

Summary
Recently, haploinsufficiency mutations in the melanocortin-4 receptor gene (MC4-R) were detected which were assumed to lead to the phenotype of extreme obesity. Previously, we detected three obese carriers among 306 index patients. Here we describe the detection of one haploinsufficiency carrier in an additional study group of 186 obese individuals. We subsequently genotyped and phenotyped 43 family members of these four index patients, two of whom were second-degree cousins. A total of 19 carriers were identified. Extreme obesity was the predominating phenotype. However, moderate obesity occurred in three of the carriers. No other specific phenotypic abnormalities were detected. Female haploinsufficiency carriers were heavier than male carriers in the respective families, a finding similar to findings in MC4-R–knockout mice. In conclusion, our data fully support the etiologic role of MC4-R haploinsufficiency mutations in obesity.
Introduction
MC4-R (MIM 155541) is a G-protein–coupled seven-transmembrane receptor primarily expressed in the hypothalamus, hippocampus, and thalamus (Gantz et al. 1993). The receptor is implicated in the central regulation of body weight: MC4-R is activated by α-melanocyte-stimulating hormone (MSH), which is derived from pro-opiomelanocortin and is antagonized by agouti-related protein. α-MSH induces weight loss, whereas the ectopic expression of agouti protein results in obesity in the agouti mice (Fan et al. 1993; Lu et al. 1994). Additional evidence for the role of MC4-R in weight regulation stems from both a knockout model in mice (Huszar et al. 1997) and haploinsufficiency mutations in humans (Vaisse et al. 1998; Yeo et al. 1998; Hinney et al. 1999). In MC4-R–knockout mice, an increased body weight was discernible by age 5 wk. By age 15 wk, homozygous mutant females were, on average, twice as heavy as their wild-type littermates, whereas homozygous mutant males were ∼50% heavier than wild-type controls. Mice heterozygous for the MC4-R knockout showed a weight gain intermediate to that seen in wild-type and homozygous mutant littermates, thus demonstrating a gene dosage effect of MC4-R ablation on body-weight regulation. The food intake of homozygous mutants was increased by ∼50% in comparison to that in wild-type sibs (Huszar et al. 1997).
Recently, mutations leading to haploinsufficiency for MC4-R were detected in obese individuals by means of systematic mutation screens of the MC4-R. Three different haploinsufficiency mutations have currently been described: Vaisse et al. (1998) screened 43 obese probands and detected a heterozygous 4-bp insertion at codon 244 in one of them. Five family members of this index patient who also harbored the mutation were all extremely obese; the LOD score was 1.5. Yeo et al. (1998) screened 63 obese probands and detected a young index patient heterozygous for a 4-bp deletion at codon 211. The father, who had a body-mass index (BMI) of 41 kg/m2, was a mutation carrier, too. Hinney et al. (1999) also observed this deletion in both an extremely obese female adolescent and her similarly obese mother. In addition, the screening of 306 extremely obese children and adolescents led to the identification of two independently ascertained index patients with a nonsense mutation at codon 35. This presumably results in a receptor protein being truncated at the extracellular domain. Both patients also harbored a missense mutation at codon 37 (Hinney et al. 1999). Whereas these three studies strongly suggest that haploinsufficiency mutations underly the obesity observed in the carriers, the causal relationship has not been proven. Thus, in contrast to the large number of obese individuals screened, only a limited number of lean controls have been screened (Hinney et al. 1999). Furthermore, because it can be expected, a priori, that either one or both parents of an extremely obese index patient are also obese, the observation of cosegregation of these mutations with the obesity phenotype in the limited number of informative meioses cannot be viewed as unequivocal evidence for a causal relationship.
Several other allelic variants of MC4-R have been identified (Gu et al. 1999; Hinney et al. 1999). In in vitro assays, one of these mutations (Ile137Thr) was found to severely impair ligand binding and signaling, raising the possibility that this mutation may contribute to the obese phenotype. However, the effect of the Ile137Thr allele seems to be recessive (Gu et al. 1999).
For the purpose of this study, we have recontacted the parents of the index patients with haploinsufficiency mutations, to genotype and phenotype additional family members. Furthermore, we extended the mutation screen to another 186 extremely obese children and adolescents to detect further mutations leading to haploinsufficiency and, if this was successful, to extend the study to the respective families. Our intention was to assess whether haploinsufficiency mutations indeed cosegregate with the phenotype obesity and, if they do, to delineate the phenotype associated with these mutations.
