

Summary
The t(11;22) is the only known recurrent, non-Robertsonian constitutional translocation. We have analyzed t(11;22) balanced-translocation carriers from multiple unrelated families by FISH, to localize the t(11;22) breakpoints on both chromosome 11 and chromosome 22. In 23 unrelated balanced-translocation carriers, the breakpoint was localized within a 400-kb interval between D22S788 (N41) and ZNF74, on 22q11. Also, 13 of these 23 carriers were tested with probes from chromosome 11, and, in each, the breakpoint was localized between D11S1340 and APOA1, on 11q23, to a region ⩽185 kb. Thus, the breakpoints on both chromosome 11 and chromosome 22 are clustered in multiple unrelated families. Supernumerary-der(22)t(11;22) syndrome can occur in the progeny of balanced-t(11;22) carriers, because of malsegregation of the der(22). There has been speculation regarding the mechanism by which the malsegregation occurs. To elucidate this mechanism, we have analyzed 16 of the t(11;22) families, using short tandem-repeat–polymorphism markers on both chromosome 11 and chromosome 22. In all informative cases the proband received two of three alleles, for markers above the breakpoint on chromosome 22 and below the breakpoint on chromosome 11, from the t(11;22)-carrier parent. These data strongly suggest that 3:1 meiosis I malsegregation in the t(11;22) balanced-translocation–carrier parent is the mechanism in all 16 families. Taken together, these results establish that the majority of t(11;22) translocations occur within the same genomic intervals and that the majority of supernumerary-der(22) offspring result from a 3:1 meiosis I malsegregation in the balanced-translocation carrier.
Introduction
The t(11;22)(q23;q11.2) is the only known recurrent, non-Robertsonian, constitutional translocation in humans (Fraccaro et al. 1980; Zackai and Emanuel 1980). Carriers of the balanced constitutional t(11;22) are phenotypically normal but are at risk of having progeny with the supernumerary-der(22)t(11;22) syndrome, as a result of malsegregation of the der(22). The affected progeny are genotypically unbalanced because they carry the der(22) as a supernumerary chromosome—either 47,XX,+der(22)t(11;22) or 47,XY,+der(22)t(11;22). Individuals with the +der(22) syndrome have a distinct phenotype, which consists of severe mental retardation, preauricular tag or sinus, ear anomalies, cleft or high-arched palate, micrognathia, microcephaly, kidney abnormalities, heart defects, and genital abnormalities in males (Zackai and Emanuel 1980; Lin et al. 1986).
Although it is generally stated that the mechanism that leads to the unbalanced karyotype seen in the supernumerary-der(22) syndrome is a 3:1 meiosis I malsegregation in the carrier parent (Lindenbaum and Bobrow 1975), there have been several reported cases that suggest alternative mechanisms. In these cases the malsegregation appears to be the result of either meiosis II or postzygotic nondisjunction of the der(22) (Lockwood et al. 1989; Abeliovich and Carmi 1990; Lurie and Podleschuk 1992; Simi et al. 1992). Furthermore, Dawson et al. (1996) reported a patient with +der(22) resulting from rescue of an adjacent-1 segregation of a de novo paternal translocation by complementation with a maternal gamete disomic for chromosome 22. Despite this single observation, rescue by complementation has not been demonstrated as a mechanism for malsegregation in other t(11;22) families. Segregation analysis of a small number of t(11;22) families, using chromosome 22 markers, has demonstrated that the proband with an unbalanced +der(22) had inherited one normal chromosome 22 from each parent, as well as the der(22) from the translocation-carrier parent (Funke et al. 1999). This result was suggestive of a 3:1 meiosis I malsegregation in the translocation-carrier parent. However, these results could also be explained by a mechanism in which a 2:2 adjacent-2 malsegregant gamete containing the normal chromosome 22 and the der(22) and missing a chromosome 11 was rescued by complementation with a gamete disomic for chromosome 11 from the noncarrier parent. This would similar to the case reported by Dawson et al. (1996). Therefore, although previous studies have been consistent with a 3:1 meiosis I malsegregation, they have not definitively ruled out other possibilities.
