

Summary
Rett syndrome (RTT) is a neurodevelopmental disorder characterized by loss of acquired skills after a period of normal development in infant girls. The responsible gene, encoding methyl-CpG binding protein 2 (MeCP2), was recently discovered. Here we explore the spectrum of phenotypes resulting from MECP2 mutations. Both nonsense (R168X and R255X) and missense (R106W and R306C) mutations have been found, with multiple recurrences. R168X mutations were identified in six unrelated sporadic cases, as well as in two affected sisters and their normal mother. The missense mutations were de novo and affect conserved domains of MeCP2. All of the nucleotide substitutions involve C→T transitions at CpG hotspots. A single nucleotide deletion, at codon 137, that creates a L138X stop codon within the methyl-binding domain was found in an individual with features of RTT and incontinentia pigmenti. An 806delG deletion causing a V288X stop in the transcription-repression domain was identified in a woman with motor-coordination problems, mild learning disability, and skewed X inactivation; in her sister and daughter, who were affected with classic RTT; and in her hemizygous son, who died from congenital encephalopathy. Thus, some males with RTT-causing MECP2 mutations may survive to birth, and female heterozygotes with favorably skewed X-inactivation patterns may have little or no involvement. Therefore, MECP2 mutations are not limited to RTT and may be implicated in a much broader phenotypic spectrum.
Introduction
Rett syndrome (RTT) is a neurological disorder characterized by a period of early normal growth and development followed by regression with loss of speech and acquired motor skills, stereotypical hand movements, and seizures. Slowing of brain growth and postnatal growth failure are frequently present (Rett 1966; Hagberg et al. 1983; Trevathan et al. 1988). Usually sporadic, RTT causes developmental impairment almost exclusively in girls. The cause of RTT has been debated in the literature for more than a decade (Hagberg et al. 1983; Martinho et al. 1990; Migeon et al. 1995). Studies of rare familial cases, including a severely affected male infant, provided evidence that RTT is caused by X-linked dominant mutations in a gene (or genes) subject to X-chromosome inactivation (XCI). Polymorphic marker typing of familial cases allowed exclusion of most regions of the X chromosome and focused the search for the RTT gene on band Xq28 (Ellison et al. 1992; Schanen et al. 1997; Schanen and Francke 1998; Sirianni et al. 1998).
The incidence of RTT, estimated at 1/10,000–15,000 females, with 99.5% of all cases being sporadic, requires a high rate of new mutations (Hagberg 1985). In analogy to recurrent de novo microdeletions mediated by recombination between highly homologous flanking sequences—as seen in Prader Willi/Angelman syndromes, Williams-Beuren syndrome, Smith-Magenis syndrome, and velocardiofacial syndrome (Lupski 1998)—we strongly considered a genomic rearrangement mechanism for RTT. A systematic search for microdeletions in the nonexcluded region of the X chromosome, however, excluded deletions >100 kb (Fan et al. 1999) and suggested a single-gene mechanism.
Our systematic mutation analyses of genes in the 10-Mb region encompassing band Xq28 excluded several candidates (Wan and Francke 1998; Amir et al., in press) before it culminated in the recent finding of missense and truncating mutations in the MECP2 gene that encodes a methyl CpG-binding protein (Amir et al. 1999). This discovery came as a surprise, since MECP2 would not appear to be a strong candidate for a primary brain disorder, because the protein is ubiquitously expressed, associated with 5-methylcytosine (5-mC)–rich heterochromatin, and potentially involved in global gene silencing (Nan et al. 1997). In addition, chimeric mice, that can be considered equivalent to XCI mosaics, made with Mecp2-deficient embryonic stem cells, for an X-linked RTT mutation, died in utero (Tate et al. 1996).
Symmetrical methylation of the C5 position of cytosines in CpG dinucleotides, as well as chromatin modification, have long been known to cause transcriptional silencing of individual genes and to mediate global epigenetic phenomena, such as mammalian XCI and gametic imprinting (Lindsay et al. 1985; Li et al. 1993). Although the two processes are functionally linked (Keshet et al. 1986; Buschhausen et al. 1987; Kass et al. 1993), the molecular mechanism for the interaction was unclear. Methylated DNA is associated with hypoacetylated histones (Eden et al. 1998). MeCP2 has recently been proposed to act as a molecular link by binding to 5-mC with its methyl-CpG–binding domain (MBD) and to the corepressor Sin3A via its transcriptional repression domain (TRD), thus recruiting histone deacetylases and other proteins to the silencing complex (Jones et al. 1998; Nan et al. 1998b; Razin 1998; Ng and Bird 1999). Deacetylation of lysine residues of the core histones H3 and H4 alters the chromatin structure and renders the methylated DNA inaccessible to the transcriptional machinery. This mechanism leads to stable transcriptional repression that is heritable in somatic cells by the action of maintenance methylase on hemimethylated DNA at the replication fork.