Material and Methods
In our initial study, 306 extremely obese German children and adolescents (mean BMI 34.4 ± 6.6 kg/m2, mean age 14.3 ± 2.4 years) were screened, by SSCP, for mutations in MC4-R, leading to the identification of 3 adolescents with haploinsufficiency (Hinney et al. 1999). For this study, we screened an additional 186 obese index patients (mean BMI 30.92 ± 5.79 kg/m2, mean age 13.19 ± 2.67 years) by SSCP. Bidirectional sequencing of PCR products showing an SSCP pattern similar to the pattern observed in the nonsense mutation carriers was performed as described elsewhere (Hinney et al. 1999).
The parents of all identified haploinsufficiency carriers were recontacted and were asked to participate in an extended family study. At the time of the original investigation of index patients, parents had given written informed consent to be recontacted in case a medically relevant molecular finding emerges. All contacted nuclear families, in turn, asked other family members to participate in the study. Participating family members gave their written informed consent in accordance with the Ethics Committee of the University of Marburg.
The family members (fig. 1; pedigrees A–C with n=21, n=18, and n=8 family members, respectively) were contacted and were examined at their homes. The following variables were measured: body weight and height; body composition, as determined with bioelectrical impedance analysis (BIA) (Data Input 2000-S); and waist and hip circumference. Age- and sex-specific BMI percentiles were calculated on the basis of the large and representative German National Nutrition Survey (Hebebrand et al. 1996). To illustrate the BMI distribution of both carriers and noncarriers of haploinsufficiency mutations in relation to the BMI distribution observed in the general population, we plotted BMIs of the ascertained family members into BMI percentile curves on the basis of anthropometric data obtained from the German National Nutrition Survey. The percentiles were calculated as described by Cole et al. (1998), for individuals aged 8–70 years. Blood sampling was performed to allow for DNA isolation and for determination of serum leptin levels with a radioimmunoassay (Blum 1997).
Figure 1.
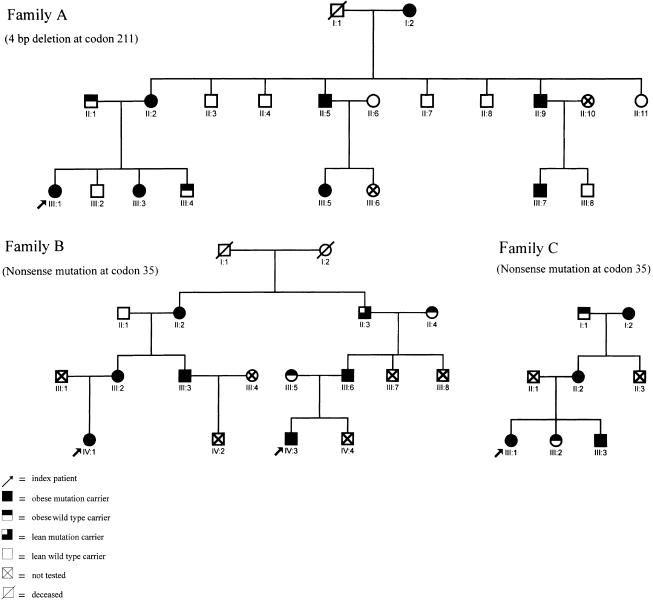
Three pedigrees ascertained via four index patients with haploinsufficiency mutations in MC4-R. A 4-bp deletion at codon 211 in pedigree A and a nonsense mutation at codon 35 in pedigrees B and C underly the haploinsufficiency (for more details see table 1).
On the basis of the findings from a semistructured interview that included the eating disorders module of the M-Composite International Diagnostic Interview (Wittchen et al. 1995), the following data were obtained from all participating family members: eating disorders, diet habits, weight history as of early childhood, and history of medical disorders, including assessment of possible effects on body weight. All family members were asked if they had been in psychological treatment related or unrelated to their obesity. Furthermore, each individual was questioned as to their subjective opinion on the cause of their obesity.
