

Summary
Hypertrophic cardiomyopathy (HCM) is an autosomal dominantly inherited disease of the cardiac sarcomere, caused by numerous mutations in genes encoding protein components of this structure. Mutation carriers are at risk of sudden cardiac death, mostly as adolescents or young adults. The reproductive disadvantage incurred may explain both the global occurrence of diverse independent HCM-associated mutations and the rare reports of founder effects within populations. We have investigated whether this holds true for two South African subpopulations, one of mixed ancestry and one of northern-European descent. Previously, we had detected three novel mutations—Ala797Thr in the β-myosin heavy-chain gene (βMHC), Arg92Trp in the cardiac troponin T gene (cTnT), and Arg645His in the myosin-binding protein C gene (MyBPC)—and two documented βMHC mutations (Arg403Trp and Arg249Gln). Here we report three additional novel mutations—Gln499Lys in βMHC and Val896Met and Δc756 in MyBPC—and the documented βMHC Arg719Gln mutation. Seven of the nine HCM-causing mutations arose independently; no conclusions can be drawn for the remaining two. However, the βMHC Arg403Trp and Ala797Thr and cTnT Arg92Trp mutations were detected in another one, eight, and four probands, respectively, and haplotype analysis in families carrying these recurring mutations inferred their origin from three common ancestors. The milder phenotype of the βMHC mutations may account for the presence of these founder effects, whereas population dynamics alone may have overridden the reproductive disadvantage incurred by the more lethal, cTnT Arg92Trp mutation.
Introduction
Hypertrophic cardiomyopathy (HCM), an autosomal dominantly inherited primary cardiac disease, historically has been defined as a rare cardiomyopathy of unknown cause, characterized by hypertrophy of the ventricular muscle, in the absence of other predisposing clinical conditions (Teare 1958). Studies initiated during the past decade have, to some extent, changed this view of the disease. It is now known that HCM is a disease of the sarcomere, caused by ⩾100 different mutations in seven genes encoding proteins constituting the cardiac contractile unit (reviewed in Bonne et al. 1998). Recent clinical observational studies performed in the general population have suggested that, far from being a rare genetic disorder, the prevalence of HCM is close to 1/500 individuals (Maron et al. 1995). Furthermore, although genotype-phenotype correlations have emphasized the varied clinical spectrum that long has been associated with this disease, they have also shown that ventricular hypertrophy is not an inevitable consequence of the presence of an HCM-causing mutation (Bonne et al. 1998). However, it appears that mutation carriers do remain at risk of sudden cardiac death, as adolescents or young adults, whatever the other clinical manifestations of their condition (Moolman et al. 1997).
Mutations that are associated with significant mortality, especially at a young age, confer a reproductive disadvantage. Consequently, within populations, recurring identical disease-causing mutations are more likely to have arisen independently, rather than to reflect a common ancestor (de la Chapelle et al. 1993). An investigation of the origin of eight different HCM-causing mutations in the β-myosin heavy-chain gene (βMHC) revealed that they had arisen independently in ⩾12 of the 14 families studied (Watkins et al. 1993b). This finding led Watkins et al. (1993b) to predict that the prevalence of HCM-causing mutations would be comparable in all populations globally and that founder mutations would be rare. The continuing identification of diverse disease-associated mutations in various sarcomeric protein–encoding genes in populations in North America, Europe, Asia, Australia, and South Africa (Bonne et al. 1998, and references therein) supports this reasoning.
Indeed, reports of evidence of founder effects in familial HCM are rare and often incidental to the main investigation. The first such report, which appeared in 1993, described two small families of Portuguese descent harboring the βMHC Gly584Arg mutation (Watkins et al. 1993b). Five years later, truncating mutations in myosin-binding protein C (MyBPC), which were likely to be identical by descent, were detected in Japanese families, with three kindreds bearing the InsG791 mutation and two bearing the Int12ASA-2G mutation (Niimura et al. 1998). In the same year, it was suggested that the Asp175Asn mutation in the α-tropomyosin gene (αTM), borne by four Finnish families, might have arisen from a common ancestor (Jääskeläinen et al. 1998).
We have undertaken investigations of the molecular spectrum of HCM-causing mutations in the seven implicated genes (reviewed in Bonne et al. 1998) in two subpopulations living in the Western and Eastern Cape provinces of South Africa—namely, persons of mixed ancestry and those of white descent. The history and population dynamics of these two groups, from the 17th century to the present, is probably responsible for the presence of several well-documented founder effects, particularly in persons of Afrikaans descent (Kotze et al. 1991; Brink et al. 1995; De Jager et al. 1996; Warnich et al. 1996). The Afrikaners are a subgroup of Afrikaans-speaking South Africans of northern-European origin, who have retained a cultural identity largely through intermarriage and who show minor differences, in allelic-gene frequencies, from the English-speaking white subpopulation (Botha and Pritchard 1972). The mixed-ancestry and white subgroups represent an ideal model with which to investigate whether the HCM-causing mutations that they carry generally arose independently and fairly recently or whether founder effects are present in a particular population context. A comparison of the HCM-associated mutational spectrum of the particular South African subpopulations studied versus that in other countries would indicate whether the mutations either have an independent origin or possibly emanate from Europe. In the case of identical mutations being detected in apparently unrelated families, haplotype analysis would ascertain whether they were of independent or founder origin, on the basis of the chromosomal background in which they are embedded.
Previously, we had reported three βMHC mutations—namely, the novel Ala797Thr (Moolman et al. 1995), the Arg249Gln (Posen et al. 1995), and the Arg403Trp (Moolman et al. 1993; Posen et al. 1995) mutations—as well as novel mutations in cTnT (Arg92Trp [Moolman et al. 1997]) and MyBPC (Arg645His [Moolman-Smook et al. 1998]).
In the present study, we describe detection of a further three βMHC mutations, two of which are novel, and detection of two previously unreported MyBPC mutations and show that the βMHC Arg403Trp mutation arose independently in South Africa and France. Thus, it appears that a high proportion of the South African HCM-causing mutations are unique to South Africa, confirming the expectation that, in particular subpopulations, HCM is caused by a high proportion of relatively new mutations. However, both unlike the mutation profile most often seen in other countries and contrary to generally held views, which have been discussed by Watkins et al. (1993b), we also report the existence of strong founder-gene effects in both of the subpopulations studied. Both the occurrence of founder-gene effects and the history of the South African subpopulations in which they were detected are discussed, together with the significance that these results have for molecular diagnosis.
