), a nonsignificant LOD score (LOD=1.44, P=5.03×10-3) is obtained in univariate analysis of D5 in extended pedigrees, and nonsignificant evidence for linkage (LOD=2.15, P=8.23×10-4) is also found with univariate analysis of Q4. When the discrete trait is examined jointly with the correlated continuous phenotype, the LOD score increases but does not quite achieve statistical significance (LOD[1]=2.87, P=1.37×10-4). Statistically significant evidence for linkage to MG5 is not detected with either phenotype when nuclear families are used.
Alternatively, one can compare the univariate LOD scores for Q4 versus the bivariate LOD scores for Q4/D5, to understand the effect of supplementing a focal quantitative trait Q4 with a correlated discrete trait D5. At MG4 on chromosome 8 (table 3), univariate analysis of Q4 in extended pedigrees gives strong evidence for linkage (LOD=5.05, P=7.10×10-7). When the quantitative trait is analyzed jointly with the correlated discrete trait D5, the additional information provided by the qualitative trait does not quite counteract the increased df in the test—the evidence for linkage decreases slightly but remains significant (LOD[1]=4.92, P=7.80×10-7). At MG5 on chromosome 9 (table 4), a nonsignificant LOD score (LOD=2.15, P=8.23×10-4) is obtained in univariate analysis of Q4; in bivariate analysis of Q4 with the correlated discrete trait D5, the LOD score increases and nearly achieves statistical significance (LOD[1]=2.87, P=1.37×10-4).
Multipoint LOD-Score Plots
Multipoint LOD-score plots for chromosomes 8 and 9 are shown in figures 2 and 3, respectively, for a representative replicate of the extended-pedigree data. Each plot shows the LOD-score curves from the univariate analyses of Q4 and D5 and from the bivariate analysis of Q4/D5; also shown is the bivariate LOD score LOD[1] after adjustment to 1 df. The location of major genes MG4 (51.2 cM on chromosome 8) and MG5 (15.7 cM on chromosome 9) are indicated, and in each figure the peak LOD score accurately localizes the major gene, to within a few centimorgans.
Figure 2.

Multipoint LOD scores for GAW10 data on chromosome 8, in univariate and bivariate linkage analyses of Q4 and D5
Figure 3.

Multipoint LOD scores for GAW10 data on chromosome 9, in univariate and bivariate linkage analyses of Q4 and D5
In figure 2, univariate analysis of Q4 easily detects linkage to MG4 (LOD=8.03, P=5.98×10-10), and, in this replication, significant evidence for linkage is also detected in univariate analysis of D5 (LOD=3.30, P=4.88×10-5). Bivariate analysis of the discrete and continuous phenotypes also gives significant evidence for linkage (LOD[1]=6.97, P=1.58×10-10). If D5 is regarded as the focal phenotype, then bivariate analysis clearly extracts markedly greater evidence for linkage of the trait to MG4. If Q4 is the focal phenotype, however, then bivariate analysis of Q4 with the correlated discrete trait leads to a slight reduction in the evidence for linkage (although the LOD score remains highly significant) as the number of df in the test increases without sufficient additional linkage information.
In the multipoint LOD-score plot of figure 3, neither univariate analysis provides significant evidence for linkage to MG5 on chromosome 9 (for Q4, LOD=2.89, P=1.32×10-4; for D5, LOD=1.09, P=.0124), but evidence for linkage is statistically significant in the bivariate analysis (LOD[1]=3.55, P=2.57×10-5). In this case, it does not matter which trait is regarded as the focal phenotype; bivariate analysis of either trait with the correlated character yields significant evidence for linkage where none is detected with univariate analysis.
Figures 2 and 3 illustrate that the evidence for linkage in a multivariate linkage analysis of positively correlated phenotypes is not necessarily the sum of the evidence for linkage in separate univariate analyses (Jiang and Zeng 1995; Korol et al. 1995). Although the multipoint LOD-score curves for the univariate and bivariate analyses can have similar profiles, the linkage evidence obtained in the joint analysis derives in part from the correlation of the discrete and continuous traits.
