

Abstract
Background
For many years, scientists believed that point mutations in genes are the genetic switches for somatic and inherited diseases such as cystic fibrosis, phenylketonuria and cancer. Some of these mutations likely alter a protein's function in a manner that is deleterious, and they should occur in functionally important regions of the protein products of genes. Here we show that disease-associated mutations occur in regions of genes that are conserved, and can identify likely disease-causing mutations.
Results
To show this, we have determined conservation patterns for 6185 non-synonymous and heritable disease-associated mutations in 231 genes. We define a parameter, the conservation ratio, as the ratio of average negative entropy of analyzable positions with reported mutations to that of every analyzable position in the gene sequence. We found that 84.0% of the 231 genes have conservation ratios less than one. 139 genes had eleven or more analyzable mutations and 88.0% of those had conservation ratios less than one.
Conclusions
These results indicate that phylogenetic information is a powerful tool for the study of disease-associated mutations. Our alignments and analysis has been made available as part of the database at http://cancer.stanford.edu/mut-paper/. Within this dataset, each position is annotated with the analysis, so the most likely disease-causing mutations can be identified.
Introduction
For many years, scientists believed that point mutations in genes are the genetic switches for somatic and inherited diseases such as cystic fibrosis, phenylketonuria and cancer. For this to be the case, disease-associated amino acid substitutions should occur in functionally important regions of the protein products of genes. While it has been shown in specific cases that disease-associated amino acid substitutions affect protein function, until now few studies have examined this across many genes. Here we provide direct evidence that disease-associated point mutations occur in functionally important regions of the genome and are not distributed equally across the coding regions of genes. This work supports recent efforts to collect disease associated mutational data in databases and suggests that many of the mutations represented in those databases are the likely underlying molecular cause of disease.
Recently there have been a number of commercial and public projects aimed at collecting and understanding human genomic variation [1]. The goal of these projects is to provide an understanding of how genotype is associated with disease, how it affects our response to drugs and how it affects the protein products of genes. Examples of these projects include the SNP Consortium, the Human Genome Mutation Database [2], many gene specific databases ([3,4], for example), and both public and private genome sequencing efforts [5]. Much of the data that is being collected are mutations annotated with their observed phenotype. Automated annotation methods based on structural and evolutionary parameters can lead to insight into the molecular basis of disease.
With more than 4,000,000 identified variations and with over 20,000 of them annotated with a phenotype, we are facing the problem of having many uncharacterized mutations. Algorithms are needed for automatically annotating these gene variations to gain insight into how they affect the gene's regulation and/or function of its protein products. Using many collection technologies, uncharacterized SNP data is being placed in public databases such as the Human Genome Mutation Database (over 20,000 entries) [2] and the National Cancer Institute's CGAP-GAI (Cancer Genome Anatomy Project Genetic Annotation Initiative) [6]. The CGAP-GAI group has identified 10,243 SNPs by examining publicly available EST (Expressed Sequence Tag) chromatograms.
Software for analyzing unannotated SNPs in known disease associated genes will be especially useful when previously unobserved mutations are discovered. Every human has genotypic differences from the standard genome approximately every thousand base pairs [7-9]. Given knowledge of how a genotype differs from the standard, it is important to be able to predict which of the variations are likely to be the cause of disease or other phenotypic differences. Evolutionary information about regulatory and coding regions of genes can be used to highlight certain mutations or groups of mutations that are attributable to a phenotype [10-12].
Early tools using phylogenetic and structural information have shown promise in predicting the functional consequences of a mutation [13]. These reports predict that anywhere between 20–36% of non-synonymous SNPs alter the function of a gene's protein product. In the report by Chasman and Adams, evolutionary information was predicted to be a useful component in determining whether a mutation is deleterious [13,14]. Disease causing mutations are also likely structurally perturbing at the protein level [15]. Ng and Henikoff have introduced SIFT, a method for predicting functional SNPs from a database of unannotated polymorphisms [16,17].
The relationship between disease-associated mutation positions and evolutionary conservation has been reported in specific cases. An analysis of the breast and ovarian cancer susceptibility gene, BRCA1, showed that disease-associated mutations tend to occur in highly conserved regions [18]. An analysis of homologous sequences in the androgen receptor has shown similar results [10]. Keratin 12, KRT12, is associated with Meesmann Corneal Epithelial Dystrophy (MCD). Reported mutations often occur in the highly conserved alpha-helix-initiation motif of rod domain 1A or in the alpha-helix-termination motif of rod domain 2B [19]. Structure based analysis methods have also been used to analyze Osteogenesis imperfecta associated COL1A1 mutations and disease-associated P53 mutations (Mooney and Klein, unpublished),[20]. Miller and Kumar have reported that disease-associated mutations are conserved in seven model genes [21].