Patients and Methods
Patient Study Group and Clinical Examination
The study was performed with the approval of the University of Stellenbosch ethics committee, with informed consent obtained either from subjects or, in the case of minors participating in the study, from parents. A panel of apparently unrelated HCM probands (denoted by the prefix “SB”), of mixed ancestry or white descent, who reside in the Western and Eastern Cape provinces of South Africa, had previously been established, all of whom were screened for reported and novel mutations. After identification of a disease-causing mutation in a proband, relatives were traced and pedigrees were established, whenever feasible. The pedigrees extended in this manner were given a pedigree number (denoted by the prefix “Ped”).
As a result of continuing prospective studies, at the time of the present study the panel consisted of 40 HCM-affected probands, including those 6 in whom an HCM-causing mutation had been detected in earlier studies; these were the probands of Ped108 with the Arg249Gln (Posen at al. 1995), Ped106 with the Arg403Trp (Posen et al. 1995), and Ped101 with the Ala797Thr (Moolman et al. 1995) mutations in βMHC, the probands of Ped100 and Ped109 with the cTnT Arg92Trp mutation (Moolman et al. 1997), and the proband of Ped136 with the MyBPC Arg654His mutation (Moolman-Smook et al. 1998). Mutations detected in the present study, those previously detected in South African families, relevant proband or extended-pedigree identification codes, and either the individual's or the family's ethnicity are shown in table 1.
Table 1.
HCM-Causing Mutations Present in Two South African subpopulations
| Gene, Mutation, and Source | Ethnic Group | Reference |
| βMHC: | ||
| Arg249Gln:a | ||
| Ped108 | Mixed ancestry | Posen et al (1995) |
| Arg403Trp:b | ||
| Ped106 | Mixed ancestry | Posen et al. (1995) |
| SB1140 | Mixed ancestry | Present study |
| Ala797Thr: | ||
| Ped101 | White | Moolman et al. (1995) |
| Ped104 | Mixed ancestry | Present study |
| Ped110 | Mixed ancestry | Present study |
| Ped124 | White | Present study |
| Ped131 | White | Present study |
| Ped138 | White | Present study |
| SB902 | White | Present study |
| SB983 | Mixed ancestry | Present study |
| SB955 | Mixed ancestry | Present study |
| Glu499Lys: | ||
| SB1112 | White | Present study |
| Arg719Gln:c | ||
| SB1159 | White | Present study |
| cTnT: | ||
| Arg92Trp: | ||
| Ped100 | Mixed ancestry | Moolman et al. (1997) |
| Ped109 | Mixed ancestry | Moolman et al. (1997) |
| Ped137 | Mixed ancestry | Present study |
| Ped139 | Mixed ancestry | Present study |
| Ped142 | Mixed ancestry | Present study |
| SB385 | Mixed ancestry | Present study |
| MyBPC: | ||
| Arg654His: | ||
| Ped136 | Mixed ancestry | Moolman-Smook et al. (1998) |
| Δc756: | ||
| SB752 | Mixed ancestry | Present study |
| Val896Met: | ||
| SB852 | White | Present study |
Clinical examination was performed as described elsewhere (Posen et al. 1995). In brief, echocardiographic diagnosis of HCM was made in the presence of a maximal ventricular-wall thickness ⩾13 mm, in the absence of contributing confounding factors. In children, diagnosis of HCM was made with reference to age-adjusted tables (Feigenbaum 1981, pp. 551–552). Electrocardiographic diagnosis of HCM was based on the presence of either left-ventricular hypertrophy or abnormal Q waves. Other echocardiographic or electrocardiographic changes previously described in familial HCM were noted for each patient.
Source of DNA and DNA Extraction
Peripheral blood for genotypic analysis was collected from all probands and family members and from healthy, unaffected, unrelated control individuals of mixed ancestry or white descent (100 individuals per control panel) entered in the study, and DNA was extracted as described elsewhere (Corfield et al. 1993). Dr. Lucy Carrier supplied DNA from French family 730 (Dausse et al. 1993).
Mutation Detection
Members of the HCM-affected panel were screened for previously reported and novel mutations in exons of βMHC (exons 3–10 and 12–23), MyBPC (exons 8, 18, 24–28, and 34), cTnT (exons 8, 9, 11, and 14–16), αTM (exons 2b, 3–5, 6b, 7, 8, and 9ab), cardiac troponin I (cTnI) (exons 7 and 8), myosin essential light chain (MELC) (exon 4), and myosin regulatory light chain (MRLC) (exons 2 and 5) (Vikstrom and Leinwand 1996; DNA Mutation Database), by PCR-SSCP analysis, and allele-specific restriction-enzyme analysis (ASREA) when applicable. Amplified exons in which mobility shifts were identified by PCR-SSCP analysis were bidirectionally sequenced as described elsewhere (Posen et al. 1995).
In the case of the βMHC Glu499Lys mutation, exon 15 of the gene was PCR-amplified by use of custom-synthesized primers (DNA Laboratory, Cape Town)—MYH15F (5′-CACCCACTTTCTGACTGC-3′) and MYH15R (5′-GAATTCAGGTGGTAAGGCC-3′)—designed from published gene sequences (Jaenicke et al. 1990). In the case of variants Δc756 and Val896Met, exons 24 and 27 (exon numbering is according to Niimura et al. 1998), respectively, of MyBPC were amplified by use of the relevant published primer sequences (Carrier et al. 1997). PCR amplification was performed in a standard reaction mixture (Moolman et al. 1993), during 30 cycles of the following cycling profile: denaturation at 95°C (30 s), annealing at either 52°C (for Glu499Lys) or 55°C (for Δc756 and Val896Met) (30 s), and extension at 72°C (30 s), by a GeneE thermocycler (Techne). The sequence changes causing the βMHC Glu499Lys and MyBPC Val896Met mutations were verified by ASREA. The βMHC Glu499Lys nucleotide substitution abolished a MnlI site, whereas the MyBPC Val896Met mutation created an additional NlaIII site, in the respective PCR-amplified products. These ASREA tests—or, in the case of MyBPC Δc756, PCR-SSCP analysis—were used to genotype 100 unrelated, unaffected control samples of the relevant ethnic group, for the presence of these mutations.
