

Summary
Atypical hemolytic uremic syndrome (HUS) presents with the clinical features of hypertension, microangiopathic hemolytic anemia, and acute renal failure. Both dominant and recessive modes of inheritance have been reported. This study describes the genetic and functional analysis of a large Bedouin kindred with autosomal recessive HUS. The kindred consists of several related nuclear families in which all parent unions of affected children are consanguineous. A previous report demonstrated that a dominant form of HUS maps to chromosome 1q and that complement factor H (CFH), a regulatory component of the complement system, lies within the region and is involved in the dominant disorder. Early-onset and persistent hypocomplementemia in this Bedouin kindred prompted us to evaluate the CFH gene. Linkage analysis was performed, demonstrating linkage between the disorder and the markers near the CFH gene. Mutation analysis of the CFH coding region revealed a single missense mutation. Functional analyses demonstrate that the mutant CFH is properly expressed and synthesized but that it is not transported normally from the cell. This is the first study reporting that a recessive, atypical, early-onset, and relapsing HUS is associated with the CFH protein and that a CFH mutation affects intracellular trafficking and secretion.
Introduction
Hemolytic uremic syndrome (HUS) is a microangiopathic disease of various etiologies. Atypical HUS is characterized by the absence of antecedent diarrhea, the tendency to relapse, a positive family history, and a poor outcome (Kaplan et al. 1975; Kaplan 1977; Kaplan and Proesmans l987; Segal and Sinai 1995). Both autosomal dominant (MIM 134370) (Farr et al. 1975) and recessive (MIM 235400) (Kaplan et al. 1975; Mattoo et al. 1989) modes of inheritance have been reported for familial HUS. Previous studies have reported an increased incidence of human-leukocyte-antigen (HLA) B40 group antigens in children with typical HUS. Both low C3 levels and hypocomplementemia have been reported (Barre et al. 1977; Gonzalo et al. 1981; Bogdanovic et al. 1988, 1997; Pichette et al. 1994). Recently, several families with familial HUS and low serum complement factor H (CFH) levels were described. CFH deficiency has been associated with an increased tendency for several diseases, including systemic lupus erythematosus (Fijen et al. 1996), pyogenic infection susceptibility (Warwicker et al. 1998), type II membranoproliferative glomerulonephritis (Jansen et al. 1995), a form of chronic collagen type III glomerulopathy (Vogt et al. 1995; West and McAdams 1998), and atypical familial HUS (Ohali et al. 1998).
We recently described the clinical pathological features and complement studies of a Bedouin kindred with autosomal recessive HUS (Ohali et al. 1998). All patients were members of one extended inbred Bedouin kindred and had low C3 and hypocomplementemia during and between relapses. Serum CFH levels were either significantly lower or absent in affected patients compared with unaffected siblings and parents, which suggests that a mutant form of this regulatory protein of the complement cascade could cause the disorder.
Material and Methods
Subjects
This study evaluated Bedouin-Arab patients from a single large kindred with atypical HUS and their unaffected siblings and parents. Atypical HUS was defined as a simultaneous occurrence of microangiopathic hemolytic anemia and acute renal insufficiency that was not preceded by either a diarrheal prodrome or an enteric infection caused by cytotoxin-producing microorganisms. Informed consent was obtained from all family members participating in the study.
Genotyping
DNA was prepared from whole blood by use of a standard protocol, and the concentration was determined by spectrophotometry at optical density 260. DNA was diluted to a final concentration of 20 ng/μl. Single-tandem-repeat polymorphisms (STRPs) were amplified in an 8.4-μl PCR mixture containing 40 ng DNA; 1.25 μl PCR buffer stock (100 mM Tris-HCl pH 8.8, 500 mM KCl, 15 mM MgCl2, and 0.01% w/v gelatin); 200 μΜ each dATP, dCTP, dGTP, and dTTP; 2.5 pmol each forward and reverse primer; and 0.25 U Taq polymerase. PCR reactions were performed under the following conditions: 35 cycles each at 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s. PCR products were electrophoresed on denaturing polyacrylamide (6%) gels (containing 7.7 M urea). The gels were visualized by silver staining. Genotyping was performed with the use of the following markers: D1S191, D1S202, D1S2757, D1S412, D1S408, D1S2794, D1S2840, GATA135F02, D1S1660, D1S1175, F13B, D1S1276, D1S1647, D1S249, and D1S245. The STRPs used in this study were developed by the Cooperative Human Linkage Center, Généthon, and the University of Utah (Sheffield et al. 1995; Utah Marker Development Group 1995; Dib et al. 1996). Genotypes were independently scored by two observers and were entered into a local database.