Although chromosome 22 only represents ∼2% of the haploid human genome (Morton 1991), many malignant diseases and developmental abnormalities are associated with recurrent rearrangements of this chromosome (reviewed in Kaplan et al. 1987). These include tumor-associated rearrangements, such as the t(11;22) of Ewing sarcoma (ES) and peripheral neuroepithelioma (NE) (ES/NE [MIM 133450]) (Aurias et al. 1984; Turc-Carel et al. 1984), as well as constitutional rearrangements of 22q, such as the t(11;22) associated with the supernumerary-der(22) syndrome, the microdeletion of 22q11, which is associated with DiGeorge syndrome (DGS [MIM 188400]) and with velocardiofacial syndrome (VCFS [MIM 192430]) (DGS/VCFS) (reviewed in Budarf and Emanuel 1997), and the supernumerary bisatellited marker chromosome of cat-eye syndrome (CES [MIM 115470]) (McDermid et al. 1986). Previous data, from us and others, on a limited number of t(11;22) families suggested the clustering of constitutional t(11;22) breakpoints on 22q11.2, between markers D22S788 (N41) and ZNF74 (Shaikh et al. 1997; Funke et al. 1999). Within 22q11, this breakpoint interval is distinct from the breakpoints of the standard 3-Mb deletion associated with DGS/VCFS (Emanuel et al. 1999) and from the CES-associated rearrangement (Mears et al. 1994; McTaggart et al. 1998). The breakpoint on chromosome 11 has not been precisely mapped but, in a single t(11;22) carrier, has been located distal to D11S144 and proximal to marker APOA1, in 11q23.2 (Budarf et al. 1989; Tunacliffe et al. 1993). This region is distal to the region, in 11q22-11q23, that is involved in multiple tumor-associated rearrangements of chromosome 11 (Arai et al. 1996) but is proximal to both the ES/NE-associated t(11;22) breakpoint and the Jacobsen syndrome (JS [MIM 147791]) breakpoints in 11q23.3-11q24.2 (Tunacliffe et al. 1999).
We have analyzed 23 t(11;22) families, using FISH with probes from chromosome 22. We have also analyzed 13 of these 23 t(11;22) families, using FISH with probes from chromosome 11. We find that, in all families tested, the breakpoints cluster within the same small intervals on both chromosome 11 and chromosome 22. Furthermore, we have performed genotype analysis of 16 of these t(11;22) families, with short tandem-repeat–polymorphism markers from both chromosome 11 and chromosome 22. We have analyzed chromosome 11 markers in addition to the chromosome 22 markers to rule out alternative mechanisms. Our results definitively demonstrate 3:1 meiosis I malsegregation in all cases. Taken together, these results establish that the majority of supernumerary-der(22) offspring result from 3:1 meiosis I malsegregation of a translocation that recurs at a similar genomic location on chromosome 11 and chromosome 22.
Subjects, Material, and Methods
Study Population
The samples used in this study were derived from patients participating in a research study of the t(11;22). Families segregating the t(11;22) were recruited through our Clinical Genetics Center at the Children's Hospital of Philadelphia (CHOP). The families were of varied ethnic and racial backgrounds and several countries of origin. The center obtained both informed consent and detailed multigenerational pedigrees by telephone interview, and the biological specimens from translocation carriers and their offspring with the supernumerary-der(22)t(11;22) syndrome were shipped to CHOP. Cytogenetic analysis was performed to confirm the karyotpes of study subjects, and, when applicable, specimens from additional family members also were obtained.
DNA Isolation
Genomic DNA from t(11;22) families was prepared directly from 3 ml of whole blood, by use of the Puregene™ DNA-isolation kit (Gentra Systems). Genomic DNA from somatic-cell hybrids and cell lines was isolated by use of the 341 Nucleic Acid Purification system (Applied Biosystems), an automated DNA extractor. Cosmid and bacterial artificial chromosome (BAC) DNA was isolated by use of Qiagen DNA-isolation kits, and YAC DNA was prepared as described elsewhere (Philippsen et al. 1991).