Previously reported mutations—namely, βMHC Arg719Gln (Consevage et al. 1994), additional βMHC Arg403Trp (Moolman et al. 1993) and Ala797Thr (Moolman et al. 1995) and cTnT Arg92Trp–mutation carriers (Moolman et al. 1997)—identified in HCM-affected probands and family members in the present study, were detected by ASREA, as described elsewhere.
Genotyping Markers at the βMHC and cTnT Loci
Members of Ped106, the French family 730, and individual SB1140 were genotyped for the βMHC Arg403Trp mutation, an intragenic marker in the 5′ UTR region of βMHC (MYH7 5′UTR), and with βMHC-flanking markers at the D14S50, MYH6, and D14S64 loci, covering a distance of ⩾8.8 cM (fig. 1A) (GeneMap '99). In addition to these markers, members of Ped101, Ped104, Ped110, Ped124, Ped131, and Ped138 and individuals SB902, SB983, and SB995, all carrying the βMHC Ala797Thr mutation (table 1), were genotyped at two other loci—D14S283 and D14S264. Members of Ped100, Ped109, Ped137, Ped139, and Ped142 and individual SB385, all carrying the cTnT Arg92Trp mutation (table 1), were genotyped with cTnT-flanking markers at the F13B and D1S53 loci, spanning a distance of 11.2 cM (Watkins et al. 1993a), and with an exon 9 cTnT intragenic RFLP marker (TnTex9) (Thierfelder et al. 1994) (fig. 1B).
Figure 1.

A, Genetic map of the region flanking the βMHC locus on chromosome 14q11-12. The order of genetic markers, as well as the genetic distances between them, used in haplotype construction in families harboring the βMHC Arg403Trp and the Ala797Thr mutations are shown, according to GeneMap '99. MYH7 = βMHC; MYH6 = αMHC. B, Genetic map of the region flanking the cTnT locus on chromosome 1q3. The order of genetic markers, as well as the genetic distances between them, used in haplotype construction in families harboring the cTnT Arg92Trp mutation are shown, as mapped by Watkins et al. (1993a).
The short tandem-repeat markers at the D14S50, D14S283, MYH7 5′UTR, MYH6, D14S64, D14S264, F13B, and D1S53 loci were analyzed by PCR-based assays, according to standard methods (Brink et al. 1995). The TnTex9 marker was genotyped by ASREA after TaqI digestion of the PCR-amplified product of exons 8 and 9, by use of a published primer sequence (Thierfelder et al. 1994). Forty members of each of the two control panels were genotyped at each marker locus analyzed in the family studies, to determine the frequency of alleles in the two subpopulations.
Haplotype and “Assumed” Haplotype Construction
Most-likely haplotypes were constructed with reference to gene and marker order on the respective chromosomes, in a family context. In the case of probands without a traced family, evidence of identity by descent was accepted if the alleles present allowed construction of a haplotype (designated as the “assumed haplotype”) identical with the disease-associated haplotype.
Results
Identification of Novel and Reported Mutations in the South African HCM-Affected Panel
During screening of the panel of HCM-affected probands, a novel guanine-to-adenine (g→a) transition was detected in exon 15 of βMHC in one individual (SB1112) (fig. 2A). This modification results in the substitution of lysine for the conserved wild-type glutamine at amino acid residue 499 in the protein (Glu499Lys), fortuitously resulting in the loss of an MnlI restriction-enzyme site, allowing mutation screening by ASREA (fig. 2B). Although it was not possible to extend the study to the proband's family, the mutation was not present in 100 white control individuals.
Figure 2.

Identification of the Glu499Lys mutation in exon 15 of βMHC. A, Partial sequence of the coding strand of exon 15 of βMHC in an individual heterozygous for the Glu499Lys mutation, showing the g→a transition, which results in the amino acid substitution of Lys for Glu at codon 499. B, Genotyping the Glu499Lys mutation by ASREA. Products of MnlI digestion of the 243-bp PCR-amplified product of exon 15 were separated on a 12% polyacrylamide gel. Internal MnlI sites generated 87-, 73-, 37-, 16-, and 15-bp fragments in unaffected individuals (lane 2). Loss of one of these sites in an HCM-affected heterozygous individual resulted in an additional, undigested 102-bp fragment (lane 1). Lane 3, Lambda PstI molecular-size marker. The 16- and 15-bp fragments were not resolved on this gel.
A further two, apparently novel mutations were also detected in MyBPC, one in each of two probands (SB752 and SB852) in the HCM panel. One of these mutations was a g→a transition in MyBPC exon 27 in SB852 (fig. 3A), which would cause the substitution of a methionine for a valine at residue 896 (Val896Met) and which, since it created an NlaIII restriction-enzyme site (fig. 3B), could be detected by ASREA. The second nucleotide variation, detected in SB752, involves the deletion of a cytosine at the first base position in codon 796 in MyBPC exon 24 (Δc756) (fig. 4A); this results in both a shift in open reading frame 3′ to codon 796 and the predicted incorporation of 65 aberrant amino acid residues before premature truncation of the protein. This deletion did not create or abolish a restriction-enzyme site, and screening for the mutation was subsequently based on PCR-SSCP analysis (fig. 4B). It was not possible to perform extended family studies in the case of the two probands with these sequence variations. However, the sequence variations were not present in 100 control individuals of the appropriate subpopulation groups screened by ASREA or PCR-SSCP analysis.
Figure 3.