Testing Pleiotropy and Coincident Linkage
When multiple traits are analyzed, the hypotheses of pleiotropy and of close linkage of independent major genes are of particular interest (Jiang and Zeng 1995; Mangin et al. 1998). These hypotheses are easily tested in the multitrait variance-components approach, by comparison of the likelihoods of appropriate nested models (Boehnke et al. 1986; Almasy et al. 1997).
In the bivariate analysis of Q4 and D5, the mechanisms of complete pleiotropy and coincident linkage are special cases of the general two-QTL genetic linkage model in which the effects due to potentially distinct QTLs (Q4σ2q and D5σ2q) are estimated and the correlation between these effects (ρq) is unconstrained. To test for complete pleiotropy, the likelihood of a two-QTL linkage model in which the correlation ρq between the QTLs is estimated is compared with the likelihood of a two-QTL linkage model in which ρq is constrained to +1 (the sign of ρq is chosen to agree with the sign of the polygenic correlation ρa). To test for coincident linkage of the traits to two independent but closely spaced genes having additive effects, the likelihood of a linkage model in which ρq is estimated is compared with the likelihood of a model in which ρq is constrained to 0.
Table 5 summarizes the likelihood-ratio tests for complete pleiotropy and coincident linkage made at the location of major genes MG4 and MG5 in the extended-pedigree replicate used to prepare figures 2 and 3. For each test, the table gives (a) the value of the likelihood-ratio statistic λ=-2ln(L0/L1), where L0 and L1 are, respectively, the likelihoods of the indicated null and alternative models; (b) the asymptotic distribution of the statistic under the null model; and (c) the corresponding P value. The asymptotic distributions for these tests were determined by use of the methods described by Self and Liang (1987). The test against complete pleiotropy does not give significant results at either location, whereas the test against coincident linkage gives significant results at both locations. For each location, the inference is that joint variation in Q4 and D5 is mediated by a common QTL, which indeed corresponds with the genetic model shown in figure 1.
Table 5.
Likelihood-Ratio Tests for Pleiotropy and Coincident Linkage at Locations of Major Genes MG4 and MG5, for Extended-Pedigree Data Used in Figures 2 and 3
|
Major Gene MG4 |
Major Gene MG5 |
||||
| Null Distribution | λ | P | λ | P | |
| Complete pleiotropy | 1/2χ21: 1/2χ20 | .00 | .5000 | .00 | .5000 |
| Coincident linkage | χ21 | 28.06 | 1.18×10-7 | 17.82 | 2.43×10-5 |
Discussion
Quantitative risk factors, disease precursors, and biological markers are often known to be correlated, causally or statistically, with qualitatively assessed disease states. Conversely, robust clinical diagnosis of a disease condition may be available to supplement the measurement of physiologically implicated quantitative phenotypes. In either situation, joint linkage analysis of the discrete and continuous traits can be used to exploit the correlational information between the disease state and the quantitative trait, in the search for mediating genetic factors.
From a statistical point of view, the nature of the relationship between the discrete trait and the quantitative character can be used to distinguish three general situations. First, the discrete trait may have been constructed by ad hoc polychotomization of an inherently continuous physiological quantity. For example, diastolic blood pressure (DBP) and systolic blood pressure (SBP) are used to classify hypertension into “mild” (DBP ⩾90 mm Hg, SBP ⩾140 mm Hg) and “severe” (DBP ⩾95 mm Hg, SBP ⩾165 mm Hg) presentations. In Western populations, body-mass index (BMI) is frequently dichotomized into “obese” (BMI >27) and “nonobese” (BMI ⩽27) phenotypes. A diagnosis of type 2 diabetes is indicated by fasting blood-glucose levels >140 mg/dl. One of the defining criteria in the diagnosis of glaucoma, in addition to examination of the optical disk and testing of the visual field, is intraocular pressure >22 mm Hg.