FISH Analysis
Metaphase spreads were prepared either from peripheral blood lymphocytes or from lymphoblastoid cell lines, by use of standard methodology. Interphase nuclei were prepared according to the method described by Trask et al. (1991). FISH was performed as described elsewhere (Holmes et al. 1997). Chromosomes were visualized by counterstaining with DAPI. Probes used for FISH were labeled by nick translation, with either biotin-16 dUTP or digoxygenin-11 dUTP, as described by Lichter et al. (1988), with minor modifications. These probes were then detected by either fluorescein-conjugated avidin or rhodamine-conjugated anti-digoxygenin respectively. The chromosome 22 FISH probes, c68a1 and c87f9, were cosmids isolated from the LL22NCO3 cosmid library. The chromosome 22 BAC, BAC 48m11, used for interphase FISH is from the CITB human BAC library (Research Genetics). The chromosome 22 PAC, PAC 181g22, used for metaphase FISH is from the RPCI13 human PAC library (Roswell Park Cancer Institute). The chromosome 11 probe (BAC 442e11 [designated as “BAC b1030” in GenBank; accession number AC007707]) used for FISH is from the RPCI11 (segment 2) human BAC library (Roswell Park Cancer Institute).
Genotype Analysis
For segregation analysis, 100 ng of genomic DNA from each individual studied was separately amplified by PCR with eight highly polymorphic genetic markers. Five of these markers were from chromosome 22 and include 46STS and 115STS (Driscoll et al. 1997), D22S941 and D22S944 (Morrow et al. 1995), and D22S264 (Marineau et al. 1992). The other three markers were from chromosome 11 and include D11S614 (Tunacliffe et al. 1993) and D11S1317 and D11S1356 (Quackenbush et al. 1995). For each marker, one of the two oligonucleotide primers was fluorescently labeled with either TET, HEX, or FAM (Applied Biosystems). PCR conditions for all markers except 115STS and D11S1356 were as follows: PCR was performed in a 25-μl PCR reaction containing 1 × PCR buffer composed of 1.5 mM MgCl2 (Boehringer-Mannheim), 1 μM each primer, 200 μM of each dNTP, and 3 units of Taq polymerase (Boehringer-Mannheim). PCR was performed with 5-min denaturation at 95°C, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 1 min. Marker 115STS required 2.0 mM MgCl2, and D11S1356 required 1.0 mM MgCl2. PCR products from all eight markers for each individual were diluted, multiplexed, and loaded into a single lane of an ABI Prism 377 DNA sequencer. The DNA fragments were sized and data were analyzed by use of ABI Prism GeneScan Analysis software.
Library Screening
A 6×coverage BAC library from Roswell Park Cancer Institute (RPCI11 segment 2) was screened with radiolabeled probe, as follows. The BAC filters were prehybridized and then hybridized in Church's buffer (0.5 M Na2PO4, pH 7.3, 7% SDS, 1 mM EDTA, pH 8), at 65°C for 4–5 h and 16–24 h, respectively. Prior to being added to the hybridization buffer, the probe was preannealed to sheared human placental DNA (250 mg/ml), at 65°C for 30 min. Final washes were performed in 0.1 × SSC, 0.1% SDS, at 65°C (twice, for 20 min each). Results were visualized by autoradiography, on exposure of Kodak X-OMAT AR film to the filters, at −70°C.
Probe Generation and PCR
A. Probe for ZNF74
The probe for ZNF74 was synthesized by PCR with primers derived from the sequence of ZNF74 gene–containing clone PAC 52f6 (GenBank accession number AC005500). The DNA template used was cosmid c87f9, and the primers were as follows: znf.for (5′-AGGGCCAATTCCTTGCTGAG-3′) and znf.rev (5′-CAGATGTTGCCCGAGGTGTG-3′). The PCR product was 414 bp and corresponded to bases 145159–145572 of PAC 52f6.
B. Probe for telomeric end of PAC 201m18
The probe for the telomeric end of PAC 201m18 was synthesized by PCR with primers derived from the sequence of PAC 201m18 (GenBank accession number AC000097). The DNA template used was PAC 201m18, and the primers were as follows: M18.for (5′-TGATGGATCCGTCATTACCAG-3′) and M18.rev (5′-CCTCACGTAACTGTAAACCAG-3′). The PCR product was 360 bp and corresponded to bases 161791–162150 of PAC 201m18.