Linkage Analysis
Linkage analysis was performed on the family with HUS, by use of the Mendel program (Lange et al. 1988), which accommodates inbreeding loops in the family. The disease was assumed to be transmitted in an autosomal recessive mode with complete penetrance. Allele frequencies were assumed to be equal for each marker.
SSCP Analysis
PCR of CFH primers (table 1) was performed with 40 ng genomic DNA in 20-μl PCR mixtures containing 2 μl PCR buffer stock (100 mM Tris-HCl pH 8.8, 500 mM KCl, 15 mM MgCl2, and 0.01% w/v gelatin); 200 mM each dATP, dCTP, dGTP, and dTTP; 2.5 pmol each forward and reverse primer; and 0.25 U Taq polymerase. PCR products were electrophoresed on nondenaturing gels (5 ml glycerol, 5 ml of 5 × Tris-borate EDTA [TBE], 12.5 ml 37.5:1 acrylamide/bis, and 77.5 ml ddH2O) for 3–4 h in 0.25 × TBE. The gels were visualized by silver staining (Bassam et al. 1991). Variants seen on the SSCP gel were sequenced and were compared with the control samples, to detect any changes from the normal sequence.
Table 1.
Primers from the CFH Coding Region for Use in SSCP
| CFH PrimerName | 5′ Sequence | Size(bp) | 3′ Sequence |
| CFHexon1 | ATGGGCTATTTCTGTAGCAG | 205 | CATTTCCTTAATGGATTAAG |
| CFHexon2 | CAGAAAAGGCCCTGTGGACA | 182 | CATATAGGAATATCATTGGT |
| CFHexon3 | GAAGTTGTGAAGTGTTTACC | 193 | ACACACTTTGGTTTCTCTTT |
| CFHexon4 | GAAATTTCATGCAAATCCCC | 169 | TCACATGAAGGCAACGGACG |
| CFHexon5 | GAAAAATCATGTGATAATCC | 170 | ACATCTCGGAGCAGGTATCC |
| CFHexon6 | ACCTTGAAACCTTGTGATTA | 197 | CATGGTACTGCTGGCGACCA |
| CFHexon7 | CTCAGAAAATGTTATTTTCC | 173 | ATGCATCTGGGAGTAGGAGA |
| CFHexon8 | CGTGTCAAAACATGTTCCAA | 210 | TGCACGTGGGTTGAGCTCAC |
| CFHexon9 | AAATCTTGTGATATCCCAGT | 178 | AACATATGGGTAAATCAGAC |
| CFHexon10 | GAAAGAGAATGCGAACTTCC | 176 | TTACATATTGGGAGGTCAGG |
| CFHexon11 | GAGCAAGTACAATCATGTGG | 185 | ACAATACACACTGGTAAAGT |
| CFHexon12 | GAGGAGAGTACCTGTGGAGA | 186 | ATTGCCACACACTGGGGAAG |
| CFHexon13 | GATAAACTTAAGAAGTGCAA | 174 | CCATTGAGCAGTTCACTTCT |
| CFHexon14 | CAAATACAATTATGCCCACC | 179 | ACACAGAGTGGTATTGACT |
| CFHexon15 | GAAAAAATTCCATGTTCACA | 182 | CACTGAGGTGGAGAACTCCA |
| CFHexon16 | GAAGGCCTTCCTTGTAAATC | 181 | TGCATGATGGAGGGTGAGAC |
| CFHexon17 | AAACAGATTGTCTCAGTTTTA | 175 | CTGCATGTTGGCCTTCCTGT |
| CFHexon18 | GACACCTCCTGTGTGAATCC | 179 | TGCATTGAGGTGGTTCCGTC |
| CFHexon19 | GATTCTACAGGAAAATGTGG | 188 | AAGCATTTTGGTGGTTCTGA |
| CFHexon2021 | CCGTGTGTAATATCCCGAGA | 211 | GCACAAGTTGGATACTCCAG |
DNA Sequencing
PCR products were electrophoresed on a 1% agarose gel and were purified with use of the Quiaquick Gel Extraction kit (Qiagen). Purified PCR product (4.5 μl) was used as a template for each sequencing reaction. Then, 1 μl 20-pmol primer and 4.5 μl terminator sequencing mix (Amersham) were added, for a final reaction volume of 10 μl. Cycling conditions were performed as specified by the manufacturer. The sequencing reactions were precipitated and were resuspended in 2 μl loading buffer. The reactions were analyzed on an ABI 377 sequencer with a 3-h run time.