With other disease traits, the qualitative state and the quantitative character may exhibit different relationships before and after clinical disease onset. If the disease radically alters patient physiology, for example, then previously correlated quantitative measures may no longer display any direct or regular relationship with the disease trait. Type 2 diabetes, although generally studied as a dichotomous trait defined on the basis of fasting blood-glucose levels, provides a good example of a complex relationship between the clinical presentation of the disease and a related continuous physiological quantity (Ghosh and Schork 1996). Insulin resistance is strongly associated with the development of type 2 diabetes, and elevated insulin concentration is a significant risk factor for the disease (Lillioja et al. 1993). Longitudinal studies have shown that elevated insulin concentrations occur prior to the onset of clinical diabetes (Lillioja et al. 1987). Once overt diabetes is established, insulin concentration often continues to increase for a time but eventually will decline as indigenous insulin production becomes compromised. In this case, a disease trait (diabetes) and a quantitative biological marker (insulin concentration) are correlated until disease onset and for some time thereafter, but the relationship between the two is not stable, and the quantitative phenotype may effectively become truncated after clinical presentation of the disease.
Yet a third situation is encountered with psychiatric disorders such as schizophrenia, alcoholism, or bipolar disorder, for which diagnosis is generally made on a binary (presence/absence) basis, at a nominal level, or, perhaps, on an ordinal scale of severity having relatively few and possibly mutually nonexclusive classes. With psychiatric disorders, there are no directly related, inherently continuous characters, as there are with obesity or diabetes, but there are often correlated quantitative biological markers that can be measured easily. For example, the amplitude of the P300 component of event-related brain potentials is significantly lower in individuals at risk for alcoholism (Begleiter et al. 1984; Polich et al. 1994; Porjesz et al. 1998), and schizophrenia is correlated with a number of altered psychophysiological paradigms (Blackwood et al. 1991; Freedman et al. 1997).
In the first of these situations involving paired discrete and continuous observations, the discrete trait is an ad hoc construct and contributes no new information. The discrete construct essentially remeasures an available quantitative trait, and, in so doing, introduces measurement error that, in a variance-components analysis, will appear as environmental variance (Falconer 1989, p. 305). When the observable phenotype is inherently continuous, methods of linkage analysis for quantitative traits are to be preferred (Comuzzie et al. 1997; Duggirala et al. 1997). For common diseases in particular, it is appropriate to examine the continuum of variation rather than to limit studies to “affected” individuals. Furthermore, the use of continuous traits does not preclude ascertainment (nonrandom sampling) to enrich the tails of the phenotypic distribution. In any event, the use of a discretized version of an available, biologically continuous phenotype for linkage analysis is plainly unnecessary and will markedly reduce the power to detect and localize genes influencing such phenotypes (Xu and Atchley 1996; Duggirala et al. 1997; Wijsman and Amos 1997).
In the second and third situations described above, it would clearly be advantageous to exploit the independent information in the discrete and the continuous traits, as well as the correlational information between them. In particular, linkage analysis to detect and localize genetic factors influencing a disease trait can benefit considerably from joint consideration of the disease and a correlated quantitative factor (Moldin 1994; Ott 1995; Almasy et al. 1997; Blangero et al. 1997; Williams et al. 1999 [in this issue]). Depending on the genetic etiology of the phenotypes and the structure of the correlation between the discrete and continuous traits, one can, in general, expect both increased power to detect linkage and improved estimation of QTL effect size and location, compared with univariate analysis of the individual phenotypes.
The ability to jointly analyze multivariate qualitative and quantitative data also suggests some interesting analytical possibilities. For example, a multivariate discrete phenotype comprising disease diagnoses under different diagnostic systems could be investigated, and the quantitative phenotype might be a vector of measurements monitoring specific physiological processes. Specific knowledge of the precise causal relationships, if any, between the discrete traits and the quantitative characters is not required in order to exploit any statistical dependence between them, but several investigators have discussed the criteria that a biological marker should meet to be useful in statistical genetic analyses of a correlated qualitative disease (Begleiter et al. 1984; Lander 1988; Blackwood et al. 1991; Moldin 1994; Porjesz et al. 1998).
In the companion paper (Williams et al. 1999 [in this issue]), we apply the method for joint qualitative-quantitative–trait linkage analysis to data, from the Collaborative Study on the Genetics of Alcoholism, on alcoholism diagnoses and event-related brain potentials (Begleiter et al. 1995). There we find that simultaneous consideration of a correlated quantitative phenotype (P300 amplitude of event-related potential) significantly increases the evidence for linkage to a discrete disease trait (alcoholism).