C. Probe for APOAI gene
The probe for the APOAI gene was synthesized by PCR with primers derived from the sequence of the APOA1 cDNA (GenBank accession number J00098). The DNA template used was YAC 785e12 (Arai et al. 1996), and the primers were as follows: Apo.for (5′-TGGTCTGGATGGAGAAACCG-3′) and Apo.rev (5′-AGGCACAGAGAGGAGCTAAA-3′). The Apo PCR product was 303 bp and corresponded to bases 1204–1506 of the APOA1 cDNA. PCR conditions were the same as those given above. The annealing temperature was 55°C for the probes for the ZNF74 gene and for the telomeric end of PAC 201m18. For the APOA1 gene probe, the annealing temperature was 57°C. The D11S1340 primers and PCR conditions were as described (Gyapay et al. 1994).
Results
Clustering of the t(11;22) Breakpoint on Chromosome 22
To determine the location of the translocation on chromosome 22, a total of 23 carriers of the balanced constitutional t(11;22) were analyzed by FISH. Each of the carriers was from a different family. The probes used for FISH include (a) c87f9, a cosmid specific for ZNF74, (b) c68a1, a cosmid specific for D22S788 (N41), and (c) a control cosmid probe that maps to the distal long arm of chromosome 22 (D22S39). The distal cosmid was used, in each experiment, to mark the telomeric end of chromosome 22 (22q13.3) and as a hybridization control. The locations of cosmids c87f9 and c68a1 are shown, along with a map of the chromosome 22q11 region flanking the t(11;22) breakpoint, in figure 1A.
Figure 1.
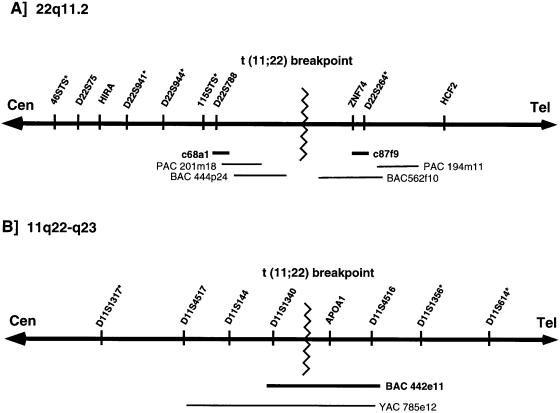
Location of t(11;22) breakpoint. Chromosomes are illustrated as thick horizontal lines with arrowheads at both ends. The orientation of the centromere (Cen) and telomere (Tel) are also indicated. The t(11;22) breakpoint is denoted by a zigzag line. STS markers used for genotype analysis are indicated by an asterisk (*). A, 22q11.2. Clones used to construct a map in the t(11;22) breakpoint region are indicated by thin lines. The location of cosmids used as FISH probes—c68a1 and c87f9—are shown as thick lines. B, 11q22-q23. FISH probe BAC 442e11 is shown as a thick line.
A typical result of FISH with c68a1 and the chromosome 22 control probe on a metaphase spread from a t(11;22) carrier is shown in figure 2A. In all 23 carriers tested, the FISH probe c68a1 always localized to the normal chromosome 22 and the der(22), indicating that c68a1—and, therefore, marker D22S788—is proximal to the t(11;22) breakpoint on 22q11. A typical result of FISH with c87f9 and the chromosome 22 control probe on a single metaphase spread is shown in figure 2B. In all 23 carriers tested, c87f9 always localized to the normal chromosome 22 and the (der)11, indicating that c87f9—and, therefore, marker ZNF74—is distal to the t(11;22) breakpoint on 22q11.
Figure 2.