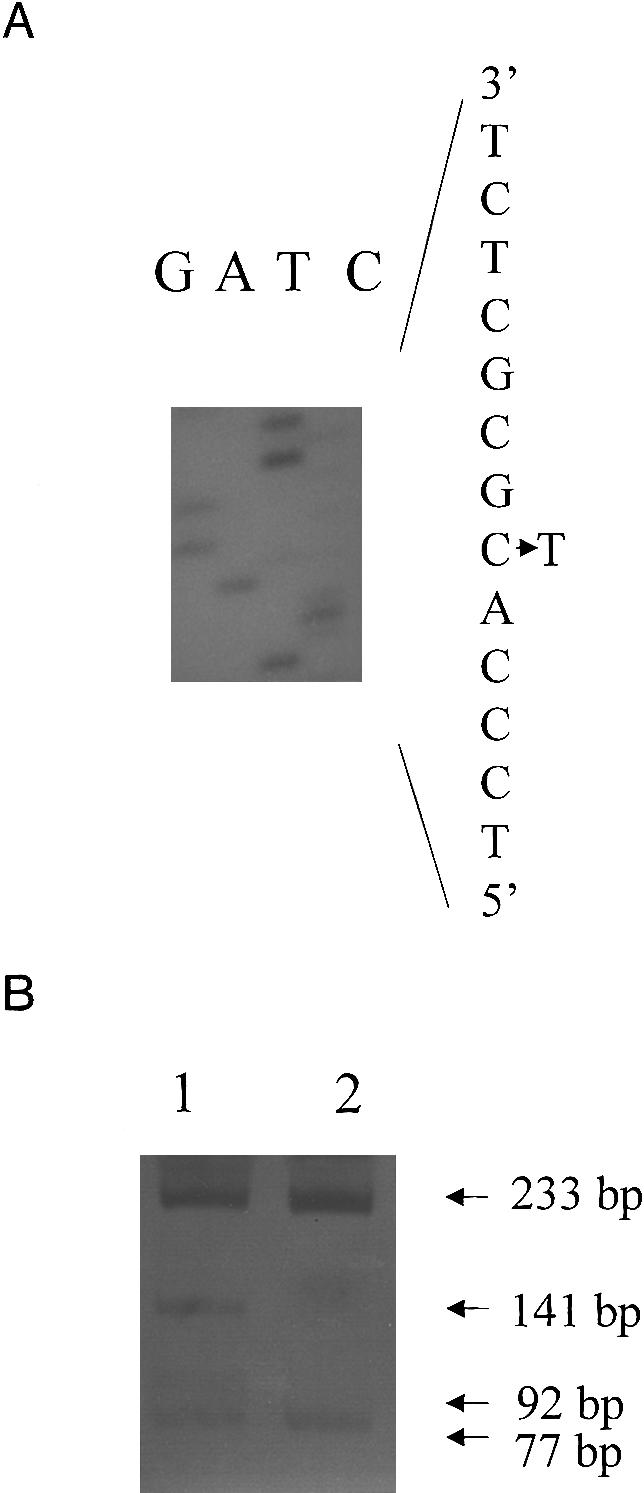
Identification of the Val896Met mutation in exon 27 of MyBPC. A, Partial sequence of the noncoding strand of exon 27 in an individual heterozygous for the Val896Met mutation, showing the c→t transition (g→a, coding strand) that results in the amino acid substitution of Met for Val at codon 896. B, Genotyping of the Val896Met mutation, by ASREA. Products of NlaIII digestion of the 310-bp PCR-amplification product of exon 27 were separated on a 12% polyacrylamide gel. An internal NlaIII site generated fragments of 233 and 77 bp in an unaffected individual (lane 2). The creation of an NlaIII site in an HCM-affected heterozygous individual resulted in additional, 141- and 92-bp fragments (lane 1).
Figure 4.

Identification of the cytosine deletion in codon 796 in exon 24 of MyBPC. A, Partial sequence of the noncoding strand of exon 24 of MyBPC in an unaffected individual (i) and a heterozygous HCM-affected individual (ii), showing the deletion of a g (c, coding strand) at nucleotide position 2298 (first position of codon 796), which results in a shift in reading frame. B, PCR-SSCP analysis of amplified products of exon 24 of MyBPC, separated on a 10% polyacrylamide gel containing 5% glycerol. The arrows indicate the mobility shifts caused by the Δc mutation at nucleotide 2298 in a heterozygous HCM-affected individual (lane 2). The single-stranded conformers of an unaffected individual are seen in lane 1.
In addition to the three novel mutations reported above, three documented βMHC mutations were detected by ASREA in the present study—the Arg403Trp substitution in an additional proband (SB1140), the Ala797Thr substitution in eight more probands, and the Arg719Gln substitution in a single proband of the HCM-affected panel. The previously reported cTnT Arg92Trp mutation was detected in a further four probands.
Thus, if the five mutations previously identified in six probands are included, a total of nine different mutations have been found in 23 of the individuals constituting the panel of 40 HCM-affected patients (table 1). In the remaining 17 probands, none of the ⩾100 previously reported mutations were present (Bonne et al. 1998), nor were any novel mutations detected in the screening of selected exons of the seven candidate sarcomeric protein–encoding genes. Of these genetically undiagnosed individuals, 60% were white and the remainder were of mixed ancestry.
Origins of the βMHC Arg403Trp Mutation
The βMHC Arg403Trp mutation (Dausse et al. 1993; Posen et al. 1995) was embedded in two distinct haplotypes in South African Ped106 and French family 730, indicating their independent origins (fig. 5). However, individual SB1140 shared alleles—and, therefore, an assumed disease-associated haplotype—with Ped106, implying the existence of a common ancestor (fig. 5). The validity of this assumption was supported by the fact that the allele occurring at each of the markers from which the disease-associated haplotype was constructed was not the one carried most commonly in the control mixed-ancestry population (fig. 6, legend). The disease-associated haplotype extended from D14S50 to D14S64, a distance of ⩾8.8 cM, with no evidence of recombination in the extended Ped106 (data not shown).
Figure 5.

Haplotypes across the β and αMHC loci associated with the βMHC Arg403Trp mutation. The disease-associated haplotypes in a representative subset of South African Ped106 and the French family 730, as well as the assumed haplotype of individual SB1140, bearing the βMHC Arg403Trp mutation are boxed. In the married-in individuals, the haplotypes were inferred on the basis of those of their children. The marker loci used in haplotype construction, covering a distance ⩾8.8 cM, are indicated to the left of the subsets, in centromeric-telomeric order, from top to bottom. The frequencies of the alleles of the markers genotyped in the control mixed-ancestry subpopulation are given in the legend to figure 6. Note that in the Ped106 subset shown there were no clinically HCM-affected mutation carriers; however, in the extended pedigree there were numerous affected individuals (Posen et al. 1995). MYH7 = βMHC; MYH6 = αMHC; R = arginine; W = tryptophan.
Figure 6.