To determine the degree to which mutation positions differ evolutionarily from other positions, we have built alignments of homologous genes for 231 disease-associated genes. These multiple alignments have then been used to assess the difference in evolutionary conservation for positions that are both disease-associated and not associated. The results show that, in general, positions with disease-associated mutations are conserved more than the average position in the alignment. This suggests the most conserved mutations are likely to be the causative agents of disease, and our data set identifies these mutations.
Results and Discussion
Our method compares the negative entropy of disease-associated columns within an alignment to other columns in that alignment. The goal of this work is to build these alignments, map the mutations to them, and show that disease-associated positions are, in general, conserved. The analysis was performed on the built alignments and the results are shown in Table 1.
Table 1.
Genes used in analysis with conservation ratio. 231 total genes were analyzed
| ABCA1 | 0.67 | CYP2C19 | 0.79 | KCNA1 | 0.40 | PROS1 | 0.98 |
| ABCA4 | 0.95 | CYP2D6 | 1.00 | KCNE1 | 0.32 | PTEN | 0.75 |
| ABCB11 | 0.75 | DES | 0.61 | KCNH2 | 0.51 | PTS | 0.70 |
| ABCC8 | 1.04 | DKC1 | 1.11 | KCNJ1 | 0.55 | RAG1 | 0.00 |
| ABCD1 | 0.61 | DMD | 1.06 | KCNQ1 | 0.69 | RAG2 | 0.31 |
| ACTA1 | 1.04 | ED1 | 1.04 | KCNQ4 | 0.18 | RB1 | 0.51 |
| ACTC | 1.01 | EGR2 | 0.62 | KRT12 | 0.38 | RDH5 | 0.76 |
| ACTN4 | 0.64 | F2 | 0.83 | L1CAM | 0.80 | RDS | 0.29 |
| ACVRL1 | 0.52 | F7 | 0.75 | LDLR | 0.96 | RGR | 1.01 |
| ADA | 0.77 | F8C | 0.85 | LIPC | 0.43 | RHO | 0.89 |
| ADSL | 1.08 | F9 | 0.88 | LMNA | 1.10 | ROM1 | 1.64 |
| ALB | 0.98 | FBN1 | 0.98 | MC2R | 0.73 | RPGR | 0.36 |
| ALDH10 | 0.66 | FBN2 | 0.91 | MEFV | 1.12 | RPS19 | 0.19 |
| ALDOB | 1.02 | FECH | 0.82 | MLH1 | 0.82 | RPS6KA3 | 0.47 |
| APC | 1.13 | FGA | 0.66 | MNGIE | 0.26 | RS1 | 0.86 |
| APP | 0.51 | FGFR3 | 1.15 | MPI | 0.77 | RUNX2 | 0.11 |
| AQP1 | 1.49 | FGG | 0.90 | MPZ | 0.40 | SCN4A | 0.64 |
| AQP2 | 0.77 | FIC1 | 0.85 | MSH2 | 0.77 | SCN5A | 0.61 |
| AR | 0.51 | FSHR | 0.54 | MSH6 | 1.09 | SCNN1B | 1.13 |
| AT3 | 0.95 | FUT1 | 0.38 | MTM1 | 0.78 | SGCA | 0.78 |
| ATM | 0.78 | G6PC | 0.46 | MYBPC3 | 0.94 | SLC12A1 | 0.56 |
| ATP2A2 | 0.64 | G6PD | 0.94 | MYH7 | 1.13 | SLC12A3 | 0.66 |
| ATP2A3 | 1.24 | G6PT1 | 0.66 | MYL2 | 1.43 | SLC17A5 | 0.75 |
| ATP2C1 | 0.59 | GAA | 0.36 | MYO7A | 0.88 | SLC22A5 | 0.61 |
| ATP6B1 | 0.57 | GALC | 0.82 | MYOC | 0.73 | SLC26A2 | 0.77 |
| ATP7A | 0.62 | GALE | 0.75 | NAGA | 1.01 | SLC2A2 | 0.58 |
| ATP7B | 0.36 | GALK1 | 1.18 | NF1 | 1.05 | SLC3A1 | 0.89 |
| AVP | 0.70 | GALNS | 0.83 | NF2 | 0.87 | SLC4A1 | 0.70 |
| AVPR2 | 0.82 | GALT | 0.81 | NOTCH3 | 0.93 | SLC5A1 | 0.66 |
| BCHE | 0.47 | GCH1 | 0.58 | NR0B1 | 0.42 | SLC5A5 | 0.76 |
| BLM | 0.65 | GCK | 0.71 | NR2E3 | 0.54 | SLC7A7 | 0.60 |
| BTK | 0.57 | GJB1 | 0.58 | NTRK1 | 0.68 | SLC7A9 | 0.43 |