In mammalian cells, the vast majority of CpG sites are methylated. The 15% that are not methylated are preferentially located in CpG islands at the promoters of housekeeping genes. In mice, 5-mC is highly enriched in pericentromeric heterochromatin regions where MeCP2 is also concentrated (Lewis et al. 1992). On the remaining chromosome arms and on rat and human chromosomes (Nan et al. 1998a), anti-MeCP2 staining reveals a uniform pattern suggesting that 5-mC is distributed throughout the genome and that binding of the 106 MeCP2 molecules per cell nucleus may function to suppress transcriptional noise rather than to suppress specific genes (Nan et al. 1997). CpG methylation is not important for the proliferation and in vitro differentiation of embryonic stem (ES) cells in which MeCP2 is not expressed. Targeted ES cells that lack de novo DNA methyltransferase (DNMT) or MeCP2 behave normally in culture (Li et al. 1992; Tate et al. 1996). Methylation becomes important, however, at the stage of organ differentiation when the specific tissue and developmental patterns of gene expression are established. CpG methylation and its associated histone modification are likely to suppress the tissue-specific genes whose activity is not required in the particular cell type. Clonal stability of CpG methylation is the mechanism for the maintenance of these patterns throughout life. CpG methylation is essential for development, since Dnmt−/− embryos die in midgestation (Li et al. 1992). Similarly, chimeras between normal and MeCP2-deficient ES cells survive, albeit with developmental defects, only if the proportion of mutant ES cells is low (Tate et al. 1996). These mouse experiments, although limited to Mecp2-targeted ES cells of a single-strain (129) origin, suggest that MeCP2 protein expression is essential for embryonic development. If that situation were analogous in humans, we would not expect to find null mutations in surviving XCI mosaics (which are comparable to mouse aggregation chimeras) or in hemizygous males. The experience with individuals with RTT who carry MECP2 mutations, therefore, may not be consistent with the mouse data or may indicate that the human mutant alleles are hypomorphs with residual function. We here expanded our study of naturally occurring MECP2 mutations and amino acid variants in humans. The results allow us to examine the essential nature of the protein and the functional importance that its various domains have for the gene-silencing process. To fully explain the disease phenotype will require the identification of target genes whose expression is dysregulated by the loss of normal MeCP2 function in the nervous system.
Patients, Materials, and Methods
Patients and DNA Samples
DNA from lymphoblastoid cell lines (LCLs) and fresh blood leukocytes from individuals diagnosed with RTT and from their affected and unaffected family members was obtained as reported elsewhere (Hofferbert et al. 1997; Schanen et al. 1997; Schanen and Francke 1998; Wan and Francke 1998). We also studied LCLs from the Brazilian family with three affected sisters reported by Sirianni et al. (1998). A girl with features of RTT and incontinentia pigmenti (IP), previously reported in an abstract (Lo et al. 1997), was included as well. Her clinical history is summarized here. At birth, she had on her right leg faint red papules that extended from the groin to the calf, in a linear fashion. At age 1 mo, the papules were replaced by hyperpigmentation. There were never any blisters or involvement of any other parts of the body. A biopsy of the lesion at age 3 years revealed prominent spongiosis and cytoid bodies in the dermis, together with superficial perivascular infiltrate, prominent pigment incontinence, and abundant eosinophils. The histopathological findings were compatible with the diagnosis of IP. Her neurodevelopment was normal for the first 18 mo. Regression of speech occurred at age ∼20 mo. In addition, she gradually lost previously learned hand skills and developed frequent hand-washing movements at age ∼2 years. She also developed autistic features with gaze avoidance and bruxism. At age 3 years 8 mo, her height was at the 3d percentile, her weight and head circumference were at the 10th percentile, and she was hypotonic. She did not fulfill all the diagnostic criteria for RTT and was considered to represent a variant. Differential diagnostic possibilities were excluded by appropriate testing.
Mutation Detection
PCR amplification of the three MECP2 coding exons from genomic DNA was performed by use of the primer pairs and conditions described elsewhere (Amir et al. 1999), with some modifications. Primers were redesigned for amplification of exon 1: forward 5′-AAAACAGATGGCCAAACCAG-3′ and reverse 5′-CAATGGGGGCTTTCAACTTA-3′, with annealing temperature of 55°C. For the most 3′ portion of the exon-3 coding region, the published forward primer 5′-GGAGAAGATGCCCAGAGGAG-3′ was used with a new reverse primer, 5′-CCAACTACTCCCACCCTGAAGC-3′, at an annealing temperature of 62°C.
PCR products were treated with 1 U exonuclease, followed by treatment with 1 U shrimp alkaline phosphatase, each at 37°C for 15 min. The enzymes were inactivated for 10 min at 80°C. The treated PCR products were sequenced directly on both strands, by use of the same PCR primers with fluorescent-dye terminators, on an ABI 377 automatic sequencer. Alternatively, PCR products were gel purified (Qiagen) prior to sequencing. For the LCL from the RTT/IP patient, PCR products were also cloned by use of a TOPO TA cloning kit (Invitrogen), and 13 clones were sequenced. Sequencing results were compared with the reference human MECP2 sequence (GenBank X89430 and AF030876) by use of Sequencher 3.0 (GeneCodes). Both strands were sequenced to confirm all the mutations detected.
Restriction-Enzyme Analysis
Restriction-enzyme digestion of PCR products was carried out to confirm mutations that generated or abolished specific cleavage sites and for evaluation of family members. Specifically, HphI was used to detect the common R168X mutation, since C502T creates a new cleavage site for HphI. The 806delG deletion abolishes a NlaIV restriction site and was detected by NlaIV digestion of PCR products. StyI digestion was used for study of the single-nucleotide polymorphism 1189G→A (E397K).
XCI
The androgen-receptor polymorphism was studied in fresh leukocytes from the RTT/IP individual, as described elsewhere (Allen et al. 1992; Schanen et al. 1997).