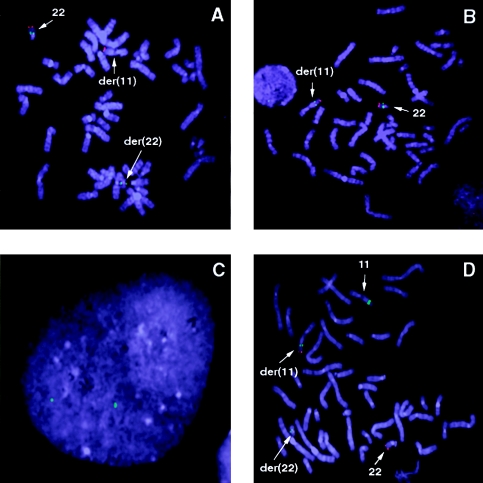
FISH analysis I. A, B, and D, Metaphase chromosomes derived from a balanced-t(11;22) carrier hybridized to different test probes—c68a1 (A), c87f9 (B), and BAC 442e11 (D). In each case, the control probe that marks the telomeric end of chromosome 22 is seen as a red signal (rhodamine), and test probes are seen as a green signal (fluorescein). Chromosomes with fluorescent signal are indicated by arrows and are labeled. C, Interphase nucleus hybridized to FISH probe BAC 442e11. Two discrete signals indicate that the clone BAC 442e11 does not contain chromosome 11–specific duplications.
In a previous study (Shaikh et al. 1997), the distance between markers D22S788 and ZNF74 had been estimated to be ⩽400 kb, on the basis of the results of FISH with c68a1 and c87f9 on interphase nuclei. Thus, the map of 22q11 in the vicinity of the t(11;22) breakpoint was extended in both directions from D22S788 and ZNF74 (fig. 1A). A 6×coverage BAC library was separately screened with a probe for the telomeric end of PAC 201m18 and with a probe for the ZNF74 gene. Among the multiple positive clones obtained for the end of PAC 201m18, BAC 444p24 extended farthest toward the t(11;22) breakpoint. Screening with the probe for the ZNF74 gene resulted in a single clone, BAC 562f10. It has been determined that the portions of both BAC 444p24 and BAC 562f10 that are closest to the t(11;22) breakpoint contain elements that are duplicated in multiple locations on 22q11 (Emanuel et al. 1998; Edelmann et al. 1999; Funke et al. 1999; Saitta et al. 1999). The presence of duplicated sequence elements in this region has made it difficult to close the gap between BAC 444p24 and BAC 562f10.
Previous attempts by us and others (Morrow et al. 1995) had failed to obtain YAC clones spanning the gap between D22S788 and ZNF74, suggesting that this region may be unstable in YACs. Therefore, we attempted to close the gap, using BAC and PAC clones. We isolated several BAC and PAC clones that appeared to close the gap between BACs 444p24 and 562f10, using, as hybridization probes, end sequences from both clones. Metaphase FISH on t(11;22) carriers with each of these clones initially suggested that they all spanned the chromosome 22 t(11;22) breakpoint, since a signal was detected on the normal chromosome 22, the der(22), and the der(11). A typical result of metaphase FISH with one such clone, PAC 181g22, is shown in figure 3A and B. However, interphase FISH with each of these clones demonstrates that they contain what appear to be chromosome-specific duplicated sequences. A typical result of interphase FISH with one such clone, BAC 48m11, is shown in figure 3C. Multiple signals are observed in interphase nuclei, suggesting that the human genomic DNA contained in BAC 48m11 is duplicated in multiple locations on chromosome 22. This would explain the misleading result of metaphase FISH, since there are copies of the duplicated sequences both above and below the t(11;22) breakpoint (Emanuel et al. 1998, and in press; Edelmann et al. 1999; Saitta et al. 1999). All of the clones that appeared to close the gap between BACs 444p24 and 562f10, including PAC 181g22 and BAC 48m11, have since been mapped to other regions on chromosome 22, by virtue of partial unique sequences contained within their inserts (University of Oklahoma Advanced Center for Genome Technology; Edelmann et al. 1999). Also, nucleotide sequences derived from all the putative gap-spanning clones, including PAC 181g22 and BAC 48m11, share only 96%–97% nucleotide-sequence identity with the sequences from BACs 444p24 and 562f10. Truly overlapping clones should exhibit >99% sequence identity with each other, even after allowance for the possibility of allelic polymorphism and the fact that the clones are from different libraries. This suggested to us that the clones that we had isolated were not from the t(11;22) breakpoint region but from other copies of the duplication on chromosome 22.