Haplotypes across the β and αMHC loci associated with the βMHC Ala797Thr mutation. The disease-associated haplotype in representative subsets of families Ped101, Ped104, Ped110, Ped124, Ped131, and Ped138, as well as the assumed haplotype of individuals SB902, SB983, and SB995, bearing the Ala797Thr mutation are boxed. The marker loci used in haplotype construction, covering a distance ⩾8.8 cM, are indicated to the left of the subsets, in centromeric-telomeric order, from top to bottom. The frequencies (white/mixed ancestry) of the alleles of the markers genotyped in the control subpopulations, listed in the order of marker-allele number, are as follows: D14S50—1 (.00/.02), 2 (.01/.00), 3 (.05/.09), 4 (.28/.26), 5 (.21/.18), 6 (.11/.11), 7 (.20/.24), 8 (.13/.08), 9 (.01/.01), and 10 (.00/.01); MYH7 5′UTR—1 (.01/.06), 2 (.03/.05), 3 (.15/.08), 4 (.38/.21), 5 (.32/.28), 6 (.11/.23), and 7 (.00/.09); MYH6—1 (.01/.00), 2 (.01/.00), 3 (.01/.00), 4 (.03/.04), 5 (.01/.00), 6 (.01/.06), 7 (.04/.01), 8 (.07/.04), 9 (.13/.13), 10 (.15/.17), 11, (.32/.21), 12 (.07/.15), 13 (.05/.06), 14 (.04/.07), 15 (.04/.04), and 16 (.01/.02); and D14S64—1 (.02/.01), 2 (.12/.08), 3 (.12/.15), 4 (.29/.06), 5 (.32/.11), 6 (.10/.36), 7 (.02/.14), 8 (.01/.07), and 10 (.00/.02). The allele frequencies at D14S283 and D14S264 were not determined in the control subpopulation groups. MYH7 = βMHC; MYH6 = αMHC; A = alanine; T = threonine.
Origin of the βMHC Ala797Thr Mutation
In the six pedigrees—Ped101, Ped104, Ped110, Ped124, Ped131, and Ped138—that harbored βMHC Ala797Thr, a common core haplotype encompassing the βMHC and αMHC loci segregated with the disease-causing mutation, suggesting descent from a shared ancestor (fig. 6). Furthermore, the two parents shown in the Ped101 subset (fig. 6) both carried the Ala797Thr mutation, embedded in the disease-associated haplotype, suggesting that they may be related. Within the families, the shared haplotype generally extended ⩾8.8 cM across the region flanking βMHC and αMHC, with evidence of recombination both between MYH7 5′UTR and the proximal D14S283 marker, in Ped138, and between MYH6 and the distal D14S64 marker, in Ped104 (fig. 6). These events reduced the extent of the conserved haplotype to ⩽8.8 cM, which, because of both the unavailability of mapped markers between MYH6 and D14S64 and the small genetic distance between MYH7 and D14S283 (fig. 1A), could not be substantially narrowed further. The alleles at the marker loci genotyped in individuals SB902, SB983, and SB995 were also compatible with that of the common extended ancestral disease-associated haplotype that was ⩾8.8 cM (fig. 6). The existence of a common ancestor was supported by the fact that the allele at the markers from which the disease-associated haplotype was formed was generally not the one carried most commonly in either of the control subpopulation groups (fig. 6, legend). The only exception was at the D14S50 locus in an individual in Ped101, in whom a recombination event had occurred. Five of the families or probands carrying the βMHC Ala797Thr mutation were members of the white subpopulation, and four were of mixed ancestry (table 1).
Origin of the cTnT Arg92Trp Mutation
In the five families that carried the cTnT Arg92Trp mutation and that could be traced (i.e., Ped100, Ped109, Ped137, Ped139, and Ped142), a common disease-associated haplotype, stretching, without recombination, between marker loci D1S53 and F13B (a distance of ∼11.2 cM) in the extended pedigrees, segregated with the mutation (fig. 7). Since genotyping was not continued beyond these markers, the size of the conserved fragment is unknown and may indeed extend further. In addition, individual SB385 possessed, at the markers genotyped, alleles that were compatible with the disease-associated haplotype. The data suggest that, in these families and this individual, all of which are members of the mixed-ancestry subpopulation, the mutation is identical by descent. The validity of this assumption was supported by the fact that the allele occurring at each of the markers from which the disease-associated haplotype was constructed was not the one carried most commonly in the control mixed-ancestry population, except at the biallelic TnTex9 marker (fig. 7, legend).
Figure 7.

Haplotypes across the cTnT locus associated with the Arg92Trp mutation. The disease-associated haplotype in representative subsets of families Ped100, Ped109, Ped137, Ped139, and Ped142, as well as the assumed haplotype of individual SB385, bearing the Arg92Trp mutation are boxed. In the married-in individual, the genotype at each marker locus is shown; a haplotype was not constructed, since the phase was unknown. The marker loci used in haplotype construction, covering a distance of 11.2 cM, are indicated to the left of the subsets, in centromeric-telomeric order, from top to bottom. The frequencies of the alleles at the markers genotyped in the control mixed-ancestry subpopulation were as follows, listed in the order of marker-allele number: F13B—1 (.28), 2 (.23), 3 (.14), 4 (.08), and 5 (.27); TnTex9—1 (.70) and 2 (.30); and D1S53—1 (.05), 2 (.09), 4 (.02), 5 (.02), 7 (.05), 8 (.02), 9 (.11), 10 (.12), 11 (.09), 12 (.06), 13 (.09), 14 (.05), 15 (.14), 16 (.02), and 17 (.07). Note that, although, in the Ped100 subset shown, there were no HCM-affected mutation carriers, in the extended pedigree there were numerous affected individuals (Moolman et al. 1997). R = arginine; W = tryptophan.
Discussion
The Spectrum and Possible Effects of HCM-Causing Mutations in South Africa
Our investigations into the molecular causes of HCM in a panel of 40 apparently unrelated South African patients revealed that previously reported mutations, occurring worldwide, were apparently rare in this country. Of these mutations, only the βMHC Arg719Gln (Consevage et al. 1994) and Arg249Gln (Watkins et al. 1992; Posen et al. 1995) substitutions were detected in two separate probands of the South African HCM panel. Additionally, the βMHC Arg403Trp mutation, which originally had been described as occurring in South Africa (Moolman et al. 1993), subsequently was reported in French and Polish families (Dausse et al. 1993; Al-Mahdawi et al. 1994). Other than the aforementioned βMHC mutations, none of the ⩾100 other mutations detected globally (Bonne et al. 1998, FHC database) in βMHC, cTnT, MyBPC, αTM, MELC, MRLC, or cTnI were present in the South African subpopulations screened. The α−cardiac actin gene, which recently had been implicated as another cause of HCM (Mogensen et al. 1999), was not screened in the present investigation.