Ascertained family members were genotyped by means of SSCP. Additionally, for the nonsense mutation, PCR was performed with allele-specific primers (forward primers MC4R-Y 5′-GTCCCTTGGAAAAGGCTAC-3′ for the wild-type allele and MC4R-STOP 5′-GTCCCTTGGAAAAGGCTAA-3′ for the 35-Stop allele, respectively, both with MC4R-1R 5′-GACAGCACTACTATCTGAGT-3′ as a reverse primer).
An exact 95% confidence interval for the frequency of haploinsufficiency mutations in German children and adolescents with extreme obesity was calculated as described in StatXact (1996). LOD scores were calculated by use of FASTLINK (Cottingham et al. 1993).
We investigated whether the median age- and sex-adjusted maximum lifetime BMI percentile in family A was larger in mutation carriers than in noncarriers. To take into account residual correlation among relatives, we performed a Monte Carlo permutation. For each sibship, 1,000,000 permutations were conducted separately. The system time was used as seed for the experiment via the GAUSS (1994) procedure RNDU, which uses a multiplicative-congruential method. The test statistic was the sum of the standardized sibship-rank sums. Means and variances were calculated under the null hypothesis. The midrank method was used to handle ties.
Mean BMIs of carriers of the nonsense mutation were compared with mean BMIs of the carriers of the 4-bp deletion by means of the t-test. This test, however, does not adjust for residual correlation between relatives, as discussed in detail by (for example) Gu et al. (1999). If these dependencies exist, they are most likely positive. This would lead to an underestimation of the variance. Hence, the results of this analysis should be interpreted carefully.
Results
The screening of 186 children and adolescents with extreme obesity led to the detection of a single index patient with a haploinsufficiency mutation, which was caused by the nonsense mutation at codon 35 of MC4-R. This individual also harbored the missense mutation at codon 37. Not totally unexpectedly, this patient, who was ascertained independently of other index patients, turned out to be related to one of the previously identified carriers of the two nonsense mutations (Hinney et al. 1999); the two index patients were second-degree cousins (fig. 1, pedigree B: IV:1, IV:3). We did not identify an individual homozygous for any of the haploinsufficiency mutations. The exact 95% confidence interval for the frequency of haploinsufficiency mutations is between 0.2% and 2.1% for German children and adolescents with extreme obesity, since we identified a total of four carriers in 492 individuals.
Cosegregation of either the deletion (fig. 1, pedigree A) or the nonsense mutation (fig. 1, pedigrees B and C) with obesity was observed in 18 of the 19 identified individuals within the three families. Only one carrier, a male aged 60 years, (pedigree B: II:3) had a current BMI (27.9 kg/m2) below the 90th percentile. However, this male had had a heart attack at age 56 years and had subsequently dieted continuously to control his weight; his maximal BMI (on the basis of recalled weight and measured height) had been 32.2 kg/m2 at age 54 years (96th percentile).
Six individuals (fig. 1, pedigree A: II:1, III:4; pedigree B: II:4, III:5; and pedigree C: I:1, III:2), who were noncarriers of one of the two haploinsufficiency mutations or other mutations in MC4-R, also had a BMI above the 90th percentile. Of these, four were spouses of mutation carriers. Two obese noncarriers were sibs of index patients (pedigree A III:4 and pedigree C III:2). Relevant phenotypic data are shown in table 1.
Table 1.