| CACNA1A | 0.73 | GJB2 | 0.74 | OCA2 | 0.82 | SOD1 | 1.00 |
| CACNA1F | 0.31 | GJB3 | 0.98 | OCRL | 0.62 | SOX9 | 0.17 |
| CACNA1S | 0.91 | GLA | 0.63 | OTC | 0.71 | SRY | 0.46 |
| CAPN3 | 0.61 | GLB1 | 0.67 | PAFAH1B1 | 0.23 | STAR | 1.25 |
| CASR | 1.42 | GNAS1 | 0.67 | PAH | 0.69 | TAT | 0.52 |
| CAV3 | 0.70 | GP1BA | 1.08 | PAX3 | 0.86 | TGFBI | 0.55 |
| CBS | 0.57 | GP9 | 0.66 | PAX6 | 0.99 | TH | 0.84 |
| CDKN2A | 0.84 | HADHA | 0.77 | PAX8 | 0.00 | THBD | 1.08 |
| CFTR | 0.51 | HADHB | 0.72 | PCCA | 0.56 | TIMP3 | 0.83 |
| CLCN1 | 0.00 | HBB | 1.00 | PCCB | 0.62 | TNFRSF1A | 1.15 |
| CLN3 | 0.44 | HEXA | 0.61 | PDE6B | 0.71 | TNFRSF6 | 0.60 |
| CNGA3 | 0.52 | HEXB | 0.66 | PEPD | 0.08 | TNFSF5 | 0.40 |
| COMP | 0.61 | HGD | 0.23 | PHEX | 0.62 | TNNI3 | 0.40 |
| CPT2 | 1.01 | HK2 | 1.22 | PKLR | 0.63 | TNNT2 | 0.74 |
| CRB1 | 0.01 | HPRT1 | 0.90 | PLP1 | 0.82 | TP53 | 0.28 |
| CRX | 0.44 | HSD11B2 | 0.74 | PMM2 | 0.54 | TPM1 | 1.32 |
| CSX | 0.48 | HSD17B3 | 0.60 | PMP22 | 0.51 | TPMT | 0.76 |
| CYBB | 0.41 | HSD17B4 | 0.15 | POU1F1 | 0.36 | TSHR | 0.49 |
| CYP11B1 | 1.10 | HSD3B2 | 0.67 | POU3F4 | 0.26 | TTR | 0.91 |
| CYP11B2 | 0.83 | IL2RG | 0.81 | PPARG | 1.29 | TYR | 0.69 |
| CYP17 | 0.15 | ITGA2 | 1.05 | PPGB | 0.73 | VMD2 | 0.66 |
| CYP19 | 0.37 | ITGA2B | 0.89 | PPOX | 0.63 | VWF | 1.01 |
| CYP1B1 | 0.87 | ITGB2 | 0.51 | PPT1 | 0.74 | WAS | 1.15 |
| CYP21A2 | 0.86 | ITGB3 | 0.94 | PRNP | 0.42 | WISP3 | 0.00 |
| CYP27A1 | 0.59 | ITGB4 | 0.79 | PROC | 0.85 | WT1 | 0.41 |
| CYP27B1 | 0.67 | JAK3 | 0.93 | PROP1 | 0.00 |
To collect the mutation data, 231 genes were used for the analysis. They were chosen because they had a reported cDNA sequence, disease-associated mutations and homologs in SWISSPROT. These genes are listed in Table 1. Each alignment consists of all the homologs in SWISSPROT as determined by a BLAST search with an e-value threshold of 10e-15. For each alignment the negative entropy for each column was calculated.
The conservation ratio parameter is defined as the average negative entropy of analyzable positions with reported mutations divided by the average negative entropy of every analyzable position in the gene sequence. Analysis was performed on 231 genes and 6185 mutations and of those we found that 84.0% had conservation ratios less than one. From those, 139 genes had more than ten analyzable mutations and, of those, 88.0% had conservation ratios less than one.
Use of evolutionary information is a promising approach to automated characterization of mutations. These results show that although conservation alone is not a perfect predictive measure, there is useful information contained in sequence alignments containing homologous genes. Approaches using conservation in a multiple alignment should work better when associated with other methods such as structural analysis, population analysis and experimental data. Knowledge of how the sequence pool clusters into families may increase the sensitivity of the method.