We previously had described three novel mutations in the South African population—namely, βMHC (Ala797Thr) (Moolman et al. 1995), cTnT (Arg92Trp) (Moolman et al. 1997), and MyBPC (Arg654His) (Moolman-Smook et al. 1998). Here, we have presented evidence of three additional novel disease-causing mutations identified in HCM-affected individuals in the Western and Eastern Cape provinces of South Africa, one in βMHC and two in MyBPC.
The βMHC Glu499Lys substitution occurred at a residue conserved in myosin proteins, across a range of species—including human, rabbit, mouse, pig, hamster, and rat—and in a region that probably forms an interface with the reactive sulfydryls (Rayment et al. 1995). The Δc756 MyBPC deletion, which occurs in a region encoding the Ig-like module-C5 domain of the protein, results in a reading frameshift, with predicted production of an aberrant and prematurely terminated molecule. The resultant loss of the carboxy-terminal residues of MyBPC is likely to affect the ability of the protein both to be incorporated into the A-band of the sarcomere and to bind to myosin and titin (Freiburg and Gautel 1996). Considering the Val896Met substitution, structurally similar nonpolar amino acid residues with aliphatic hydrocarbon side chains occur at position 896 in MyBPC, across isoforms. Valine, which has a branched side chain, occurs in human cardiac MyBPC at residue 896 and differs, by a single methyl group, from the isoleucine residue found in human fast and slow, chicken cardiac, and rat MyBPC isoforms at this position. It can be surmised that replacement of this valine by methionine, with its longer, unbranched side chain containing a sulfur group, will affect the conserved fibronectin type 3 structure of MyBPC module C7 (Gautel et al. 1995). The latter domain, along with modules C8–C10, is necessary for incorporation of the protein into the A-band of the sarcomere (Freiburg and Gautel 1996).
It was not possible to follow the segregation of either these described variants or the disease, in the families of the three probands. However, their occurrence in conserved positions and/or regions postulated to be functionally important protein domains of βMHC and MyBPC, as well as their absence in control panels of both subpopulation groups studied, implicates them as disease-causing mutations.
Independent Origin of the South African Mutations
In the 17th century, French Huguenots settled in the Western Cape region of South Africa (Botha and Beighton 1983), so the possibility existed that the βMHC Arg403Trp mutation in Ped106 and French family 730 were identical by descent. The mild phenotypic expression associated with this mutation on both continents (Dausse et al. 1993; Posen et al. 1995) also suggests that it might have spread by migration, since reproductive fitness might be unaffected in families harboring it. However, a comparison of the haplotypes associated with the mutation in these two kindreds provides evidence in favor of their independent origin (fig. 5). The βMHC Arg403Trp mutation has also been reported in Polish families (Al-Mahdawi et al. 1994), and the βMHC Arg249Gln and Arg719Gln mutations identified in the South African HCM-affected panel also occur in Canadian and Hispanic families, respectively (Watkins et al. 1992; Consevage et al. 1994). We do not know whether the South African versions of these mutations are identical by state or identical by descent, to their counterparts in the other populations. It is possible that French immigrants to Canada and South Africa may share common ancestry for the Arg249Gln mutation. However, since immigrants from Poland and Spain have not made a major contribution to the South African gene pool, it is likely that Arg403Trp and Arg719Gln are identical by state, rather than identical by descent, to those in these two countries.
Six of the nine HCM-causing mutations detected in South Africa are novel and, when the βMHC Arg403Trp data presented here also are considered, at least one of the other three also has an independent origin, whereas the source of the other two mutations could not be investigated. No other of the disease-associated mutations reported worldwide occurs in the South African groups studied. These data support the proposal that the profile of HCM-causing mutations will be unique in different geographic areas and that it is the result of numerous nascent mutations.
Founder Effects in South African HCM-Affected Families
In addition to the evidence supporting the independent occurrence of several HCM-causing mutations in probands originating from the Western and Eastern Cape provinces, three of the mutations were found in more than one apparently unrelated affected individual. The βMHC Arg403Trp and Ala797Thr mutations were found in two and nine probands, respectively, and the cTnT Arg92Trp mutation was found in six apparently unrelated affected individuals. Haplotype analysis across the βMHC and cTnT loci in relevant families and individuals (table 1) revealed that all the individuals who carried the same mutation also shared either a disease-associated haplotype or a haplotype that was assumed to be disease associated (figs. 5–7). The haplotype was conserved over a region of ⩽8.8 cM of the βMHC locus in persons harboring the Ala797Thr mutation and over a region of ⩾11.2 cM of the cTnT locus in individuals carrying Arg92Trp. These data are consistent with the possibility that each of the three recurring mutations is identical by descent—because of the existence, in each case, of a common ancestor—rather than being identical by state.
This report is the first that substantiates the existence of strong founder effects as the cause of HCM in generally homogeneous population groups. Jääskeläinen et al. (1998) have suggested that the occurrence of the αTM Asp175Asn mutation in four unrelated Finnish kindreds might reflect the presence of a common ancestor. This finding would not be unexpected, since the genetically homogeneous population of Finland is known for its “Finnish disease heritage” (de la Chapelle 1993, p. 857). However, the proposal was based only on the fact that all the HCM-affected relatives shared the same allele at an αTM intragenic marker, and the frequency of this allele in this homogeneous population was not given. In the two Cape subpopulations studied, the likely existence of two founder individuals for the βMHC Ala797Thr and cTnT Arg92Trp mutations was based on extended haplotype data.
The presently reported exception to the general finding of the independent nature of HCM-causing mutations is not unanticipated. Founder-gene effects are not uncommon in South Africa, especially among the Afrikaner subgroup of the white population (Botha and Beighton 1983; Kotze et al. 1991; Brink et al. 1995; De Jager et al. 1996; Warnich et al. 1996). These effects are probably due to bottlenecks in gene transmission, caused by rapid expansion of the white population in the Western Cape after the initial European colonization (Botha and Beighton 1983). During the period 1691–1791, the colony's population increased from 1,000 to 13,028 (Botha and Beighton 1983), mainly because of natural expansion, since only 766 new European immigrant surnames were tabulated during this period (Theal 1922; Cameron and Spies 1992, pp. 11–139). At the same time, a group of ethnic admixture originated by intermingling of the European immigrants, mostly with the resident Khoi-khoi and San peoples (the Khoisan) but also with native black Africans and, later, with slaves transported from Malaysia and other East Indian countries (Cameron and Spies 1992, pp. 11–139). This group has become a distinct genetic entity, herein referred to as “mixed ancestry” (often referred to as “colored”), which has largely been maintained by intercultural matings (Botha and Pritchard 1972; May and Du Toit 1989). The history of the white and mixed-ancestry subpopulations of the Western Cape also explains the presence of the βMHC Ala797Thr mutation in both subpopulations. After the initial colonization of the Western Cape, changing economic circumstances resulted in the migration of individuals to the Eastern Cape (Botha and Beighton 1983).