Phenotypic Data of Carriers and Noncarriers of Haploinsufficiency Mutations in MC4-R[Note]
|
Current BMI |
Maximum BMI |
||||||||||||
| Family and Patient | Mutation Carriera | Sexb | Age (years) | Weight (kg) | Height (cm) | Absolute (kg/m2) | Percentile | Absolute (kg/m2)c,d | Percentilee | Waist/Hip Ratiof | Percent Body Fat (%)f | Leptin (ng/ml)f | Lifetime Positive Diet Historyc,g |
| A | |||||||||||||
| I:2 | +/− | F | 81 | 76 | 155 | 31.6 | 100 | NI | NC | NM | NM | 20.7 | NI |
| II:1 | +/+ | M | 49 | 93 | 175 | 30.4 | 93 | NI | NC | .91 | 28.5 | 4.1 | NI |
| II:2 | +/− | F | 45 | 103 | 163 | 38.8 | 99 | 40.7 | 100 | .90 | 45.5 | 58.1 | + |
| II:3 | +/+ | M | 59 | 76 | 176 | 24.5 | 26 | 25 | 26 | .88 | 28.1 | 20.7 | − |
| II:4 | +/+ | M | 46 | 70 | 175 | 22.9 | 17 | 23 | 17 | .81 | 27.4 | 2.8 | − |
| II:5 | +/− | M | 50 | 97 | 171 | 33 | 97 | 33 | 97 | .99 | 33.9 | 29.9 | − |
| II:6 | +/+ | F | 39 | 75 | 164 | 28.1 | 86 | 29 | 94 | .77 | 39.6 | 16.8 | + |
| II:7 | +/+ | M | 42 | 72 | 180 | 22.2 | 16 | 22.5 | 59 | .81 | 21.4 | NM | + |
| II:8 | +/+ | M | 39 | 79 | 177 | 25.2 | 53 | NR | NC | .90 | 26.3 | 5.3 | + |
| II:9 | +/− | M | 40 | 94 | 178 | 29.7 | 92 | 29.7 | 92 | .97 | NM | 8.7 | − |
| II:11 | +/+ | F | 61 | 63 | 167 | 22.6 | 18 | NR | NC | .93 | 35.5 | 14.9 | − |
| III:1 | +/− | F | 22 | 117 | 171 | 40 | 100 | 42.8 | 100 | .89 | 46.6 | 63.1 | + |
| III:2 | +/+ | M | 17 | 72 | 185 | 21 | 37 | NR | NC | .80 | 17.6 | 1.1 | − |
| III:3 | +/− | F | 14 | 79 | 165 | 29 | 99 | 29 | 99 | .86 | 37.4 | 40.9 | − |
| III:4 | +/+ | M | 12 | 81 | 162 | 30.9 | 100 | 31 | 100 | 1.02 | 35.8 | 94.1 | − |
| III:5 | +/− | F | 21 | 78 | 168 | 27.7 | 98 | 28 | 98 | .75 | 39.7 | 65.8 | + |
| III:7 | +/− | M | 20 | 110 | 194 | 29.2 | 98 | NI | NC | NM | NM | 24.9 | NI |
| III:8 | +/+ | M | 18 | 66 | 178 | 21 | 37 | 21 | 37 | .79 | NM | 1.6 | − |
| B | |||||||||||||
| II:1 | +/+ | M | 68 | 88 | 173 | 29.4 | 86 | 38.4 | 100 | 1.07 | 30.9 | 1.9 | − |
| II:2 | +/− | F | 64 | 106 | 153 | 45.3 | 100 | 45 | 100 | .77 | 58.3 | 91.9 | − |
| II:3 | +/− | M | 60 | 82 | 171 | 27.9 | 67 | 32.2 | 96 | .79 | 31.3 | 39.9 | + |
| II:4 | +/+ | F | 58 | 87 | 168 | 30.6 | 93 | 31 | 93 | NM | 43.1 | NM | − |
| III:2 | +/− | F | 39 | 115 | 167 | 41.2 | 100 | 41.2 | 100 | .88 | 52.6 | 47.3 | + |
| III:3 | +/− | M | 41 | 98 | 176 | 31.6 | 93 | NI | NC | .94 | 40.3 | 86.2 | NI |
| III:5 | +/+ | F | 32 | 90 | 168 | 31.9 | 96 | NI | NC | NM | 33.5 | 41.6 | NI |
| III:6 | +/− | M | 40 | 110 | 181 | 33.6 | 99 | 34 | 99 | .88 | 33.4 | 15 | − |
| IV:1 | +/− | F | 19 | 139 | 165 | 51.1 | 100 | 40.4 | 100 | .82 | 55.3 | NM | + |
| IV:3 | +/− | M | 8 | 53 | 143 | 25.9 | 97 | 26 | 97 | .96 | 40.1 | 88.9 | + |
| C | |||||||||||||
| I:1 | +/+ | M | 59 | 97 | 173 | 32.4 | 96 | 45.1 | 100 | .88 | 33.3 | 2.2 | − |
| I:2 | +/− | F | 63 | 154 | 165 | 56.6 | 100 | 56.6 | 100 | .82 | 54.1 | 128 | + |
| II:2 | +/− | F | 35 | 144 | 171 | 49.2 | 100 | 49 | 100 | .81 | 51.6 | 123 | + |
| III:1 | +/− | F | 14 | 114 | 168 | 40.4 | 100 | 40 | 100 | .85 | 46.5 | 56.5 | + |
| III:2 | +/+ | F | 12 | 59 | 154 | 24.9 | 97 | 25 | 97 | .84 | 34.5 | 16.8 | − |
| III:3 | +/− | M | 10 | 58 | 140 | 29.8 | 100 | 30 | 100 | NM | NM | 45.9 | − |
Note.—Carriers were identified by means of four index patients, who belonged to three different families (A–C) (see fig. 1).