Western Blot Assay
Serum CFH analysis was performed by western blotting, as described elsewhere (Vogt et al. 1995). In brief, serum samples (20 μl serum diluted 50-, 100-, and 200-fold with saline) were exposed to SDS PAGE and were transferred onto a nitrocellulose membrane. CFH on the membrane was visualized by serial incubations with specific monoantibodies, then with second antibodies conjugated with peroxidase and then with diaminobenzidine as substrate.
Pulse-Chase Assay
Skin-derived fibroblasts from affected individuals and from normal control individuals were grown to confluence, in multiwell tissue-culture plates, in Dulbecco's minimal essential medium (DMEM) containing 10% FCS; they were washed to remove serum and spent medium and were cultured, for 16–24 h, either in DMEM with 1 mg BSA/ml alone or in medium containing BSA and various concentrations of interleukin-1 and tumor necrosis factor. Biosynthetic experiments were performed by incubation of the cells for 2 h in methionine-free DMEM medium containing [35S]-methionine (300 μCi/ml), as described elsewhere (Pichette et al. 1994). Total protein synthesis was estimated from incorporation of [35S]-methionine into trichloracetic acid–insoluble protein. Cell lysates and extracellular medium were prepared for immunoprecipitation, were preabsorbed, and then were immunoprecipitated with formalin-fixed Staphylococcus aureus. Immunoprecipitates were subjected to SDS-PAGE, and gels were fixed, impregnated with autofluor, dried, and exposed, at −70°C, to X-ray film. Incorporation of [35S]-methionine into the individual immunoprecipitated proteins was determined in gel slices, after digestion with 15% hydrogen peroxide for 16 h at 65°C and after addition of Ultima Gold (Packard). The effect of the stimuli on protein synthesis was normalized after correction of the specific protein counts in the gel slices for total protein synthesized in each sample. Newly synthesized CFH proteins were detected by use of specific antibody.
Northern Blot Assay
Fibroblasts were grown to confluence in 162-cm2 flasks and were stimulated with interferon-γ (100 ng/ml) and tumor necrosis factor (10 ng/ml) 20 h prior to RNA isolation. Approximately 108 cells were lysed, and RNA was isolated by use of the method described by Chirgwin et al. (1979). Total RNA (10 μg) was denatured and was subjected to electrophoresis in a 1% agarose/formaldehyde gel. The RNA was transferred to a nylon membrane (Armesham Life Sciences) and was processed as described by Vicra et al. (1990). The nylon-bound mRNA was hybridized to nick-translated [32P]-labeled DNA coding for the CFH gene. Human β-actin probe was used as an internal control.
Results
Clinical Evaluation
All patients were members of an extended inbred Bedouin kindred (fig. 1). The median age at presentation was 2 wk (range 1–20 wk). Of the 11 patients, 10 died. One is alive and is undergoing dialysis. The clinical and histopathologic features of these patients have been reported by Ohali et al. (1998). Thrombotic microangiopathy with a predominant early arteriolar involvement and subsequent development of ischemic glomerular changes were found by renal biopsy. All patients had low complement-component levels during and between relapses. The complement deficiency could not be normalized by repeat plasma transfusions.
Figure 1.