The ethnicity of the three ancestors in whom the founder mutations arose cannot be ascertained; however, since our data suggest that it is unlikely that the mutations originated in Europe, it can be assumed that they arose in Africa. Since HCM-causing mutation screening has not been performed in the Khoisan or black African subpopulations, the latters' contribution to the pool of disease-associated alleles cannot be established. It is possible that the βMHC Ala797Thr mutation arose in the white population, postcolonization, and thereafter was introduced into the mixed-ancestry group. However, both the mild phenotype generally accompanying this mutation (authors' unpublished observations) and the disease-associated haplotype of ⩽8.8 cM, allow speculation that this is an older mutation, which arose in the indigenous Khoisan people, whose presence in the Western Cape predated migration of black Africans from north of the Cape Province (Cameron and Spies 1992, pp. 11–139). Its presence in families of white descent would reflect the blurring of ethnic distinctions, because of earlier admixture among the subpopulations living in the Western Cape. In contrast, the reduced life expectancy previously reported to be associated with the cTnT Arg92Trp mutation (Moolman et al. 1997), together with the extensive conserved haplotype in which it is embedded in so many families, suggests a recent mutational event. Since this variant has been detected only in persons of mixed ancestry, it can be speculated that it arose in an ancestor of this subgroup and then, despite its malignant phenotype, increased in frequency during a time of rapid population expansion. The decreased survival associated with this cTnT mutation was most pronounced in male family members (Moolman et al. 1997). Therefore, it is possible that inheritance of the mutant allele was either through female carriers in the generally large families seen to this day in this subpopulation group or through those male carriers who did survive to reproductive age. Rationally, pronounced founder effects are not expected to be associated with mutations resulting in significant mortality during reproductive years (de la Chapelle 1993; Watkins et al. 1993b). We propose that the population dynamics seen in the South African subpopulations studied here have overridden the reproductive disadvantage conferred by these mutations.
The prevalence of founder effects in a population may have an impact on the success of any molecular diagnosis program. In the Western and Eastern Cape provinces, the βMHC Ala797Thr mutation alone accounts for ∼25% of the HCM cases in the panel, whereas the cTnT Arg92Trp is the causative mutation in 15%, and the βMHC Arg403Trp in 5%, of affected individuals. Three other βMHC mutations—namely, Arg249Gln, Arg719Gln, and Glu499Lys—collectively account for another 7.5% of disease cases. Therefore, in the South African subpopulations studied here, five βMHC mutations are collectively responsible for ∼37.5% of HCM. This finding is very different from the profiles seen in most North American and European referral centers, where numerous “private” βMHC mutations are reported to account for ∼30%–40% of HCM. Consequently, we have adopted a mutation-screening program in which any patient referred for molecular diagnosis is first screened for these three founder mutations, by ASREA, and more extensive mutation screening is performed only in their absence. Such a focused molecular diagnosis program saves both time and money.
Moreover, the presence of large cohorts of affected individuals who are members of relatively homogeneous subpopulations bearing the same βMHC Ala797Thr or cTnT Arg92Trp mutations will make it possible to develop a clearer picture of the true genotype-phenotype correlations. This is especially relevant, since it is increasingly being realized that the clinical expression of a disease is context dependent and is influenced not only by the presence of a recognized major-locus mutation but also by the genetic and environmental background in which it occurs (Bonne et al. 1998). In a previous study based on two families (Ped100 and Ped109), the cTnT Arg92Trp mutation was shown to be associated with only moderate hypertrophy but with a high risk of sudden cardiac death (Moolman et al. 1997). A similar clinical picture was evident in the additional families bearing this mutation (our unpublished observations). However, although the initial report of the βMHC Ala797Thr mutation seen in one nuclear family suggested a poor prognosis (Moolman et al. 1995), continuing investigations have suggested a milder clinical disease course (authors' unpublished observations). It is anticipated that further extensive genotype-phenotype correlations will facilitate more-accurate prognostication and thereby improve patient management and counseling of this clinically insidious condition.