Results
Mutations Leading to Premature Termination of Translation (PTT)
The 502C→T transition changes a CGA arginine to a TGA stop at codon 168 in exon 3 (table 1 and fig. 1A). This mutation, which occurs at a CpG dinucleotide, was found in six unrelated sporadic cases. The mutation, in five white individuals and one Japanese individual diagnosed with classic RTT, was not present in any of the mothers or in the available fathers. The same mutation was identified in a family with three affected sisters, from Brazil, that had been previously studied for exclusion mapping by haplotype analysis (Sirianni et al. 1998; Webb et al. 1998). As shown in figure 1B, this R168X mutation creates an HphI site and was present in the LCLs derived from two affected sisters and their phenotypically normal mother and was not present in LCLs from the two unaffected sisters. As previously reported, the mother had a completely skewed X–activation pattern (Sirianni et al. 1998). Codon 168 is located between the MBD and the TRD. Since the R168X mutation occurs in the final exon, it is not expected to cause nonsense-mediated decay of the mutant mRNA (Carter et al. 1996). A putative truncated protein may retain its ability to bind to 5-methyl CpG. However, since the nuclear-localization signal (NLS) resides within the TRD (Lewis et al. 1992), the truncated protein of 167 amino acids, lacking the NLS, may remain largely in the cytoplasm. Indeed, the product of a transfected mutant construct containing the N-terminal 173 amino acids of the rat Mecp2 gene was located predominantly in the cytoplasm (Kudo 1998).
Table 1.
MECP2 Mutations and Normal Variants
| Mutation Type and Exon | Domain | Nucleotide Changea | Amino Acid Change | Restriction Siteb (+/−) | CpG Hotspotc | Recurrencec | Reference |
| Missense: | |||||||
| 2 | MBD | 316C→T | R106W | NlaIII (+) | + | + (2×) | Amir et al. (1999); Present study |
| 3 | MBD | 397C→T | R133C | None | + | − | Amir et al. (1999) |
| 3 | MBD | 464T→C | F155S | TfiI (+), HinfI (+) | − | − | Amir et al. (1999) |
| 3 | MBD | 473C→T | T158M | NlaIII (+) | + | − | Amir et al. (1999) |
| 3 | TRD | 916C→T | R306C | HhaI (−), HinP (−) | + | − | Present study |
| PTT: | |||||||
| 3 | MBD | 411delG | L138X | None | − | − | Present study |
| 3 | … | 502C→T | R168X | HphI (+) | + | + (7×) | Present study |
| 3 | … | 620insT | E235X | BsgI (−) | − | − | Amir et al. (1999) |
| 3 | TRD | 763C→T | R255X | None | + | + (2×) | Amir et al. (1999); Present study |
| 3 | TRD | 806delG | V288X | NlaIV (−) | − | − | Present study |
| Variant: | |||||||
| 3 | … | 1189G→A | E397K | StyI (+), MnlII (−) | +(AS) | + (2×) | Present study |
Numbered from the ATG initiator codon.
+ = Generated; − = abolished.
+ = Present; − = absent; AS = antisense strand.
Figure 1.
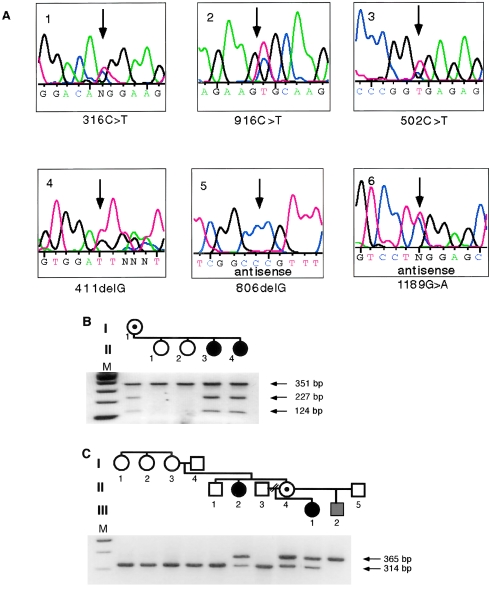
Sporadic and familial mutations in MECP2. A, Sequence tracings of six mutations, including the normal variant 1189G→A. B, R168X mutation, identified by a mutation-induced cleavage at a HphI site. The mother and two affected daughters are heterozygotes. C, 806delG frameshift mutation, identified by loss of an NlaIV site. Two females with RTT, the obligate carrier, and the affected male are positive for the diagnostic 365-bp fragment. There is no evidence for somatic mosaicism in I-3, who transmitted the mutation to her two daughters.
Frameshifts due to single-nucleotide deletions were identified in two cases. The 411delG deletion at codon 137 creates a TGA stop codon immediately downstream (L138X). This truncation mutation within the MBD was found in the individual with incomplete diagnostic features of RTT and highly localized skin findings resembling incontinentia pigmenti (IP). The mutation was seen in the heterozygous state in PCR products and in 7 of 13 clones in a bacterial vector. Since the IP2 locus is also assigned to Xq28, this patient's cells were initially scrutinized for the presence of a genomic rearrangement that might inactivate both loci and lead to the identification of both genes. The discovery of the MECP2 mutation in this patient is consistent with a diagnosis of RTT. XCI studies at the androgen-receptor locus were performed in different laboratories on several occasions. In this patient's blood, there was preferential inactivation of the paternal allele, with frequencies of 70%–80%. Her LCLs had a 100%-skewed XCI pattern. Female heterozygotes for IP have completely skewed XCI patterns in all tissues, presumably because of selection against cell-lethal alleles (Migeon et al. 1989; Parrish et al. 1996). The highly localized cutaneous findings could suggest mosaicism for a postzygotic mutation in the IP2 gene. One should consider, however, that L138X is the closest to a loss-of-function mutation of all 10 MECP2 mutations identified so far. The putative truncated product missing half of the MBD would no longer be able to bind methyl-CpG. A truncated protein made from an expression construct containing amino acids 1–156 did not bind a CpG-methylated oligonucleotide in a southwestern DNA-binding assay (Nan et al. 1993). Therefore, the possibility that this mutation may cause a more complex phenotype cannot be excluded at this time.