Genotype +/− indicates a haploinsufficiency carrier; +/+ indicates a wild type carrier.
F = female, M = male.
NI = Not interviewed.
NR = Not recalled.
NC = Not calculated.
NM = Not measured.
Plus sign (+) = positive finding; minus sign (−) = negative finding.
Current and maximal lifetime BMIs and the corresponding percentiles revealed that not all mutation carriers are or had been extremely obese (figs. 2A and 2B). Of the 19 carriers, 16 had a current BMI percentile ⩾95, which is used as a cutoff for super-obesity (Cronk and Roche 1982). Nine had a BMI exceeding that of the maximum BMI observed in the population-based control group, which encompasses ∼150–200 individuals for each sex- and age-matched group (Hebebrand et al. 1996). The female carriers (n=11) had a higher mean BMI than did the male carriers (40.9 vs. 30.0 kg/m2; P<.005, ignoring the correlation within pedigrees). BMIs of four of the five female noncarriers also exceeded the 85th percentile. Half of the 10 male noncarriers had a BMI below the 50th percentile; three had a BMI above the 85th percentile.
Figure 2.
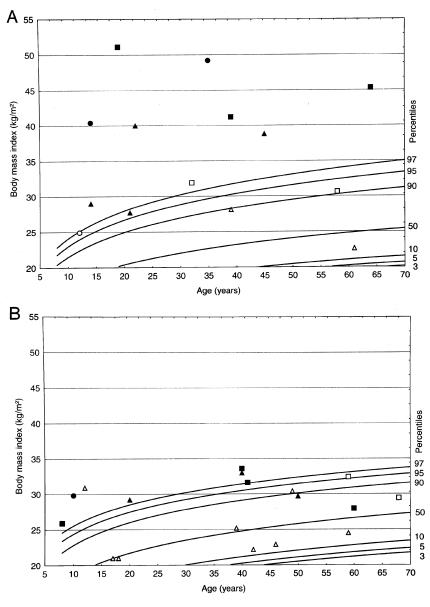
Percentile plots (A, females; B, males) for BMI developed on the basis of a representative sample of the German population (Hebebrand et al. 1996) and smoothed with the approach of Cole et al. (1998) for ages 8–70 years; current BMIs of family members were plotted into the percentile curves. Blackened symbols represent mutation carriers; unblackened symbols indicate wild-type carriers. Members of families A, B, and C are represented by triangles, squares, and circles, respectively. A single obese female, aged 81 years, with a current BMI of 31.6 kg/m2, was omitted from the plot, because percentile curves are only available for ages ⩽70 years.
On the basis of the observation that not all carriers were extremely obese, we calculated a LOD score post hoc, assuming autosomal dominant inheritance and a recombination fraction of zero and using a lifetime BMI ⩾85th percentile to define affected individuals. This percentile represents the cutoff for obesity (Cronk and Roche 1982). We used lifetime prevalences of obesity of 15% and 25%. Furthermore, we assumed a disease-allele frequency of 1% and complete penetrance. We calculated the phenocopy rate from these data. On the basis of these assumptions, LOD scores equivalent to 3.79 and 3.35, respectively, were obtained. The median age- and sex-adjusted maximal lifetime BMI percentile in family A was larger in mutation carriers than in noncarriers (P=.0054, ignoring the correlation within pedigree A).
The waist-hip ratio (WHR) was <1 in all male mutation carriers. Three females had a WHR slightly >0.85, which is considered the threshold value for identification of abdominal fat accumulation in females (WHO 1998). Serum leptin levels were within the expected range; nine carriers had a leptin level below the median for their age, BMI, and sex-matched reference range (Blum 1997). Menarche occurred between ages 10 and 14 years in the 11 female carriers. Eight female and two male carriers had a positive diet history. Three of the four index patients had been ascertained as inpatients in hospitals specializing in the treatment of extreme early-onset obesity. Individual III:1 of pedigree A had had three inpatient treatments, the longest of which lasted for a total of 9 mo, after which her BMI had transiently decreased to 37.28 kg/m2.