Figure 3.
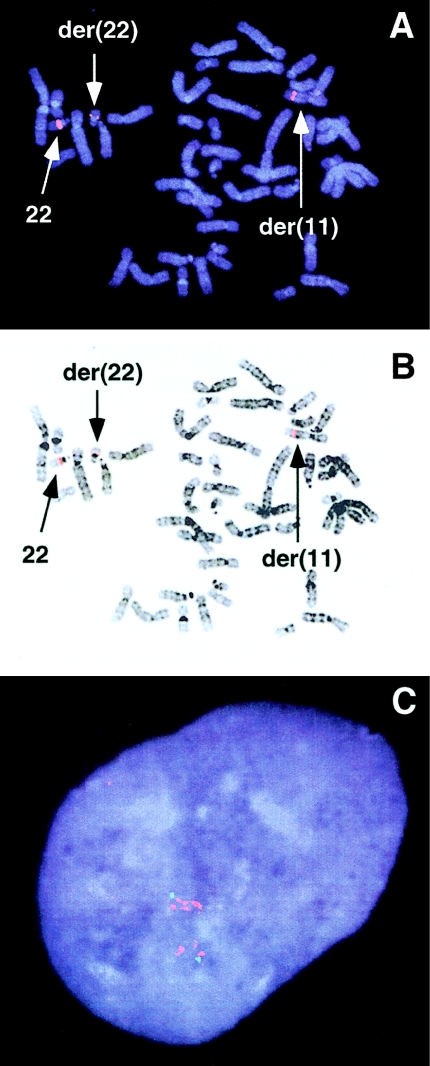
FISH analysis II. A, Metaphase chromosomes derived from a balanced-t(11;22) carrier hybridized to test probe PAC 181g22 seen as a red signal (rhodamine). B, DAPI-counterstained image in A, inverted to gray scale. Chromosomes with fluorescent signal are indicated by arrows and are labeled. C, Interphase nucleus hybridized to FISH probe BAC 48m11 seen as multiple red signals (rhodamine), indicating chromosome 22–specific duplicated sequences. The green signal (fluorescein) is from probe c16e8, a unique chromosome 22 cosmid probe containing marker D22S66 (ph 160b), which maps between markers D22S944 and 115STS (see fig. 1A) within the 22q11.2 deletion.
Thus, the region containing the t(11;22) breakpoint on chromosome 22, between BACs 444p24 and 562f10, appears to be underrepresented in the YAC, BAC, and PAC libraries screened by us and others (Funke et al. 1999). It is anticipated that the completed sequence data from all the clones in this region will help to map clones to this region more accurately in the future. Except for 87f9, all the chromosome 22 clones reported here either already have been or are currently being sequenced at University of Oklahoma Advanced Center for Genome Technology. In summary, it was determined that, in 23 unrelated balanced-t(11;22) carriers, the breakpoint on chromosome 22 clusters between markers D22S788 and ZNF74, in a region that, because of chromosome 22 duplications, has proved difficult to traverse by chromosome walking. Thus, we were unable to unambiguously identify a BAC that crosses the breakpoint on chromosome 22.
Clustering of the t(11;22) Breakpoint on Chromosome 11
Because of the difficulties in identification of clones that span the t(11;22) breakpoint on chromosome 22, we shifted our attention to the analysis of the breakpoint on chromosome 11. The t(11;22) breakpoint on chromosome 11 localizes in a region of 11q23 that has not been mapped as extensively as 22q11. Previous analysis (Budarf et al. 1989; Tunacliffe et al. 1993) of a somatic-cell hybrid containing, as its only relevant human component, the der(22) from a constitutional-t(11;22) carrier had localized the t(11;22) breakpoint on 11q23, between markers D11S144 and APOA1. To determine whether the t(11;22) breakpoints on chromosome 11 cluster similarly to those on chromosome 22, it was necessary to obtain probes that could be used for FISH. A YAC map across 11q23 has been published elsewhere (Arai et al. 1996). In this map the two markers D11S144 and APOA1, as well as a third marker, D11S1340, are present within YAC 785e12 (fig. 1B). Thus, by use of the same somatic-cell hybrid Cl 4/GB (Budarf et al. 1989) that was used to map the positions of D11S144 and APOA1, marker D11S1340 was determined to be proximal to the t(11;22) breakpoint on 11q23 (data not shown).