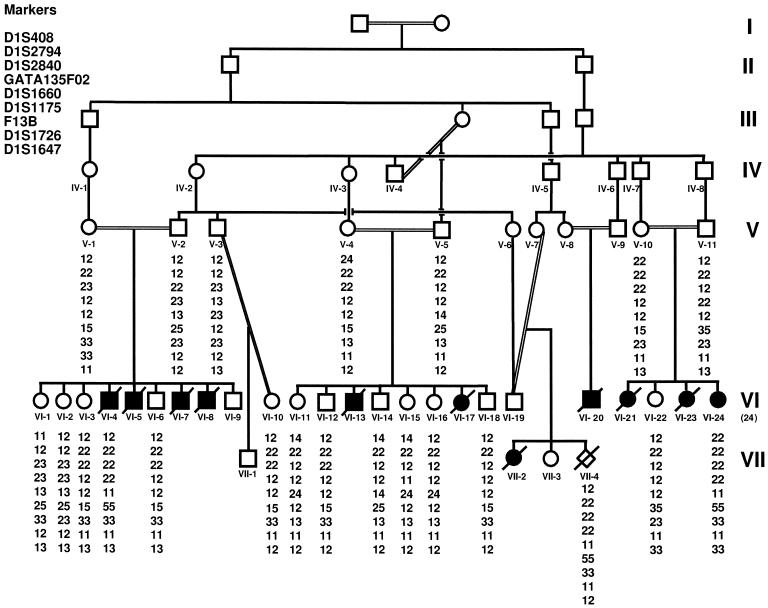
Family pedigree and genotypes. Blackened symbols denote affected individuals; unblackened symbols, unaffected individuals; unblackened diamond, CVS. I–VII denote generation. The ID numbers for each member of generations V–VII are listed below each symbol. Genotypes of both flanking and linked markers are shown below ID numbers. Note that all the affected individuals are homozygous for the markers between flanking markers D1S408 and D1S1647. Only the markers defining the critical region are listed.
Genetic Mapping
DNA samples were available for 22 family members, including 20 unaffected individuals and 2 affected individuals (VI-4 and VI-24), and 1 chorionic villus sample (CVS) (VII-4) was available. Genotyping was performed with 15 STRP markers on chromosome 1q32. The two affected individuals were homozygous for the same allele, for markers across the region flanked by D1S408 and D1S1647 (fig. 2). Genotyping of the CVS indicated an affected fetus. The CVS data were not used in the linkage analysis. Using the Mendel program, we obtained a maximum LOD score (Zmax) of 3.38, at recombination fraction (θ) 0, for the fully informative marker D1S1175 (table 2). The flanking markers define a critical interval of 6 cM.
Figure 2.
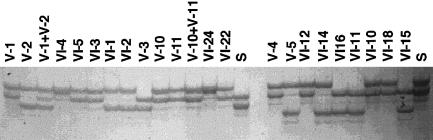
Representative genotypes with marker D1S1160. Twenty-three DNA samples from the HUS kindred were analyzed by PCR with D1S1660. The ID numbers for each lane are shown. The unaffected parents are individuals V-1 and V-2, V-10 and V-11, and V-4 and V-5. Lanes labeled with an “S” represent the siblings who do not show in the pedigree but were included in the genotyping. Two affected individuals genotyped are VI-4 and VI-24. All affected individuals are homozygous for marker D1S1160. Similar results were obtained with other linked STRPs (data not shown).
Table 2.
Pairwise Linkage Data between HUS and Markers on Chromosome 1q32
|
LOD
Score at θ
= |
|||||||||
| Marker | .000 | .025 | .050 | .100 | .200 | .300 | .400 | θ | Zmax |
| D1S408 | −∞ | −.161 | .125 | .329 | .343 | .224 | .100 | .150 | .368 |
| D1S2794 | .752 | .716 | .680 | .606 | .457 | .306 | .155 | .000 | .752 |
| D1S2840 | 1.504 | 1.419 | 1.330 | 1.142 | .753 | .401 | .144 | .000 | 1.504 |
| GATA135F02 | 2.263 | 2.497 | 2.305 | 1.933 | 1.233 | .634 | .221 | .000 | 2.693 |
| D1S2660 | 3.036 | 2.806 | 2.581 | 2.146 | 1.336 | .656 | .215 | .000 | 3.036 |
| D1S1175 | 3.383 | 3.152 | 2.925 | 2.483 | 1.648 | .907 | .346 | .000 | 3.383 |
| F13B | 1.564 | 1.472 | 1.379 | 1.194 | .831 | .500 | .220 | .000 | 1.564 |
| D1S1726 | .752 | .716 | .680 | .606 | .457 | .306 | .155 | .000 | .752 |
| D1S1647 | −∞ | −1.774 | −1.223 | −.733 | −.339 | −.170 | −.068 | .500 | .500 |
Mutation Analysis
After linkage of HUS was determined in the Bedouin kindred, the candidate gene, CFH, was screened for mutations, with use of the primers listed in table 1. The entire 20-exon coding region was analyzed for mutations by use of both SSCP and sequencing. SSCP analysis detected a bandshift in exon 20. A single C→T transition, resulting in a Ser→Lys (S1191L) change, was identified at codon 3645 (fig. 3A and B). The numbers used for nucleotides and amino acids were based on a previously published sequence (Ripoche et al. 1988). This transition was found to be a homozygous change in the available affected individuals and a heterozygous change in obligate carriers. That this variant was not found in 96 control individuals suggests that this is a disease-causing mutation. No other sequence variants were identified, by DNA sequencing, in the complete coding region of the gene in members of this family.