Only a single mutation carrier reported the lifetime occurrence of eating binges fulfilling the DSM-IV research criteria for a binge eating disorder, which lasted for 3 years, ending at age 16 years. Whereas none of the noncarriers had sought psychological treatment, five of the mutation carriers (including four females) reported psychological treatment episodes, all of which were related to their obesity. Age at onset of obesity among the mutation carriers was recalled as lying between infancy and 18 years.
Of the 13 carriers who were questioned about their subjective opinions on the causes of their obesity, 11 replied that they enjoyed eating and that they considered this aspect as a contributing factor. Otherwise, no clear pattern emerged. Thus, some carriers stated that stress at work (n=4) or at home (n=5) or frustrating experiences (n=4) repeatedly led to overeating and thus contributed to their obesity. Only three carriers stated that they currently controlled their weight; several of the carriers had done this in the past but eventually had given up.
Discussion
Our family study clearly substantiates that haploinsufficiency of the MC4-R leads to obesity that tends to be of an early-onset type. Our data indicate a sex effect similar to findings in MC4-R–knockout mice. In particular, the degree of adiposity in males was not as severe as that in the females. All female carriers had a current BMI ⩾97th percentile. However, in contrast to findings in the knockout mice, we did not find evidence for an effect of the haploinsufficiency on body height. Thus, within a sibship, the adult mutation carriers were not of a greater height than noncarriers of the same sex and age (table 1).
The pedigrees clearly illustrate problems inherent in the genetic analysis of the quantitative phenotype body weight. Not unexpectedly, some obese mutation carriers had obese spouses, presumably explaining why noncarrier sibs can also be obese (fig. 1, pedigree A: III:4). The BMIs of female noncarriers were high, in comparison to the BMI distribution in the general population; four topped the 85th percentile (fig. 2A). Quite obviously, other genetic and/or environmental factors, possibly including mutations in the regulatory region of MC4-R, are operative in these families. Accordingly, we cannot assess the quantitative impact of the haploinsufficiency mutations on body weight.
These considerations are further substantiated by the fact that, in family A, which includes all of the identified noncarriers with a BMI below the 50th percentile, the BMIs of the carriers are also lower than those in the other two families (figs. 2A and 2B). Our LOD-score calculation was only possible on a post hoc basis, after discovery of mutation carriers with moderate obesity. Therefore, it should be considered merely exploratory. Despite these methodological problems, the data are completely compatible with a cosegregation of the haploinsufficiency mutations with both moderate and severe obesity.
We cannot exclude the possibility that the two underlying mutations have different phenotypic implications. Thus, the six female carriers of the nonsense mutation in which the receptor is truncated in the extracellular domain were currently heavier (families B and C; mean BMI: 33.4 vs. 47.3 kg/m2; nominal P<.005, ignoring the aforementioned correlation between relatives) than the five females with the deletion that presumably results in a receptor truncated in the fifth transmembrane domain (fig. 2A and 2B). No difference was found for the male carriers (P=.68, ignoring the correlation between relatives).
The phenotypic assessment of individuals with haploinsufficiency mutations revealed the following interesting aspects: (1) no overt evidence of diabetes mellitus or hypertension was detected, despite extensive questioning about these frequent comorbid disorders. All carriers appeared quite healthy, with the exception of one man, who had had a heart attack at age 56 years. (2) Most mutation carriers had a gynoid pattern of fat distribution, possibly explaining the absence of comorbid disorders. (3) Despite the extreme degree of obesity observed in female carriers, only one female fulfilled lifetime criteria for a diagnosis of binge eating disorder. (4) Not unexpectedly, four females had sought psychological treatment for their extreme obesity. (5) Whereas ten carriers reported past efforts to reduce their weight, renewed weight gain had ensued. Most carriers had given up efforts to control their weight. (6) Serum leptin levels were within the expected range.