Deletion of one of four consecutive guanines (806delG) causes a frameshift that, after 19 missense amino acids, leads to a stop codon at position 288 within the TRD. This mutation was identified in all affected members of a two-generation family previously studied for exclusion mapping of the RTT gene on the X chromosome (Schanen et al. 1997). As shown in figure 1C, the aunt and niece diagnosed with classic RTT, the transmitting female with mild neurological symptoms (poor motor coordination, apraxia, and fine tremor), an IQ of 71, and favorably skewed XCI, and her son, who died from a neonatal encephalopathy (Schanen and Francke 1998), were all positive for this truncating mutation. By sequencing, as well as by NlaIV digestion, the mutation was not detected in LCLs or fresh leukocyte DNA from the grandparents in generation I, four of the grandmother's sisters (only two are shown), and their mother (not shown). Taking into account the haplotyping results for markers in Xq28 (Schanen et al. 1997) that indicate grandmaternal origin of the mutant MECP2 allele, we postulate germline mosaicism in individual I-3 as the origin of this mutation. A random XCI pattern in this phenotypically normal individual is consistent with lack of somatic mosaicism for the mutation. The mutant mRNA was detected by RT-PCR and northern blot in three different tissues from the affected male (data not shown). Our finding of a truncating MECP2 mutation in this family confirms that hemizygous males may be born alive and that skewed XCI mosaicism can lead to mild involvement in females heterozygous for a mutation that causes classic RTT when associated with random XCI.
Mutations Causing Amino Acid Substitutions
The 316C→T transition leading to the R106W substitution in the MBD was previously found in affected half-sisters but not in their common mother (Amir et al. 1999). We identified the same mutation in an unrelated sporadic case (table 1 and fig. 1A). In another sporadic case, a 916C→T transition changes an arginine to a cysteine residue. This mutation, R306C, is the first missense mutation identified in the TRD. The substituted arginine residues in both locations are conserved in MECP2, from mammals to Xenopus laevis. Sequence analysis of both sets of parents confirmed the de novo occurrence of these two missense mutations, in agreement with their designation as disease-causing mutations in classic RTT.
In contrast, a 1189G→A transition leading to a lysine-for–glutamic acid substitution is unlikely to cause disease, for several reasons. The E397K mutation was identified in the heterozygous state in an RTT individual and in her unaffected sister, as well as in the hemizygous state in the normal father. The mutated glutamic-acid residue is outside the functional domains and is not conserved in evolution—in fact, the Xenopus MECP2 sequence carries a lysine at the orthologous site. Since this mutation creates a cleavage site for the enzyme StyI, we used restriction-enzyme analysis to screen for the mutation in a panel of unrelated male and female control individuals of similar ethnic background. One of the 78 X chromosomes screened carried the 1189A (397K) allele. In that family, the variant was inherited from the normal mother. We conclude that E397K is a rare amino acid variant, so far identified in two unrelated white families. Since the mutation consists of a C→T transition at a CpG dinucleotide on the antisense strand, it could potentially result from independent mutational events in the two families.
Discussion
Spectrum of Mutations
With the discovery of mutations in the MECP2 gene, RTT became the first human disorder caused by genetic defects in a component of the epigenetic silencing machinery (Amir et al. 1999). Including the new mutations reported here, the spectrum now encompasses five missense and five nonsense or frameshift mutations leading to PTT (table 1). All missense mutations involve highly conserved amino acids in functional domains. The four amino acid substitutions in the MBD may reduce or abolish methyl-CpG binding. Replacement of arginine by cysteine in the TRD may cause abnormal folding and/or affect interactions with other proteins of the Sin3A/histone deacetylase silencing complex.
Our studies of 18 unrelated families identified only 10 different mutations because of independent de novo recurrences (fig. 2). With one recurrence each for R106W and R255X, the striking finding of six recurrences of R168X in two different laboratories (i.e., those of U.F. and N.C.S.) points to a true mutational hotspot (table 1). Thus, the majority of RTT individuals (10/12 families in this study and 12/18 total families) have PTT, rather than missense, mutations. Four of the five PTT mutations predict a truncated protein leaving the MBD intact. Although nonsense-mediated mRNA decay should not be an issue, it remains to be determined whether truncated proteins are synthesized and have retained methyl-CpG–binding activity. If they can no longer bind to Sin3A because of lack of the TRD, what could be their residual function? Full-length MeCP2 binds to single methyl-CpG sites and protects up to 12 nucleotides (Nan et al. 1993). Binding of truncated protein molecules to methyl-CpG sites could provide steric hindrance to the transcription complex. Inactive promoters usually contain a higher density of CpGs than there is room for MeCP2 molecules to bind (Nan et al. 1997). Therefore, smaller MBD-containing mutant protein molecules bound to multiple CpGs in CpG islands might accomplish some degree of silencing, either by recruiting a silencing complex by a TRD-independent mechanism or by interfering with transcription-factor binding directly.
Figure 2.
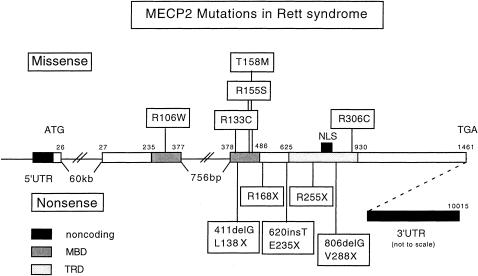
MECP2 mutations in Rett syndrome. The structure of the human MECP2 gene is derived from the genomic sequence (GenBank AF030876). The functional domains were defined by Nan et al. (1993, 1997).