Acknowledgments
This report has benefited greatly from discussions with Michael Mahaney, Braxton Mitchell, John Rice, Stephen Iturria, Tony Comuzzie, and Ravindranath Duggirala. The comments of the anonymous reviewers were valuable in clarifying our arguments and overall presentation. We are grateful to Vanessa Olmo for preparing figure 1. The development of the statistical methods used in solar (sequential oligogenic linkage analysis routines) was supported by National Institutes of Health (NIH) grants MH59490, DK44297, HL45522, HL28972, GM31575, and GM18897. The Genetic Analysis Workshop is supported by NIH grant GM31575. Information on the analysis package solar is available from The Southwest Foundation for Biomedical Research.
Electronic-Database Information
The URL for data in this article is as follows:
- Southwest Foundation for Biomedical Research, The, http://www.sfbr.org
References
- Aitken AC (1934) Note on selection from a multivariate normal population. Proc Edinb Math Soc Bull 4:106–110 [Google Scholar]
- Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasy L, Dyer TD, Blangero J (1997) Bivariate quantitative trait linkage analysis: pleiotropy versus co-incident linkages. Genet Epidemiol 14:953–958 [DOI] [PubMed]
- Amos CI (1994) Robust variance-components approach for assessing genetic linkage in pedigrees. Am J Hum Genet 54:535–543 [PMC free article] [PubMed]
- Amos CI, Elston RC, Bonney GE, Keats BJB, Berenson GS (1990) A multivariate method for detecting genetic linkage, with application to a pedigree with an adverse lipoprotein phenotype. Am J Hum Genet 47:247–254 [PMC free article] [PubMed]
- Amos CI, Laing AE (1993) A comparison of univariate and multivariate tests for genetic linkage. Genet Epidemiol 10:671–676 [DOI] [PubMed]
- Amos CI, Zhu DK, Boerwinkle E (1996) Assessing genetic linkage and association with robust components of variance approaches. Ann Hum Genet 60:143–160 [DOI] [PubMed]
- Andrews DF, Gnanadesikan R, Warner JL (1971) Transformations of multivariate data. Biometrics 27:825–840 [Google Scholar]
- Beaty TH, Liang KY (1987) Robust inference for variance components models in families ascertained through probands. I. Conditioning on the proband's phenotype. Genet Epidemiol 4:203–210 [DOI] [PubMed]
- Beaty TH, Liang KY, Seerey S, Cohen BH (1987) Robust inference for variance components models in families ascertained through probands. II. Analysis of spirometric measures. Genet Epidemiol 4:211–221 [DOI] [PubMed]
- Beaty TH, Self SG, Liang KY, Connolly MA, Chase GA, Kwiterovich PO (1985) Use of robust variance components models to analyse triglyceride data in families. Ann Hum Genet 49:315–328 [DOI] [PubMed]
- Begleiter H, Porjesz B, Bihari B, Kissin B (1984) Event-related brain potentials in boys at risk for alcoholism. Science 225:1493–1496 [DOI] [PubMed]
- Begleiter H, Reich T, Hesselbrock V, Porjesz B, Li T-K, Schuckit MA, Edenberg HJ, et al (1995) The collaborative study on the genetics of alcoholism. Alcohol Health Res World 19:228–236 [PMC free article] [PubMed] [Google Scholar]
- Blackwood DHR, St Clair DM, Muir WJ, Duffy JC (1991) Auditory P300 and eye tracking dysfunction in schizophrenic pedigrees. Arch Gen Psychiatry 48:899–909 [DOI] [PubMed]
- Blangero J (1993) Statistical genetic approaches to human adaptability. Hum Biol 65:941–966 [PubMed]
- ——— (1995) Genetic analysis of a common oligogenic trait with quantitative correlates: summary of GAW9 results. Genet Epidemiol 12:689–706 [DOI] [PubMed]
- Blangero J, Almasy L, Williams JT, Porjesz B, Reich T, Begleiter H, COGA Collaborators (1997) Incorporating quantitative traits in genomic scans of psychiatric diseases: alcoholism and event-related potentials. Am J Med Genet 74:602 [Google Scholar]