Figure 3.
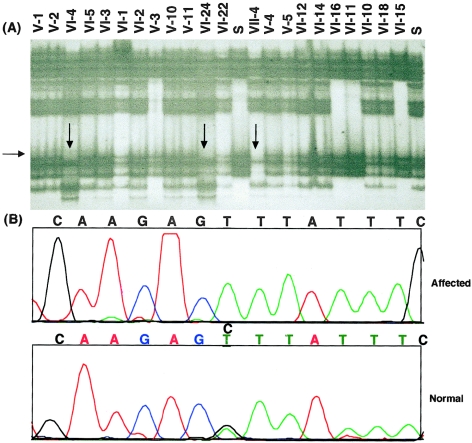
A, SSCP of exon 20 in family members with HUS. DNA samples were screened for mutations, by use of SSCP with primers from the CFH coding region. The ID numbers for each lane are shown. A CVS was included in lane 16 (VII-4). Note the shifted bands representing a mutation in the CFH gene. B, Partial chromatograms for the exon 20 codon sequence for affected individuals (VI-4) and for unaffected individuals (VI-15). Note the mutation in affected individuals (C→T change).
Serum CFH Analysis
A 155-kD band, which corresponded to the correct size of the CFH protein, was detected by western blotting in three affected individuals (VII-2, VI-4, and VI-24), for whom serum was available, and in three unaffected individuals (VI-19, V-7, and VII-13). However, the detectable signal was much weaker in affected individuals, when compared with that observed in control subjects at multiple serum dilutions (1:200–1:50) (fig. 4). Complement factors I and C3 were also tested. Normal factor-I levels were demonstrated (data not shown). A correlation between C3 and CFH levels was observed in the asymptomatic siblings and in the parent (r=0.67, P<.05) (fig. 5).
Figure 4.
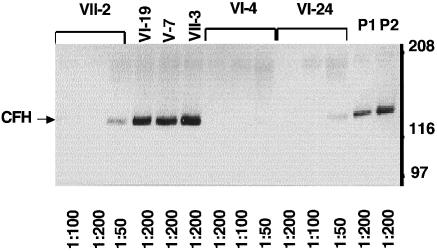
Western blot analysis of serum CFH from affected individuals VII-2 (lanes 1–3), VI-4 (lanes 7–9), and VI-24 (lanes 10–12); from unaffected parents VI-19 (lane 4) and V-7 (lane 5); and from sibling VII-3 (lane 6). P1 and P2 are the pools of unaffected individuals corresponding to VII-2 and VI-4, respectively. The blot was probed with antisera specific for CFH. Size markers are shown. The serum dilutions are indicated below each lane.
Figure 5.
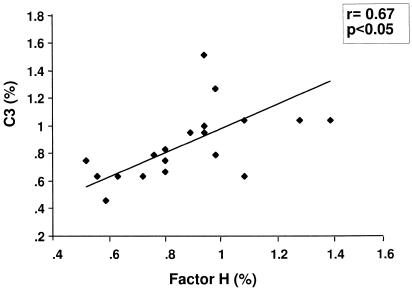
The correlation between serum CFH and C3. Immunodiffusion assay of serum from peripheral blood of unaffected parents and siblings. CFH and C3 were measured with the use of monospecific goat antibodies to human C complement. The protein counts were expressed as a percentage of pooled normal sera. Correlation: r=0.67, P<.05.
CFH mRNA Analysis
To determine whether CFH deficiency is the result of abnormal levels of mRNA, RNA from fibroblasts was analyzed on northern blots by use of a CFH cDNA probe. No difference in CFH mRNA levels was detected between the control individuals and the affected individuals (VII-2 and VI-4), with both basal and stimulated conditions (Fig. 6). This result indicates that the S1191L variant does not affect CFH mRNA levels.
Figure 6.
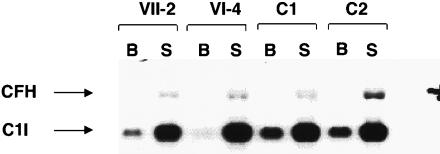
CFH mRNA: northern blot analysis of fibroblasts from affected individuals VII-2 (lanes 1 and 2), VI-4 (lanes 3 and 4) and from unaffected control individuals C1 (lanes 5 and 6) and C2 (lanes 7 and 8), in both the basal state (B) and the stimulated state (S). The blot was probe with a CFH cDNA probe. An internal control probe (C1I [complement 1 inhibitor]) was included. Size markers are not shown.