Mechanism of Mutagenesis
Seven of the eight single-nucleotide substitutions in table 1 are C→T transitions at CpG sites. Even though CpG dinucleotides are underrepresented in vertebrate genomes, they are hypermutable and represent common sites of germline and somatic mutations (Rideout et al. 1990). The proposed mechanism involves 5-methylation of cytosine by a methyltransferase and spontaneous deamination of 5-methylcytosine to thymine. This type of mutation was observed in 10 of the 12 families in this report (or in 13 of the 18 RTT families total). C→T or G→A (when the 5-methylcytosine deamination occurs on the antisense strand) transitions overall constitute ∼55% of single-nucleotide substitutions in a human genetic-disease mutation database (Krawczak and Cooper 1996), but, for some genes, the frequencies are much higher. Almost all cases of achondroplasia are due to a G→A transition at a single CpG site in the FGFR3 gene (Bellus et al. 1995). CpG hypermutatibility implies that the site is methylated in the germline and thus is prone to deamination. Male germ cells have high levels of CpG methylation, and the X chromosome, in particular, is completely inactivated. As a consequence, the MECP2 gene is likely to be methylated, since it is not expressed in male germ cells. Its expression level in female germ cells is not known; however, the DNA in oocytes is markedly undermethylated (Monk et al. 1987). Germline CpG transition mutations in the factor IX gene that cause hemophilia B show an 11-fold male predominance (Ketterling et al. 1993). Therefore, we hypothesize that the majority of de novo MECP2 mutations leading to RTT arise from a 5mC→T deamination in a male germ cell where the MECP2 gene is methylated. Although the parental origin will have to be established by haplotyping, we expect to find a preferential paternal origin for these mutations. Such a hypothesis has previously been proposed to explain the sex-limited occurrence of RTT in females (Thomas 1996).
In an attempt to focus our future mutation search on CpG hotspots, we analyzed the MECP2 coding sequence and identified a total of 63 CpGs. At 28 of those sites, a C→T transition on the coding strand would be silent, creating a synonymous codon. The remaining 35 CpGs are potential mutational hotspots (see table A1 in the Appendix, which appears only in the online edition)(table A1). Two of the five potential R→X mutations were already identified, in patients with RTT, as recurrent nonsense mutations (see above). Of nine potential amino acid substitutions in the MBD, three have been identified in patients with RTT, as has one of the six potential substitutions in the TRD (table 1). All amino acid residues in the MBD and TRD that are potentially mutable by C→T transitions are conserved in the Xenopus MECP2 sequence. Outside the MBD and TRD domains, nine potential substitutions affect conserved residues, and seven affect nonconserved residues. Mutations in these latter sites may cause minor functional impairment of the mutant protein and may not lead to RTT.
When C→T transitions at CpG sites occur on the antisense strand, they result in G→A transitions on the coding strand. For the human MECP2 sequence, all possible events of this kind could give rise to nine different types of amino acid substitutions at a total of 51 different codons. Many of these are exchanges between similar amino acids such as Val and Ile, or Asp and Asn. Only one of these predicted substitutions was observed in our study: E397K. It appears to be a rare normal variant in the population.
Mutation Detection
When mutations create or abolish restriction sites, samples can be screened by restriction-enzyme digestion (fig. 1). A stepwise protocol for mutation screening could start with restriction digests of PCR products to look for the common R168X mutation and the potential R270X mutation in the TRD. Two of the observed missense mutations and five potential arginine substitutions in the MBD also change restriction sites, as do four arginine substitutions in the TRD, one of which has already been identified.
PCR amplification of overlapping genomic fragments has detected MECP2 mutations in ∼50% of patients with RTT. So far, we analyzed only the coding region. MECP2 mutations could also involve the highly conserved regions within the 8.5-kb 3′UTR (Coy et al. 1999). The finding of de novo disease-causing mutations in the 3′UTR would be a major step toward understanding the posttranscriptional regulation of MECP2. If complete screening of the gene fails to reveal de novo mutations in a significant number of probands with RTT, the possibility of a second RTT locus should be considered. Since the families that were used to narrow the search for the RTT gene to Xq28 have mutations in MECP2, as reported here, a putative second RTT locus could reside anywhere on the X chromosome.
Effects of MECP2 Mutations on the Phenotype
MECP2 is expressed during organogenesis throughout the embryo and, later, most strongly in the hippocampus (Coy et al. 1999). Mutations in this gene, therefore, might impair several organ systems. There are many potential reasons for the RTT phenotype not being more pleiotropic. In tissues other than the brain, the function of a mutant MeCP2 could be taken over by related proteins that do exist. A database search led Hendrich and Bird (1998) to three related genes that share a similar MBD but have no sequence similarity outside this domain and no identifiable TRD. Of the three loci—called “MBD2,” “MBD3,” and “MBD4”—all of which were characterized and mapped to autosomes in mouse and human, that for MBD4 is the most closely related to that for MECP2 (Hendrich et al. 1999a). As was reported recently, MBD4 functions as a thymine glycosylase that corrects mismatches generated by C→T transitions (Hendrich et al. 1999b). As the components of the histone deacetylase complex are identified, other MBD-containing proteins have been found to carry out the methyl-CpG binding function (Wade et al. 1999). The MeCP1 complex containing MBD2 and MBD3 could be involved in silencing at sites with ⩾12 methylated CpGs (Meehan et al. 1989). In HeLa cells, which lack the MeCP2 protein, methylated genes are silenced by this complex (Ng and Bird 1999).
DNA methylation–dependent silencing is a known mechanism for maintenance of XCI and gametic imprinting patterns. Is there any evidence for abnormalities in these processes in individuals with RTT? Escape from XCI causing biallelic overexpression of X-linked genes has been considered early on as a possible causative mechanism for RTT that could never involve males, unless they were XXY. No data in support of this notion have been published. Numerous investigators have used the androgen-receptor polymorphism for XCI studies in individuals with RTT (Allen et al. 1992; Schanen et al. 1997). Reactivation of the inactive AR allele would lead to complete HpaII digestion of both alleles. To our knowledge, this has not been observed. Despite this preliminary negative evidence, a careful study of the expression of X-chromosomal genes that are subject to XCI might be in order.