- Blangero J, Williams-Blangero S, Kammerer CM, Towne B, Konigsberg LW (1992) Multivariate genetic analysis of nevus measurements and melanoma. Cytogenet Cell Genet 59:179–181 [DOI] [PubMed]
- Boehnke M, Lange K (1984) Ascertainment and goodness of fit of variance component models for pedigree data. Prog Clin Biol Res 147:173–192 [PubMed]
- Boehnke M, Moll PP, Lange K, Weidman WH, Kottke BA (1986) Univariate and bivariate analyses of cholesterol and triglyceride levels in pedigrees. Am J Med Genet 23:775–792 [DOI] [PubMed]
- Bonney GE, Lathrop GM, Lalouel J-M (1988) Combined linkage segregation analysis using regressive models. Am J Hum Genet 43:29–37 [PMC free article] [PubMed]
- Borecki IB, Lathrop GM, Bonney GE, Yaouanq J, Rao DC (1990) Combined segregation and linkage analysis of genetic hemochromatosis using affection status, serum iron, and HLA. Am J Hum Genet 47:542–550 [PMC free article] [PubMed]
- Clifford CA, Hopper JL, Fulker DW, Murray RM (1984) A genetic and environmental analysis of a twin family study of alcohol use, anxiety, and depression. Genet Epidemiol 1:63–79 [DOI] [PubMed]
- Comuzzie AG, Hixson JE, Almasy L, Mitchell BD, Mahaney MC, Dyer TD, Stern MP, et al (1997) A major quantitative trait locus determining serum leptin levels and fat mass is located on human chromosome 2. Nat Genet 15:273–276 [DOI] [PubMed]
- Crittenden LB (1961) An interpretation of familial aggregation based on multiple genetic and environmental factors. Ann N Y Acad Sci 91:769–780 [DOI] [PubMed] [Google Scholar]
- Czerwinski SA, Mahaney MC, Williams JT, Almasy L, Blangero J. Genetic analysis of personality traits and alcoholism using a mixed discrete-continuous trait variance component model. Genet Epidemiol (in press) [DOI] [PubMed] [Google Scholar]
- De Andrade M, Thiel TJ, Yu LP, Amos CI (1997) Assessing linkage on chromosome 5 using components of variance approach: univariate versus multivariate. Genet Epidemiol 14:773–778 [DOI] [PubMed]
- Ducrocq V, Besbes B (1993) Solution of multiple trait animal models with missing data on some traits. J Anim Breed Genet 110:81–92 [DOI] [PubMed] [Google Scholar]
- Duggirala R, Williams JT, Williams-Blangero S, Blangero J (1997) A variance component approach to dichotomous trait linkage analysis using a threshold model. Genet Epidemiol 14:987–992 [DOI] [PubMed]
- Elston RC, Namboodiri KK, Glueck CJ, Fallat R, Tsang R, Leuba V (1975) Study of the genetic transmission of hypercholesterolemia and hypertriglyceridemia in a 195 member kindred. Ann Hum Genet 39:67–87 [DOI] [PubMed]
- Falconer DS (1965) The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann Hum Genet 29:51–76 [Google Scholar]
- ——— (1989) Introduction to quantitative genetics, 3d ed. John Wiley & Sons, New York [Google Scholar]
- Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, et al (1997) Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci USA 94:587–592 [DOI] [PMC free article] [PubMed]
- Fulker DW, Cherny SS, Cardon LR (1995) Multipoint interval mapping of quantitative trait loci, using sib pairs. Am J Hum Genet 56:1224–1233 [PMC free article] [PubMed]
- Ghosh S, Schork NJ (1996) Genetic analysis of NIDDM: the study of quantitative traits. Diabetes 45:1–14 [DOI] [PubMed]
- Gnanadesikan R (1977) Methods for statistical data analysis of multivariate observations. John Wiley & Sons, New York [Google Scholar]
- Goldgar DE (1990) Multipoint analysis of human quantitative genetic variation. Am J Hum Genet 47:957–967 [PMC free article] [PubMed]