To more precisely map this region of 11q23 and to obtain FISH probes flanking the t(11;22) breakpoint on chromosome 11, a 6×coverage BAC library was screened with a probe for the APOA1 gene. Multiple BAC clones were obtained and were analyzed by PCR, for multiple markers on chromosome 11. Interestingly one of these BACs, BAC 442e11 (BAC b1030 in GenBank; accession number AC007707), was found, by PCR, to be positive for both D11S1340 and APOA1 (data not shown). Since somatic cell–hybrid analysis had demonstrated that these two markers flank the t(11;22) breakpoint on chromosome 11, the implication was that BAC 442e11 should span the breakpoint.
BAC 442e11 was first tested by FISH to interphase nuclei, to determine whether it contained any sequences duplicated on chromosome 11 or elsewhere in the genome. BAC 442e11 consistently appeared as two discrete signals on G0-arrested interphase nuclei (fig. 2C), suggesting that the genomic region contained within this BAC was unique and did not harbor duplicated sequences. Multiple markers present within BAC 442e11 were PCR amplified from the BAC, multiple overlapping clones, and human genomic DNA. The PCR products were sequenced and showed 100% sequence identity. Sequence heterogeneity between PCR products amplified either from different clones or from human genomic DNA would suggest the presence of duplicated sequences within BAC 442e11. Therefore, the absence of heterogeneity indicates that there is no evidence for duplicated sequences within the BAC 442e11. BAC 442e11 was then used as a FISH probe on 13 of the 23 t(11;22) carriers who had been tested for chromosome 22 breakpoint clustering. A typical result of FISH with BAC 442e11 and the chromosome 22 control probe on a metaphase spread of one of the t(11;22) carriers is shown in figure 2D. In all 13 carriers, a signal from BAC 442e11 was detected on the normal chromosome 11, the der(11), and the der(22) (fig. 2D), suggesting that BAC 442e11 spans the t(11;22) breakpoint on chromosome 11 in multiple t(11;22) carriers. The genomic-DNA insert in BAC 442e11 was determined to be ∼185 kb. This further indicates that multiple t(11;22) chromosome 11 breakpoints cluster within a 185-kb region on 11q23.
Thus, it was determined that, in 13 unrelated balanced-t(11;22) carriers, the breakpoint on chromosome 11 clusters between markers D11S1340 and APOA1, within BAC 442e11. In contrast to the 22q11 region involved in the t(11;22) translocation, this region of 11q23 appears to be relatively unique.
The Supernumerary der(22): The Result of 3:1 MI Malsegregation
The genotypes of 16 families with the constitutional t(11;22) translocation were analyzed to determine the meiotic-malsegregation mechanism that leads to the supernumerary-der(22) syndrome. For each of the 16 families the proband and both parents were included in the analysis. Eight highly polymorphic short tandem-repeat–polymorphism markers—five from chromosome 22 and three from chromosome 11—were used for the genotype analysis. Chromosome 22 markers 46STS, D22S941, D22S944, and 115STS are proximal, and D22S264 is distal, to the t(11;22) breakpoint on 22q11 (fig. 1A). Chromosome 11 marker D11S1317 is proximal, and markers D11S614 and D11S1356 are distal, to the t(11;22) breakpoint (fig. 1B).