Acknowledgments
We wish to thank the patients and their families for participating in this study, as well as the physicians who referred patients and collected samples. This work was supported by the South African Medical Research Council and the Harry and Doris Crossley Fund of the University of Stellenbosch.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- DNA Mutation Database, http://www.angis.org.au/Databases/Heart/heartbreak.html (for familial HCM)
- GeneMap '99, http://www.ncbi.nlm.nih.gov/genemap/map.cgi?CHR=14
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim (for HCM [MIM 192600])
References
- Al-Mahdawi S, Chamberlain S, Chojmowska L, Machalak E, Nihoyannopoulos P, Ryan M, Kusnierczyk B, et al (1994) The electrocardiogram is a more sensitive indicator than echocardiography of hypertrophic cardiomyopathy in families with a mutation in the MYH7 gene. Br Heart J 72:105–111 [DOI] [PMC free article] [PubMed]
- Bonne G, Carrier L, Richard P, Hainque B, Schwartz K (1998) Familial hypertrophic cardiomyopathy from mutations to functional defects. Circ Res 83:580–593 [DOI] [PubMed]
- Botha MC, Beighton P (1983) Inherited disorders in the Afrikaner population of southern Africa. I. Historical and demographic background, cardiovascular, neurological, metabolic and intestinal conditions. S Afr Med J 64:609–612 [PubMed]
- Botha MC, Pritchard J (1972) Blood group frequencies: an indication of the genetic constitution of population samples in Cape Town. S Afr Med J Suppl 46:1–27 [PubMed]
- Brink PA, Ferreira A, Moolman JC, Weymar HW, Van der Merwe P-L, Corfield VA (1995) Gene for progressive familial heart block type I maps to chromosome 19q13. Circulation 91:1633–1640 [DOI] [PubMed]
- Cameron T, Spies T (1992) Nuwe geskiedenis van Suid-Afrika, rev ed. Human & Rousseau, Cape Town [Google Scholar]
- Carrier L, Bonne G, Bährend E, Yu B, Richard P, Niel F, Hainque B, et al (1997) Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res 80:427–434 [PubMed]
- Consevage MW, Salada GC, Baylen BG, Ladda RL, Rogan PK (1994) A new missense mutation, Arg719Gln, in the β-cardiac heavy chain myosin gene of patients with familial hypertrophic cardiomyopathy. Hum Mol Genet 3:1025–1026 [DOI] [PubMed]
- Corfield VA, Moolman JC, Martel R, Brink PA (1993) Polymerase chain reaction-based detection of MN blood group-specific sequences in the human genome. Transfusion 33:119–124 [DOI] [PubMed]
- Dausse E, Komajda M, Fetler L, Dubourg O, Dufour C, Carrier L, Wisnewsky C, et al (1993) Familial hypertrophic cardiomyopathy: microsatellite haplotyping and identification of a hot spot for mutations in the β-myosin heavy chain gene. J Clin Invest 92:2807–2813 [DOI] [PMC free article] [PubMed]
- De Jager T, Corbett CH, Badenhorst JCW, Brink PA, Corfield VA (1996) Evidence of a long QT founder gene with varying phenotypic expression in South African families. J Med Genet 33:567–573 [DOI] [PMC free article] [PubMed]
- de la Chapelle A (1993) Disease gene mapping in isolated human populations: the example of Finland. J Med Genet 30:857–865 [DOI] [PMC free article] [PubMed]
- Feigenbaum H (1981) Echocardiography, 3d ed. Lea-Febigery, Philadelphia [Google Scholar]
- Freiburg A, Gautel M (1996) A molecular map of the interactions of titin and myosin-binding protein C: implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem 235:317–323 [DOI] [PubMed]
- Gautel M, Zuffardi O, Freiburg A, Labeit S (1995) Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J 14:1952–1960 [DOI] [PMC free article] [PubMed]
- Jääskeläinen P, Soranta M, Miettinen R, Saarinen L, Pihlajamäki J, Silvennoinen K, Tikanoja T, et al (1998) The cardiac β-myosin heavy chain is not the predominant gene for hypertrophic cardiomyopathy in the Finnish population. J Am Coll Cardiol 32:1709–1716 [DOI] [PubMed]
- Jaenicke T, Diederich KW, Haas W, Schleich J, Lichter P, Pfordt M, Bach A, et al (1990) The complete sequence of human beta-myosin heavy chain gene and a comparative analysis of its product. Genomics 8:194–206 [DOI] [PubMed]
- Kotze MJ, Langenhoven E, Warnich L, Du Plessis L, Retief AE (1991) The molecular basis and diagnosis of familial hypercholesterolaemia in South African Afrikaners. Ann Hum Genet 55:115–121 [DOI] [PubMed]
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA Study. Circulation 92:785–789 [DOI] [PubMed]
- May RM, Du Toit ED (1989) Blood group gene frequencies of four population groups in the western Cape. S Afr Med J 76:647–650 [PubMed]
- Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, Gregersen N, et al (1999) Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest 103:R39–43 [DOI] [PMC free article] [PubMed]
- Moolman JC, Brink PA, Corfield VA (1993) Identification of a new missense mutation at Arg403, a CpG mutation hotspot in exon 13 of the β-myosin heavy chain gene in hypertrophic cardiomyopathy. Hum Mol Genet 2:1731–1732 [DOI] [PubMed]
- ——— (1995) Identification of a novel Ala797Thr mutation in exon 21 of MYH7. Hum Mutat 6:197–198 [DOI] [PubMed]
- Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, Watkins H (1997) Sudden death due to troponin T mutations. J Am Coll Cardiol 29:549–555 [DOI] [PubMed]
- Moolman-Smook JC, Mayosi B, Brink P, Corfield VA (1998) Identification of a new missense mutation in MyBP-C associated with hypertrophic cardiomyopathy. J Med Genet 35:253–254 [DOI] [PMC free article] [PubMed]
- Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, et al (1998) Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 338:1248–1257 [DOI] [PubMed]
- Posen BM, Moolman JC, Corfield VA, Brink PA (1995) Clinical and prognostic evaluation of familial hypertrophic cardiomyopathy in two South African families with different cardiac β myosin heavy chain gene mutations. Br Heart J 74:40–46 [DOI] [PMC free article] [PubMed]
- Rayment I, Holden HM, Sellers JR, Fananapazir L, Epstein ND (1995) Structural interpretation of the mutations in the β-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc Natl Acad Sci USA 92:3864–3868 [DOI] [PMC free article] [PubMed]
- Teare RD (1958) Asymmetrical hypertrophy of the heart in young adults. Br Heart J 20:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theal GMcC (1922) History of South Africa. Vol 3. Struik, Cape Town [Google Scholar]
- Thierfelder L, Watkins H, Macrae C, Lamas R, McKenna W, Vosberg H-P, Seidman JG, et al (1994) α-Tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 77:701–712 [DOI] [PubMed]
- Vikstrom KL, Leinwand LA (1996) Contractile protein mutations and heart disease. Curr Opin Cell Biol 8:97–105 [DOI] [PubMed]
- Warnich L, Kotze MJ, Groenewald IM, Groenewald JZ, Van Brakel MG, Van Heerden CJ, De Villiers NP, et al (1996) Identification of three mutations and associated haplotypes in the protoporphyrinogen oxidase gene in South African families with variegate porphyria. Hum Mol Genet 5:981–984 [DOI] [PubMed]
- Watkins H, MacRae C, Thierfelder L, Chou YH, Frenneaux M, McKenna W, Seidman JG, et al (1993a) A disease locus for familial hypertrophic cardiomyopathy maps to chromosome 1q3. Nat Genet 3:333–337 [DOI] [PubMed]
- Watkins H, Rosenzweig T, Hwang D-S, Levi T, McKenna W, Seidman CE, Seidman JG (1992) Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med 326:1108–1114 [DOI] [PubMed]
- Watkins H, Thierfelder L, Anan R, Jarcho J, Matsumori A, McKenna W, Seidman JG, et al (1993b) Independent origin of identical β cardiac myosin heavy-chain mutations in hypertrophic cardiomyopathy. Am J Hum Genet 53:1180–1185 [PMC free article] [PubMed]