- Goldin LR, Bailey-Wilson JE, Borecki IB, Falk CT, Goldstein AM, Suarez BK, MacCluer JW (eds) (1997) Genetic Analysis Workshop 10: detection of genes for complex traits. Genet Epidemiol 14:549–1152 [Google Scholar]
- Hasstedt SJ (1993) Variance components/major locus likelihood approximation for quantitative, polychotomous, and multivariate data. Genet Epidemiol 10:145–158 [DOI] [PubMed]
- Hopper JL, Mathews JD (1982) Extensions to multivariate normal models for pedigree analysis. Ann Hum Genet 46:373–383 [DOI] [PubMed]
- Iturria SJ, Williams JT, Almasy L, Dyer TD, Blangero J. An empirical test of the significance of an observed QTL effect that preserves additive genetic variation. Genet Epidemiol (in press) [DOI] [PubMed] [Google Scholar]
- Jiang C, Zeng Z-B (1995) Multiple trait analysis of genetic mapping for quantitative trait loci. Genetics 140:1111–1127 [DOI] [PMC free article] [PubMed]
- Korol AB, Ronin YI, Kirzhner VM (1995) Interval mapping of quantitative trait loci employing correlated trait complexes. Genetics 140:1137–1147 [DOI] [PMC free article] [PubMed]
- Lalouel JM, Le Mignon L, Simon M, Fauchet R, Bourel M, Rao DC, Morton NE (1985) Genetic analysis of idiopathic hemochromatosis using both qualitative (disease status) and quantitative (serum iron) information. Am J Hum Genet 37:700–718 [PMC free article] [PubMed]
- Lander ES (1988) Splitting schizophrenia. Nature 336:105–106 [DOI] [PubMed]
- Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048 [DOI] [PubMed]
- Lange K (1978) Central limit theorems for pedigrees. J Math Biol 6:59–66 [Google Scholar]
- ——— (1997) Mathematical and statistical methods for genetic analysis. Springer-Verlag, New York [Google Scholar]
- Lange K, Boehnke M (1983) Extensions to pedigree analysis. IV. Covariance components models for multivariate traits. Am J Med Genet 14:513–524 [DOI] [PubMed]
- Lange KL, Little RJA, Taylor JMG (1989) Robust statistical modeling using the t distribution. J Am Stat Assoc 84:881–896 [Google Scholar]
- Lange K, Westlake J, Spence MA (1976) Extensions to pedigree analysis. III. Variance components by the scoring method. Ann Hum Genet 39:485–491 [DOI] [PubMed]
- Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E, Knowler WC, et al (1993) Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus: Prospective studies of Pima Indians. N Engl J Med 329:1988–1992 [DOI] [PubMed]
- Lillioja S, Mott DM, Zawadzki JK, Young AA, Abbott WGH, Knowler WC, Bennett PH, et al (1987) In vivo insulin action is familial characteristic in nondiabetic Pima Indians. Diabetes 36:1329–1335 [DOI] [PubMed]
- MacCluer JW, Blangero J, Dyer TD, Speer MC (1997) GAW10: simulated family data for a common oligogenic disease with quantitative risk factors. Genet Epidemiol 14:737–742 [DOI] [PubMed]
- Mangin B, Thoquet P, Grimsley N (1998) Pleiotropic QTL analysis. Biometrics 54:88–99 [Google Scholar]
- Mendell NR, RC Elston (1974) Multifactorial qualitative traits: genetic analysis and prediction of recurrence risks. Biometrics 30:41–57 [PubMed]
- Moldin SO (1994) Indicators of liability to schizophrenia: perspectives from genetic epidemiology. Schizophr Bull 20:169–184 [DOI] [PubMed]
- Moldin SO, Rice JP, Van Eerdewegh P, Gottesman II, Erlenmeyer-Kimling L (1990) Estimation of disease risk under bivariate models of multifactorial inheritance. Genet Epidemiol 7:371–386 [DOI] [PubMed]
- Morton NE, MacLean CJ (1974) Analysis of family resemblance. III. Complex segregation of quantitative traits. Am J Hum Genet 26:489–503 [PMC free article] [PubMed]
- Morton NE, Yee S, Elston RC, Lew R (1970) Discontinuity and quasi-continuity: alternative hypotheses of multifactorial inheritance. Clin Genet 1:81–94 [Google Scholar]