CFH and C3 Biosynthesis and Secretion Analysis
We next conducted pulse-chase experiments in which intracellular and extracellular CFH and C3 protein levels were measured. Fibroblasts both from patients (VII-2 and VI-4) and from control subjects were grown in culture, and immunoprecipitated proteins were detected from fibroblast lysates and culture media, by use of antibodies specific to CFH and C3. Gel slices containing the proteins were quantified. Intracellular CFH was properly synthesized in fibroblasts, both in affected individuals and in controls. However, at the 2-h time point, the extracellular CFH levels were significantly reduced in patients compared with controls. The low extracellular CFH levels were further evident after 8 h (fig. 7A). In contrast to CFH, C3 levels in the medium were not significantly different between affected individuals and control individuals (fig. 7B). The results indicate that both CFH and C3 can be synthesized normally in the affected fibroblasts; however, CFH is not secreted normally by fibroblasts from affected individuals. In contrast, C3 secretion is normal.
Figure 7.
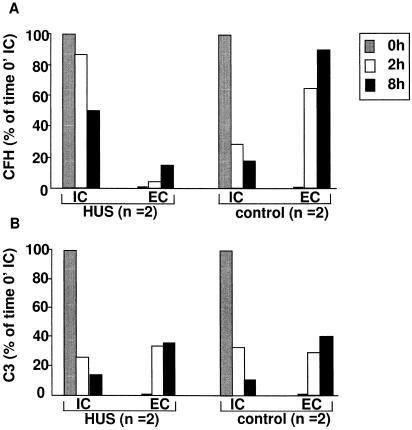
CFH and C3 biosynthesis and secretion. Pulse-chase experiments of CFH (A) and C3 (B) from skin fibroblasts from two patients (denoted by “HUS”) (VI-4 and VII-2) and from two healthy controls (denoted by “control”). Intracellular biosynthesis or extracellular secretion rate at 0 h is denoted by gray bars; at 2 h, by unblackened bars; and at 8 h, by blackened bars. IC = inside fibroblasts; EC = in medium.
Discussion
In the present study, linkage between the CFH gene and autosomal recessive atypical HUS was demonstrated in a large Bedouin kindred. A single missense mutation (S1191L) was found in the CFH gene and was shown to segregate with the disease. Northern blot analysis indicates that affected individuals produce normal levels of mRNA. However, low levels of serum CFH are present in affected individuals. Immunoprecipitation studies indicate that the CFH deficiency is the result of abnormal secretion of the mutant protein. Together, these findings provide strong molecular evidence of CFH involvement in autosomal recessive familial atypical HUS.
Recently, Warwicker et al. (1998) reported two heterozygous CFH mutations (R1197G and a 4-bp exon-1 deletion) in an autosomal dominant familial HUS pedigree and in an isolated HUS proband, respectively. Low serum C3 and CFH levels were not evident in affected individuals (Warwicker et al. 1998). In the present study, a different point mutation (S1191L) was found in the homozygous state, resulting in low serum CFH in all affected individuals. Obligate heterozygotes for the S1191L mutation appear to be clinically normal. These findings suggest a different disease mechanism for autosomal recessive and autosomal dominant HUS. The autosomal recessive disease seen in this study appears to result from a deficiency in serum CFH caused by abnormal secretion of the mutant protein, whereas the dominant disease may result from the presence of an abnormal CFH protein secreted into the plasma.
CFH, a serum glycoprotein with a molecular weight of 155 kD, is synthesized abundantly in the liver. It is also synthesized in endothelium, macrophages, platelets, leukocytes, and several other cell types (Sim et al. 1993). CFH is a down-regulating protein of the alternative pathway in the complement system. CFH controls the alternative pathway in the complement system by binding C3b (C3 convertase), thereby preventing the formation of the C3 convertase complex and accelerating the dissociation of Bb from the active C3 convertase (Weiler et al. 1976). CFH is also a cofactor for factor I (C3b inactivator) (Pangburn et al. 1977) and can distinguish between activator and nonactivator surfaces (Meri et al. 1990).