The role of MeCP2 in keeping imprinted genes silent is also unclear. Imprinted genes that are misexpressed in dmnt-deficient mouse cells are normally expressed in Mecp2-deficient cells (Hendrich and Bird 1998). A diagnostic workup of patients suspected of having RTT routinely includes testing for Angelman syndrome, because of phenotypic overlap—first by FISH, to look for 15q11-q13 deletions, and then by restriction analysis of allele-specific DNA-methylation imprints. No unexpected results have been reported. In addition, a large number of individuals with classic RTT had normal DNA methylation at the SNRPN locus (A. Beaudet and H. Y. Zoghbi, unpublished data). Normal allele-specific methylation patterns, however, do not imply proper silencing if the MeCP2 protein is absent or defective. Therefore, the possibility of dysregulation of imprinted genes in RTT will need to be evaluated at the transcriptional or translational level, accompanied by histone-acetylation studies at imprinted loci (Coffee et al. 1999). Since methyl-CpGs required for uniparental gene expression may also be located in introns of the expressed alleles, both over- and underexpression of imprinted genes may be anticipated as a consequence of MECP2 mutations. Our future research will focus on a global assessment of gene-expression levels, using high-density cDNA microarrays that will soon represent all human genes. This screening procedure will generate over- and underexpressed candidate genes, for more detailed studies that are needed to understand the pathogenetic steps leading to neuronal dysfunction.
The Phenotypic Spectrum of MECP2 Mutations and Indications for Diagnostic Testing
The two extremes of the phenotypic spectrum are lethality and normality. We identified a MECP2 mutation in a hemizygous male with congenital encephalopathy who had multiple respiratory arrests and survived for 1 year (Schanen and Francke 1998; Schanen et al. 1998). That same mutation caused classic RTT in his sister and aunt but only mild neurological symptoms and learning problems in his mother (Schanen et al. 1997). Another nonsense MECP2 mutation was found in a completely normal woman who passed it on to three daughters, who had RTT, and, possibly, to one son, who died during the neonatal period (Sirianni et al. 1998). We conclude that, regardless of the type of mutation, the XCI pattern is the major determinant of the phenotype in females. We propose the following model: individuals who meet the diagnostic criteria for RTT (Trevathan et al. 1988) are likely to have XCI ratios of 50/50–80/20, whereas those on the fringes of the bell-shaped distribution would not meet criteria. At one end of the spectrum, there are severely affected females who lack the criterion of normal early development and follow a clinical course more similar to that of hemizygous males (Schanen et al. 1998). At the other end, a mildly affected group will include partially manifesting “forme fruste” females (Hagberg 1995), as well as women with nonspecific neurologic and learning deficits, for whom a diagnosis of RTT would never be considered. Alternatively, incomplete clinical manifestations in females with milder variant forms of RTT could also be caused by missense mutations that have a subtle effect on MeCP2 function. The indications for mutation testing, therefore, have to include older females who do not meet diagnostic criteria for RTT and female infants with neurodevelopmental problems—with or without a period of normal development—as well as males with unexplained death during the neonatal period.
Acknowledgments
We thank the families with RTT for their participation, B. Foellmer, V. Meyers, and R. F. Pilotto for technical contributions, S. Fulmer-Smentek for critical reading of the manuscript, and K. Redman for administrative assistance. We are grateful to the Genome Analysis Group at the Institute for Molecular Biotechnology in Jena, Germany, for depositing the genomic MECP2 sequence in the public database. This work was supported by the Howard Hughes Medical Institute (support to U.F., S.S.J.L., X.Z., and H.Y.Z.); by NIH grants HD24234 (to H.Y.Z., U.F., and M.W.), HD24448 and RR00052 (to S.N.), HD034610 (to N.C.S. and H.-R.S.), and HD01103 (to N.C.S); by a Lynn Marie Chandler Postdoctoral Fellowship (to M.W.) by the Research for Rett Syndrome Foundation (support to S.N.), and by the International Rett Syndrome Association (support to R.E.A. and I.H.-M).
Table A1
Table A1.