Our measured parameter, the conservation ratio, is a quantity that measures the usefulness of a multiple alignment for characterizing mutations in a gene sequence. Knowledge of more mutations in a gene does not necessarily lower the conservation ratio. We expect that knowledge of more mutations in a gene will increase the statistical significance of the conservation ratio. This is the likely underlying cause of the result showing that genes with 10 or mutations increases are more likely to have a conservation ratio less than one.
The alignments and BLAST searches are integrated on the website, http://cancer.stanford.edu/mut-paper/.
Conclusions
In conclusion, there are estimated to be 30,000 non-synonymous differences between an individual and the draft genome [23,5,7-9]. Determination of which positions are likely to be disease associated is a challenging and important problem. The finding that disease-associated mutations occur in positions of functional importance supports recent efforts for the building of methods to predict which positions are likely to be disease associated [14,13,16,17]. These methods are likely to incorporate protein structure, the amino acid identity of the mutation and phylogenetic information. In an interesting twist, this observation also suggests that this data may be useable as a functional genomics tool for understanding the function of the protein products of genes on a molecular level. Such a method would use the inherent functional information contained in a phenotypically annotated polymorphism to infer functional importance within a gene.
Methods
Non-synonymous mutations were acquired from the Human Genome Mutation Database http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html. 231 genes were chosen with known disease-association each having SWISSPROT homologs, a cDNA sequence and mutations in the coding region. For each of those genes, all known non-synonymous mutations were then downloaded with the cDNA sequence for that gene.
Each cDNA sequence was then translated and placed in a FASTA formatted file. For each of the resultant files a BLAST [24] search was performed against the SwissProt database. All sequences from the returned hits were then stored in FASTA format files. For each of the genes that returned BLAST results with e-value scores smaller than 1e-15, ClustalW [25] was used to build a sequence alignment.
For each amino acid in the position of interest, the negative entropy was determined using the following formula [26]: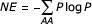
Where the Pi are the probabilities of finding a particular amino acid at that position. For this analysis, gapped positions, "-", were considered independent amino acids.
For each known mutation, the negative entropy of the column it occupies was tabulated. The average negative entropy for each mutation within a gene was compared to the average entropy of all columns satisfying the criteria for analysis. Mutations outside of the coding region or mutations encoding termination codons were discarded.
The list of genes was then sorted by average negative entropy of the mutations. We then calculated the conservation entropy, CE, using:
CE = average NE of mutation positions/average NE of all positions in the gene sequence
Table 2.
Genes with average conservation ratios of zero
| Gene | Disease Association | # Mutations |
|---|---|---|
| CLN3 | Batten Disease | 5 |
| PAX8 | Thyroid Hypoplasia | 2 |
| PROP1 | Pituitary Hormone Deficiency | 2 |
| RAG1 | Omenn Syndrome, Immunodeficiency | 7 |
| WISP3 | Pseudorheumatoid Dysplasia | 2 |
The mutations in these genes are perfectly conserved by our method. See text for details.
Contributor Information
Sean D Mooney, Email: mooney@smi.stanford.edu.
Teri E Klein, Email: teri.klein@stanford.edu.
Acknowledgements
The authors thank Irene Liu and Professor Russ Altman at Stanford University for their helpful suggestions and UCSF Computer Graphics Laboratory for use of resources. Funding provided by NIH Grant AR47720-01 (Teri E. Klein, PI) and Nitt Grant LM05652 (Russ B. Altman, PI).