CFH is a 1,301-amino-acid peptide coded for a gene on chromosome 1q32 (Ripoche et al. 1988). It contains 20 short consensus repeats (SCRs) and is homologous to C4-binding protein (Skerka et al. 1995). The SCR domains are thought to be small independent-folding units, and each represents an exon in the CFH gene. It is speculated that a mutation in CFH may prevent proper SCR folding, resulting in abnormal CFH transport. CFH has at least three C3b-binding domains (Sharma and Pangburn 1996); one of the domains resides within the SCR 16–20. The low C3 level and its correlation with CFH deficiency in our study can be explained as secondary to increased consumption of C3 rather than as a primary C3 deficiency.
Mutations in CFH were previously reported to affect protein secretion. Ault and coworkers (1997) demonstrated two mutations in SCR9 and SCR16 that caused abnormal CFH secretion and intracellular catabolism in chronic hypocomplementemic renal disease, with CFH deficiency. Both mutations affect conserved cysteine residues characteristic of SCR modules. In the present study, we described a point mutation resulting in a Ser→Lys (S1191L) change. Serine is an amino acid with an uncharged polar side chain, and its substitution by a charged lysine residue may affect proper SCR folding and C3b binding, thereby resulting in abnormal transportation and secretion.
No therapy, including numerous transfusions of fresh frozen plasma or plasmapheresis, proved to be effective in our patients. The lack of response to plasma therapy could be the result of either insufficient amounts of CFH in the plasma used for transfusions or the presence of other components that exacerbate the relapses. Isolated CFH replacement or a more specific C-activation inhibition, such as the circulating C receptor, may serve as alternatives to whole plasma. Enhancement of CFH secretion could also be considered as possible treatment.
Acknowledgments
We thank the family members for their cooperation in this study. We thank Robin Hockey for technical assistance.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Cooperative Human Linkage Center, http://lpg.nci.nih.gov/CHLC/ (for STRPs)
- Généthon, http://www.genethon.fr/ (for STRPs)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for autosomal dominant HUS [MIM 134370] and recessive HUS [MIM 235400])
References
- Ault BH, Schmidt BZ, Fowler NL, Kashtan CE, Ahmed AE, Vogt BA, Colten HR (1997) Human factor H deficiency: mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem 272:25168–25175 [DOI] [PubMed]
- Barre P, Kaplan BS, de Chadarevian J-P, Drummond KN (1977) Hemolytic uremic syndrome with hypocomplementemia, serum C3NeF, and glomerular deposits of C3. Arch Pathol Lab Med 101:357–361 [PubMed]
- Bassam BJ, Caetano-Anolles G, Gresshoff PM (1991) Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal Biochem 196:80–83 [DOI] [PubMed]
- Bogdanovic R, Cvoric A, Nikolic V, Sindjic M (1988) Recurrent hemolytic-uraemic syndrome with hypocomplementaemia: a case report. Pediatr Nephrol 2:236–238 [DOI] [PubMed]
- Bogdanovic R, Stankovic I, Jojic N, Ognjanovic M, Zlatkovic M, Popovic O, Nikolic V (1997) Recurrent hemolytic uremic syndrome with hypocomplementemia and intestinal lymphangiectasia. Nephron 76(4):481–484 [DOI] [PubMed]
- Chirgwin JM, Przbyla AE, McDonald RJ, Rutter WJ (1979) Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18:5294–5299 [DOI] [PubMed]
- Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, et al (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380:152–154 [DOI] [PubMed]
- Farr MJ, Roberts S, Morley AR, Dewar DF, Uldall PR (1975) The hemolytic-uremic syndrome: a family study. Q J Med 44:161–188 [PubMed]
- Fijen CA, Kuijper EJ, Te Bulte M, van de Heuvel MM, Holdrinet AC, Sim RB, Daha MR, et al (1996) Heterozygous and homozygous factor H deficiency states in a Dutch family. Clin Exp Immunol 105:511–516 [DOI] [PMC free article] [PubMed]