Predicted Mutations Resulting from C→T Transitions at CpG Dinucleotides in the MECP2 Coding Sequence
| Cytosinea | Domaina | Mutation (Codon Change)a | Restriction Site Changeb | Xenopusc |
| 215 | … | P72L (CCG→CTG) | BstNI (+) | NC |
| 224 | … | P75L (CCG→CTG) | … | C |
| 250 | MBD | R84W (CGG→TGG) | PflMI (+) | C |
| 253 | MBD | R85C (CGC→TGC) | HaeII (−) | C |
| 265 | MBD | R89C (CGT→TGT) | FokI (−) | C |
| 271 | MBD | R91W (CGG→TGG) | BsrI (+) | C |
| 316 | MBD | (R106W CGG→TGG) | NlaIII (+) | C |
| 343 | MBD | R115C (CGC→TGC) | BbvI (+) | C |
| 397 | MBD | R133C (CGC→TGC) | … | C |
| 419 | MBD | A140V (GCG→GTG) | … | C |
| 473 | MBD | T158M (ACG→ATG) | NlaIII (+) | C |
| 499 | … | R167W (CGG→TGG) | EcoRII (+) | C |
| 502 | … | R168X (CGA→TGA) | HphI (+) | C |
| 562 | … | R188W (CGG→TGG) | MspI (−) | C |
| 568 | … | R190C (CGC→TGC) | HgaI (−) | C |
| 590 | … | T590M (ACG→ATG) | NlaIII (+) | NC |
| 602 | … | A602L (GCG→GTG) | MscI (+) | NC |
| 608 | … | T203M (ACG→ATG) | MaeII (−) | NC |
| 686 | TRD | S229L (TCG→TTG) | … | NC |
| 748 | TRD | R250C (CGC→TGC) | … | C |
| 763 | TRD | R255X (CGA→TGA) | … | C |
| 802 | TRD | R268W (CGG→TGG) | PflMI (+) | C |
| 808 | TRD | R270X (CGA→TGA) | NlaIV (−) | C |
| 815 | TRD | R272L (CCG→CTG) | AluI (+) | C |
| 880 | TRD | R294X (CGA→TGA) | … | NC |
| 916 | TRD | R306C (CGC→TGC) | HhaI (−) | C |
| 925 | TRD | R309W (CGG→TGG) | XmaI (−) | C |
| 1030 | … | R344W (CGG→TGG) | AciI (−) | NC |
| 1060 | … | R354C (CGC→TGC) | BsgI (+) | NC |
| 1316 | … | A439L (GCG→GTG) | SacII (−) | C |
| 1334 | … | T445M (ACG→ATG) | EagI (−) | NC |
| 1357 | … | R453X (CGA→TGA) | DdeI (+) | C |
| 1372 | … | R458C (CGC→TGC) | HhaI (−) | C |
| 1432 | … | R478W (CGG→TGG) | PvuII (+) | C |
| 1436 | … | T479M (ACG→ATG) | FokI (+) | C |
Bold: Mutation observed (see table 1).
+ = generated; − = abolished.
C = amino acid conserved; NC = amino acid not conserved.
References
- Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW (1992) Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51:1229–1239 [PMC free article] [PubMed]
- Amir RE, Roth Dahle E, Toniolo D, Zoghbi HY. Candidate gene analysis in Rett syndrome and the identification of twenty-one SNPs in Xq. Am J Med Genet (in press) [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188 [DOI] [PubMed]
- Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, Kaitila I, et al (1995) Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet 56:368–373 [PMC free article] [PubMed]
- Buschhausen G, Wittig B, Graessmann M, Graessmann A (1987) Chromatin structure is required to block transcription of the methylated herpes simplex virus thymidine kinase gene. Proc Natl Acad Sci USA 84:1177–1181 [DOI] [PMC free article] [PubMed]
- Carter MS, Li S, Wilkinson MF (1996) A splicing-dependent regulatory mechanism that detects translation signals. EMBO J 15:5965–5975 [PMC free article] [PubMed]
- Coffee B, Zhang F, Warren ST, Reines D (1999) Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet 22:98–101 [DOI] [PubMed]
- Coy JF, Sedlacek Z, Bachner D, Delius H, Poustka A (1999) A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3′-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum Mol Genet 8:1253–1262 [DOI] [PubMed]
- Eden S, Hashimshony T, Keshet I, Cedar H, Thorne AW (1998) DNA methylation models histone acetylation. Nature 394:842 [DOI] [PubMed]
- Ellison KA, Fill CP, Terwilliger J, DeGennaro LJ, Martin GA, Anvret M, Percy AK, et al (1992) Examination of X chromosome markers in Rett syndrome: exclusion mapping with a novel variation on multilocus linkage analysis. Am J Hum Genet 50:278–287 [PMC free article] [PubMed]
- Fan F, Amir RE, Zhang X, Dahle EJ, Zoghbi H, Francke U (1999) Search for X-chromosomal microdeletions in Rett syndrome. Am J Hum Genet Suppl 65:A294 [Google Scholar]
- Hagberg B (1985) Rett's syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr Scand 74:405–408 [DOI] [PubMed]
- ——— (1995) Clinical delineation of Rett syndrome variants. Neuropediatrics 26:62 [DOI] [PubMed]
- Hagberg B, Aicardi J, Dias K, Ramos O (1983) A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol 14:471–479 [DOI] [PubMed]
- Hendrich B, Abbott C, McQueen H, Chambers D, Cross S, Bird A (1999a) Genomic structure and chromosomal mapping of the murine and human Mbd1, Mbd2, Mbd3, and Mbd4 genes. Mamm Genome 10:906–912 [DOI] [PubMed]
- Hendrich B, Bird A (1998) Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol 18:6538–6547 [DOI] [PMC free article] [PubMed]
- Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A (1999b) The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 401:301–304 [DOI] [PubMed]
- Hofferbert S, Schanen NC, Budden SS, Francke U (1997) Is Rett syndrome caused by a triplet repeat expansion? Neuropediatrics 28:179–183 [DOI] [PubMed]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, et al (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19:187–191 [DOI] [PubMed]
- Kass SU, Goddard JP, Adams RL (1993) Inactive chromatin spreads from a focus of methylation. Mol Cell Biol 13:7372–7379 [DOI] [PMC free article] [PubMed]
- Keshet I, Lieman-Hurwitz J, Cedar H (1986) DNA methylation affects the formation of active chromatin. Cell 44:535–543 [DOI] [PubMed]
- Ketterling RP, Vielhaber E, Bottema CD, Schaid DJ, Cohen MP, Sexauer CL, Sommer SS (1993) Germ-line origins of mutation in families with hemophilia B: the sex ratio varies with the type of mutation. Am J Hum Genet 52:152–166 [PMC free article] [PubMed]