In conclusion, our family data are fully compatible with autosomal dominant inheritance of obesity resulting from haploinsufficiency mutations in the MC4-R. However, in contrast to the initial observations (Vaisse et al. 1998; Yeo et al. 1998; Hinney et al. 1999), moderate obesity can also be associated with these mutations. It appears that this applies mainly to males. Because several noncarriers in the three ascertained families also had BMIs in the upper range of the general population, the extent of the quantitative effect of the mutation on body weight is uncertain. On the basis of our phenotypic evaluation of 19 carriers, it is possible that this subtype of obesity is only infrequently associated with comorbid disorders. The exact 95% confidence interval suggests that haploinsufficiency mutations are infrequent—but not exceedingly rare—among German children and adolescents with extreme obesity. Frequency rates of such mutations among individuals with moderate obesity remain undetermined. Obesity caused by MC4-R haploinsufficiency mutations should be considered a diagnosable subgroup. Possibly, if more mutations in MC4-R (Gu et al. 1999; Hinney et al. 1999) turn out to be functionally relevant, the clinical importance of this diagnostic subgroup will increase, especially if our previous speculation holds true that carriers of functionally relevant mutations might profit from MC4-R agonists (Hinney et al. 1999).
Acknowledgments
We thank all probands and their relatives for their participation. We thank Gerti Gerber for her excellent technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft.
Electronic-Database Information
The accession number and URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for melanocortin-4 receptor [MIM 155541])
References
- Blum WF, Juul A (1997) Reference ranges of serum leptin levels according to body mass index, gender, and developmental stage. In: Blum WF, Kiess W, Rascher W (eds) Leptin—the voice of adipose tissue. Johann Ambrosius Barth Verlag, Heidelberg and Leipzig, pp 318–326 [Google Scholar]
- Cole TJ, Freeman JV, Preece MA (1998) British 1990 growth reference centiles for weight, height, body mass index and height circumference fitted by maximum penalized likelihood. Stat Med 17:407–429 [PubMed]
- Cronk CE, Roche AF (1982) Race- and sex-specific reference data for triceps and subscapular skinfolds and weight/stature. Am J Clin Nutr 35:347–354 [DOI] [PubMed]
- Cottingham RW Jr, Idury RM, Schäffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed]
- Fan W, Bosten BA, Kesterson RA, Hruby VJ, Cone RD (1997) Role of melanocortinergic neurons in feeding and the agouti obesity syndrom. Nature 385:165–168 [DOI] [PubMed]
- Gantz I, Miwa H, Konda Y, Tashiro T, Shimoto Y (1993) Molecular cloning, and gene localization of a fourth melanocortin receptor. J Biol Chem 268:15174–15179 [PubMed]
- GAUSS (1994) Gauss. Vol 2. Command reference. Aptech Systems, Maple Valley, WA [Google Scholar]
- Gu W, Tu Z, Kleyn PW, Kissebah A, Duprat L, Lee J, Chin W, et al (1999) Identification and functional analysis of novel human melanocortin-4 receptor variants. Diabetes 48:635–639 [DOI] [PubMed]
- Hebebrand J, Himmelmann GW, Heseker H, Schäfer H, Remschmidt H (1996) Use of percentiles for the body mass index in anorexia nervosa: diagnostic, epidemiological and therapeutic considerations. Int J Eat Disord 19:359–369 [DOI] [PubMed]
- Hinney A, Schmidt A, Nottebom K, Heibült O, Becker I, Ziegler A, Gerber G, et al (1999) Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. J Clin Endocrinol Metab 84:1483–1486 [DOI] [PubMed]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, et al (1997) Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88:131–141 [DOI] [PubMed]
- Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, et al (1994) Agouti protein is an antagonist of the melanocyte-stimulating hormone receptor. Nature 371:799–802 [DOI] [PubMed]
- StatXact (1996) StatXact 3 for Windows: user manual. Cytel Software, Cambridge, MA [Google Scholar]
- World Health Organization (1998) Obesity—preventing and managing the global epidemic. World Health Organization, Geneva [PubMed] [Google Scholar]
- Wittchen HU, Kessler RC, Zhao S, Abelson J (1995) Reliability and clinical validity of UM-CIDI DSM-III-R generalized anxiety disorder. J Psychiatr Res 29:95–110 [DOI] [PubMed]
- Vaisse C, Clement K, Guy-Grand B, Froguel P (1998) A frameshift mutation in human MC4-R is associated with a dominant form of obesity. Nat Genet 20:113–114 [DOI] [PubMed]
- Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O'Rahilly S (1998) A frameshift mutation in MC4-R associated with dominantly inherited human obesity. Nat Genet 20:111–112 [DOI] [PubMed]