- Gonzalo A, Mampaso F, Gallego N, Bellas C, Segui J, Ortuno J (1981) Hemolytic uremic syndrome with hypocomplementemia and deposits of IgM and C3 in the involved renal tissue. Clin Nephrol 16:193–199 [PubMed]
- Jansen JH, Hogasen K, Grondahl AM (1995) Porcine membranoproliferative glomerulonephritis type II: an autosomal recessive deficiency of factor H. Vet Rec 137:240–244 [DOI] [PubMed]
- Kaplan BS (1977) Hemolytic uremic syndrome with recurrent episodes: an important subset. Clin Nephrol 8:495–498 [PubMed]
- Kaplan BS, Chesney RW, Drummond KN (1975) Hemolytic uremic syndrome in families. N Engl J Med 292:1090–1093 [DOI] [PubMed]
- Kaplan BS, Proesmans W (l987) The hemolytic uremic syndrome of childhood and its variants. Semin Hematol 24:148–160 [PubMed]
- Lange K, Weeks D, Boehnke M (1988) Programs for pedigree analysis: MENDEL, FISHER, and dGENE. Genet Epidemiol 5:471–472 [DOI] [PubMed]
- Mattoo TJ, Mahmood MA, Al-Harbi MS, Mikail I (1989) Familial, recurrent hemolytic-uremic syndrome. J Pediatr 114:814–816 [DOI] [PubMed]
- Meri S, Pangburn MK (1990) Discrimination between activators and nonactivators of the alternative pathway: regulation via a sialic acid/polyanion binding site on factor H. Proc Nat Acad Sci USA 87:3982–3986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohali M, Shalev H, Schlesinger M, Katz Y, Kachko L, Carmi R, Sofer S, et al (1998) Hypocomplementemic autosomal recessive hemolytic uremic syndrome associated with decreased factor H. Pediatric Nephrology 12:619–624 [DOI] [PubMed]
- Pangburn MK, Schreiber RD, Muller-Eberhard HJ (1977) Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J Exper Med 146:257–270 [DOI] [PMC free article] [PubMed]
- Pichette V, Querin S, Schurch W, Brun G, Lehner-Netsch G, Delage JM (1994) Familial hemolytic-uremic syndrome and homozygous factor H deficiency. Am J Kidney Dis 24:936–941 [DOI] [PubMed]
- Ripoche J, Day AJ, Harris TJ, Sim RB (1988) The complete amino acid sequence of human complement factor H. Biochem J 249:593–602 [DOI] [PMC free article] [PubMed]
- Segal N, Sinai L (1995) Familial hemolytic uremic syndrome. Harefuah 128:677–680 [PubMed]
- Sharma AK, Pangburn MK (1996) Identification of three physically and functionally distinct binding sites for C3b in human complement factor H by deletion mutagenesis. Proc Natl Acad Sci USA 93:10996–11001 [DOI] [PMC free article] [PubMed]
- Sheffield VC, Weber JL, Buetow KH, Murry JC, Even DA, Wiles K, Gastier JM, et al (1995) A collection of tri- and tetranucleotide repeat markers used to generate high quality, high resolution human genome-wide linkage maps. Hum Mol Genet 4:1837–1844 [DOI] [PubMed]
- Sim RB, Kolble K, McAleer MA, Dominguez O, Dee VM (1993) Genetics and deficiencies of the soluble regulatory proteins of the complement system. Int Rev Immunol 10:65–86 [DOI] [PubMed]
- Skerka C, Moulds JM, Taillon-Miller P, Hourcade D, Zipfel PF (1995) The human factor H-related gene 2 (FHR2): structure and linkage to the coagulation factor XIIIb gene. Immunogenetics 42:268–274 [DOI] [PubMed]
- Utah Marker Development Group (1995) A collection of ordered tetranucleotide-repeat markers from the human genome. Am J Hum Genet 57:619–628 [PMC free article] [PubMed]
- Vicra GD, Northemann W, Shiels BR, Widera G, Broome S (1990) Simplified northern blot hybridization using 5% sodium dodecyl sulfate. Biotechniques 8:370–371 [PubMed]
- Vogt VA, Wyatt RJ, Burke BA, Simonton SC, Kashtan CE (1995) Inherited factor H deficiency and collagen type III glomerulopathy. Pediatr Nephrol 9:11–15 [DOI] [PubMed]
- Warwicker P, Goodship THJ, Donne RL, Pirson Y, Nicholls A, Ward RM, Turnpenny P, et al (1998) Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int 53:836–844 [DOI] [PubMed]
- Weiler JM, Daha MR, Austen KF, Fearon DT (1976) Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci USA 73:3268–3272 [DOI] [PMC free article] [PubMed]
- West CD, McAdams AJ (1998) Glomerular deposits and hypoalbuminemia in acute post-streptococcal glomerulonephritis. Pediatr Nephrol 12:471–474 [DOI] [PubMed]