- Krawczak M, Cooper DN (1996) Single base-pair substitutions in pathology and evolution: two sides to the same coin. Hum Mutat 8:23–31 [DOI] [PubMed]
- Kudo S (1998) Methyl-CpG-binding protein MeCP2 represses Sp1-activated transcription of the human leukosialin gene when the promoter is methylated. Mol Cell Biol 18:5492–5499 [DOI] [PMC free article] [PubMed]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A (1992) Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69:905–914 [DOI] [PubMed]
- Li E, Beard C, Jaenisch R (1993) Role for DNA methylation in genomic imprinting. Nature 366:362–365 [DOI] [PubMed]
- Li E, Bestor TH, Jaenisch R (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69:915–926 [DOI] [PubMed]
- Lindsay S, Monk M, Holliday R, Huschtscha L, Davies KE, Riggs AD, Flavell RA (1985) Differences in methylation on the active and inactive human X chromosomes. Ann Hum Genet 49:115–127 [DOI] [PubMed]
- Lo IFM, Tang WYM, Wong TW, Lam STS (1997) A girl with features of two X-linked dominant disorders: Rett syndrome and incontinentia pigmenti. Am J Hum Genet Suppl 61:A105 [Google Scholar]
- Lupski JR (1998) Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 14:417–422 [DOI] [PubMed]
- Martinho PS, Otto PG, Kok F, Diament A, Marques-Dias MJ, Gonzalez CH (1990) In search of a genetic basis for the Rett syndrome. Hum Genet 86:131–134 [DOI] [PubMed]
- Meehan RR, Lewis JD, McKay S, Kleiner EL, Bird AP (1989) Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 58:499–507 [DOI] [PubMed]
- Migeon BR, Axelman J, de Beur SJ, Valle D, Mitchell GA, Rosenbaum KN (1989) Selection against lethal alleles in females heterozygous for incontinentia pigmenti. Am J Hum Genet 44:100–106 [PMC free article] [PubMed]
- Migeon BR, Dunn MA, Thomas G, Schmeckpeper BJ, Naidu S (1995) Studies of X inactivation and isodisomy in twins provide further evidence that the X chromosome is not involved in Rett syndrome. Am J Hum Genet 56:647–653 [PMC free article] [PubMed]
- Monk M, Boubelik M, Lehnert S (1987) Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development 99:371–382 [DOI] [PubMed]
- Nan X, Campoy FJ, Bird A (1997) MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88:471–481 [DOI] [PubMed]
- Nan X, Cross S, Bird A (1998a) Gene silencing by methyl-CpG-binding proteins. In: Chadwick DJ, Cardew G (eds) Epigenetics. John Wiley & Sons, New York, pp 6–16 [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A (1998b) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386–389 [DOI] [PubMed]
- Nan X, Meehan RR, Bird A (1993) Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res 21:4886–4892 [DOI] [PMC free article] [PubMed]
- Ng HH, Bird A (1999) DNA methylation and chromatin modification. Curr Opin Genet Dev 9:158–163 [DOI] [PubMed]
- Parrish JE, Scheuerle AE, Lewis RA, Levy ML, Nelson DL (1996) Selection against mutant alleles in blood leukocytes is a consistent feature in incontinentia pigmenti type 2. Hum Mol Genet 5:1777–1783 [DOI] [PubMed]
- Razin A, (1998) CpG methylation, chromatin structure and gene silencing—a three-way connection. EMBO J 17:4905–4908 [DOI] [PMC free article] [PubMed]
- Rett A (1966) Uber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonamie im Kindesalter. Wien Med Wochenschr 116:723–738 [PubMed]
- Rideout WMI, Coetzee GA, Olumi AF, Jones PA (1990) 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 249:1288–1290 [DOI] [PubMed]
- Schanen C, Francke U (1998) A severely affected male born into a Rett syndrome kindred supports X-linked inheritance and allows extension of the exclusion map. Am J Hum Genet 63:267–269 [DOI] [PMC free article] [PubMed]
- Schanen NC, Kurczynski TW, Brunelle D, Woodcock MM, Dure LSt, Percy AK (1998) Neonatal encephalopathy in two boys in families with recurrent Rett syndrome. J Child Neurol 13:229–231 [DOI] [PubMed]
- Schanen NC, Roth Dahle EJ, Capozzoli F, Holm VA, Zoghbi HY, Francke U (1997) A new Rett syndrome family consistent with X-linked inheritance expands the X chromosome exclusion map. Am J Hum Genet 61:634–641 [DOI] [PMC free article] [PubMed]
- Sirianni N, Naidu S, Pereira J, Pillotto RF, Hoffman EP (1998) Rett syndrome: confirmation of X-linked dominant inheritance, and localization of the gene to Xq28. Am J Hum Genet 63:1552–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate P, Skarnes W, Bird A (1996) The methyl-CpG binding protein MeCP2 is essential for embryonic development in the mouse. Nat Genet 12:205–208 [DOI] [PubMed]
- Thomas GH (1996) High male:female ratio of germ-line mutations: an alternative explanation for postulated gestational lethality in males in X-linked dominant disorders. Am J Hum Genet 58:1364–1368 [PMC free article] [PubMed]
- Trevathan E, Moser HW, Opitz JM, Percy AK, Naidu S, Holm VA, Boring CC, et al (1988) Diagnostic criteria for Rett syndrome. Ann Neurol 23:425–428 [DOI] [PubMed]
- Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP (1999) Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet 23:62–66 [DOI] [PubMed]
- Wan M, Francke U (1998) Evaluation of two X chromosomal candidate genes for Rett syndrome: glutamate dehydrogenase-2 (GLUD2) and rab GDP-dissociation inhibitor (GDI1). Am J Med Genet 78:169–172 [PubMed]
- Webb T, Clarke A, Hanefeld F, Pereira JL, Rosenbloom L, Woods CG (1998) Linkage analysis in Rett syndrome families suggests that there may be a critical region at Xq28. J Med Genet 35:997–1003 [DOI] [PMC free article] [PubMed]