- Ott J (1995) Linkage analysis with biological markers. Hum Hered 45:169–174 [DOI] [PubMed]
- Pearson K (1903) I. Mathematical contributions to the theory of evolution. XI. On the influence of natural selection on the avriability and correlation of organs. Philos Trans R Soc Lond [A] 200:1–66 [Google Scholar]
- Polich J, Pollock VE, Bloom FE (1994) Meta-analysis of P300 amplitude from males at risk for alcoholism. Psychol Bull 115:55–73 [DOI] [PubMed]
- Porjesz B, Begleiter H, Reich T, Van Eerdewegh P, Edenberg HJ, Foroud T, Goate A, et al (1998) Amplitude of visual P3 event-related potential as a phenotypic marker for a predisposition to alcoholism: preliminary results from the COGA project. Alcohol Clin Exp Res 22:1317–1323 [PubMed]
- Rice J, Reich T, Cloninger CR, Wette R (1979) An approximation to the multivariate normal integral: its application to multifactorial qualitative traits. Biometrics 35:451–459 [Google Scholar]
- Ronin YI, Kirzhner VM, Korol AB (1995) Linkage between loci of quantitative traits and marker loci: multi-trait analysis with a single marker. Theor Appl Genet 90:776–786 [DOI] [PubMed] [Google Scholar]
- Schork NJ (1993) Extended multipoint identity-by-descent analysis of human quantitative traits: efficiency, power, and modeling considerations. Am J Hum Genet 53:1306–1319 [PMC free article] [PubMed]
- Searle SR (1971) Linear models. John Wiley & Sons, New York [Google Scholar]
- Self SG, Liang K-Y (1987) Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J Am Stat Assoc 82:605–610 [Google Scholar]
- Tiwari HK, Elston RC (1997) Linkage of multilocus components of variance to polymorphic markers. Ann Hum Genet 61:253–261 [DOI] [PubMed]
- Tong YL (1990) The multivariate normal distribution. Springer-Verlag, New York [Google Scholar]
- Van Eerdewegh P (1982) Statistical selection in multivariate systems with applications in quantitative genetics. PhD diss, Department of Mathematics, Washington University, St Louis
- Weller JI, Wiggans GR, VanRaden PM, Ron M (1996) Application of a canonical transformation to detection of quantitative trait loci with the aid of genetic markers in a multitrait experiment. Theor Appl Genet 92:998–1002 [DOI] [PubMed] [Google Scholar]
- Wijsman EM, Amos CI (1997) Genetic analysis of simulated oligogenic traits in nuclear and extended pedigrees: summary of GAW10 contributions. Genet Epidemiol 14:719–735 [DOI] [PubMed]
- Williams JT, Begleiter H, Porjesz B, Edenberg HJ, Foroud T, Reich T, Goate A, et al (1999) Joint multipoint linkage analysis of multivariate qualitative and quantitative traits. II. Alcoholism and event-related potentials. Am J Hum Genet 65:1148–1160 (in this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Blangero J (1999) Comparison of variance components and sibpair-based approaches to quantitative trait linkage analysis in unselected samples. Genet Epidemiol 16:113–134 [DOI] [PubMed]
- Williams JT, Duggirala R, Blangero J (1997) Statistical properties of a variance components method for quantitative trait linkage analysis in nuclear families and extended pedigrees. Genet Epidemiol 14:1065–1070 [DOI] [PubMed]
- Wright S (1934a) An analysis of variability in number of digits in an inbred strain of guinea pigs. Genetics 19:506–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (1934b) The results of crosses between inbred strains of guinea pigs, differing in number of digits. Genetics 19:537–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Atchley WR (1996) Mapping quantitative trait loci for complex binary diseases using line crosses. Genetics 143:1417–1424 [DOI] [PMC free article] [PubMed]