References
- Sachidanandam R, Weissman D, Schmidt S, Kakol J, Stein L, Marth G, Sherry S. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409(6822):928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Krawczak M, Cooper D. The Human Gene Mutation Database. Trends in Genetics. 1997;13:121–122. doi: 10.1016/S0168-9525(97)01068-8. [DOI] [PubMed] [Google Scholar]
- Gottlieb B, Lehvaslaiho H, Beitel L, Lumbroso R, Pinksy L, Trifiro M. The Androgen Receptor Gene Mutations Database. Nucleic Acids Research. 1998;26:234–238. doi: 10.1093/nar/26.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgleish R. The Human Type I Collagen Mutation Database. Nucleic Acids Research. 1997;25:181–187. doi: 10.1093/nar/25.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter J, Adams M, MW E, Li P, Mural R, Sutton G, Smith H. The Sequence of the Human Genome. Science. 2001;291:1304. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Clifford R, Edmonson M, Hu Y, Nguyen C, Scherpbier T, Beutow K. Expression-based Genetic/Physical Maps of Single-Nucleotide Polymorphisms Identified by the Cancer Genome Anatomy Project. Genome Research. 2000;10:1259–1265. doi: 10.1101/gr.10.8.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A, Hawkins J. DNA Variation And THe Future Of Human Genetics. Nature Biotechnology. 1998;16:33–39. doi: 10.1038/5412. [DOI] [PubMed] [Google Scholar]
- Collins F, Guyer M, Chakravarti A. Variations On A Theme: Cataloging Human DNA Sequence Variation. Science. 1997;278:1580–1581. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- Cooper D, Smith B, Cooke H, Niemann S, Schmidtke J. An Estimate of Unique DNA Sequence Heterozygosity In The Human Genome. Human Genetics. 1995;69:201–205. doi: 10.1007/BF00293024. [DOI] [PubMed] [Google Scholar]
- Thornton J, Kelley D. Evolution of the Androgen Receptor: Structure Function Implications. Bioessays. 1998;20:860–869. doi: 10.1002/(SICI)1521-1878(199810)20:10<860::AID-BIES12>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Krawczak M, Edward B, Cooper D. Neighboring-Nucleotide Effects on the Rates of Germ-Line Single-Base-Pair Substitution in Human Genes. American Journal of Human Genetics. 1998;63:474–488. doi: 10.1086/301965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Bork P. SNP frequencies in human genes: an excess of rare alleles and differing modes of selection. Trends in Genetics. 2000;16:335–337. doi: 10.1016/S0168-9525(00)02058-8. [DOI] [PubMed] [Google Scholar]
- Chasman D, Adams R. Predicting the Functional Consequences of Non-synonymous Single Nucleotide Polymorphisms: Structure-based Assessment of Amino Acid Variation. Journal of Molecular Biology. 2001;307:683–706. doi: 10.1006/jmbi.2001.4510. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe WI, Kondrashov A, Bork P. Prediction of Deleterious Human Alleles. Human Molecular Genetics. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Wang Z, Moult J. SNPs, Protein Structure, and Disease. Human Mutation. 2001;17:263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- Ng P, Henikoff S. Prediting Deleterious Amino Acid Substitutions. Genome Research. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P, Henikoff S. Accounting fo Human Polymorphisms Predicted to Affect Protein Function. Genome Research. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel K, Xu J, Yin G, Lyons R, Meisler M, Weber B. Mouse Brca1: Localization Sequence Analysis and Identification of Evolutionary Conserved Domains. Human Molecular Genetics. 1995;4:2265–2273. doi: 10.1093/hmg/4.12.2265. [DOI] [PubMed] [Google Scholar]
- Nishida K, Honma Y, Dota A, Kawasaki S, Adachi W, Nakamura T, Quantock A. Isolation and Chromosomal Localization of a Cornea-Specific Human Keratin 12 Gene and Detection of Four Mutations in Meesmann Corneal Epithelial Dystrophy. American Journal of Human Genetics. 1997;61:1268–1275. doi: 10.1086/301650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacey A, Cooper D, Liney D, Hovig E, Krawczak M. Perturbational Effects of Amino Acid Substitutions in the DNA-binding Domain of p53. Human Genetics. 1999;104(1):15–22. doi: 10.1007/s004390050904. [DOI] [PubMed] [Google Scholar]
- Miller M, Kumar S. Understanding human disease mutations through the use of interspecific genetic variation. Human Molecular Genetics. 2001;10:2319–2328. doi: 10.1093/hmg/10.21.2319. [DOI] [PubMed] [Google Scholar]
- Henikoff S. Scores For Sequence Searches And Alignments. Current Opinion In Structural Biology. 1996;6:353–360. doi: 10.1016/S0959-440X(96)80055-8. [DOI] [PubMed] [Google Scholar]
- Lander E, Linton L, Birren B, Nusbaum C, Zody M, Baldwin J, Devon K. Initial sequencing and analysis of the human genome. Nature. 2002;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D. Gapped BLAST and PSI-BLAST: A New Generation Of Protein Database Search Tools. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, Higgins D, Gibson T. CLUSTAL W: Improving The Sensitivity Of Progressive Multiple Sequence Alignment Through Sequence Weighting, Positions-specific Penalties and Weight Matrix Choice. Nucleic Acids Research. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkin P, Erman B, Mastrandrea L. Information-theoretical Entropy As A Measure Of Sequence Variability. Proteins. 1991;11:297–313. doi: 10.1002/prot.340110408. [DOI] [PubMed] [Google Scholar]