In each of the 16 families tested the proband was deduced to be trisomic for the chromosome 22 markers proximal to the t(11;22) breakpoint as a result of 3:1 meiosis I malsegregation (fig. 1A and table 1). The criterion used for demonstration of this mode of inheritance was the presence of three different alleles, two of which are derived from the carrier parent. In all cases, at least one of the four markers proximal to the breakpoint on chromosome 22 was informative. Each of the 16 probands received two of their three alleles from the translocation-carrier parent and one from the noncarrier parent, for 46STS, D22S941, D22S944, and 115STS. Marker D22S264, which resides within the cosmid c87f9, is distal to the t(11;22) breakpoint, on the basis of FISH studies with c87f9 (figs. 1A and 2B). Thus, D22S264 should be dizygous in the +der(22) probands, and, by virtue of biparental inheritance, it was informative for 12 of the 16 families (table 1). In each of these families the proband was heterozygous for D22S264 (table 1) and had received one allele from each parent. These results suggest that the proband had received the normal chromosome 22, as well as the der(22), from the carrier parent and had received a normal chromosome 22 from the noncarrier parent, indicative of a 3:1 meiosis I malsegregation in the carrier parent.
Table 1.
Genotypes of t(11;22) Families
|
Genotype ofa |
||||||||
| Individual | 46STS | D22S941 | D22S944 | 115STS | D22S264 | D11S1317 | D11S614 | D11S1356 |
| Family 1: | ||||||||
| Father | 1,1 | 7,8 | 4,6 | 2,2 | 1,7 | 3,7 | 1,6 | 1,3 |
| Motherb | 2,4 | 3,9 | 3,3 | 4,5 | 1,4 | 4,7 | 5,8 | 4,8 |
| Proband | 1,2,4 | 3,7,9 | 3,(3),4 | 2,4,5 | 4,7 | 7,(7) | 1,5,8 | 3,4,8 |
| Family 2: | ||||||||
| Father | 3,4 | 3,10 | 3,3 | 2,8 | 4,7 | 2,5 | 2,6 | 3,8 |
| Motherb | 1,3 | 7,9 | 2,6 | 2,2 | 4,6 | 7,8 | 2,7 | 2,9 |
| Proband | 1,3,4 | 3,7,9 | 2,3,6 | 2,(2),8 | 4,6 | 2,7 | 2,6,7 | 2,8,9 |
| Family 3: | ||||||||
| Father | 1,2 | 3,8 | 2,4 | 7,7 | 8,8 | 1,6 | 2,5 | 8,8 |
| Motherb | 1,4 | 7,7 | 3,6 | 2,6 | 8,8 | 2,9 | 5,6 | 5,7 |
| Proband | 1,(1),4 | 3,7,(7) | 3,4,6 | 2,6,7 | 8,(8) | 2,6 | 2,5,6 | 5,7,8 |
| Family 4: | ||||||||
| Father | 1,2 | 1,6 | 2,4 | 6,8 | 1,6 | 5,7 | 2,9 | 3,3 |
| Motherb | 1,1 | 7,8 | 3,3 | 2,7 | 1,8 | 4,8 | 8,9 | 7,8 |
| Proband | 1,(1),(1) | 6,7,8 | 3,(3),4 | 2,6,7 | 1,8 | 7,8 | 2,8,9 | 3,7,8 |
| Family 5: | ||||||||
| Father | 1,3 | 3,7 | 3,4 | 2,2 | 1,9 | 4,5 | 5,9 | 3,7 |
| Motherb | 2,2 | 7,8 | 2,6 | 1,8 | 7,8 | 7,7 | 1,2 | 4,7 |
| Proband | 1,2,(2) | 3,7,8 | 2,4,6 | 1,2,8 | 7,9 | 5,7 | 1,2,9 | 3,4,7 |
| Family 6: | ||||||||
| Father | 1,5 | 3,3 | 4,4 | 6,6 | 1,6 | 3,4 | 6,8 | 1,7 |
| Motherb | 1,1 | 7,10 | 4,5 | 2,4 | 1,7 | 5,7 | 3,5 | 1,8 |
| Proband | 1,(1),5 | 3,7,10 | 4,(4),5 | 2,4,6 | 6,7 | 3,5 | 3,5,8 | 1,7,8 |
| Family 7: | ||||||||
| Father | 1,1 | 5,7 | 4,4 | 2,4 | 1,2 | 2,6 | 5,5 | 2,4 |
| Motherb | 2,3 | 9,9 | 3,6 | 2,7 | 6,8 | 5,7 | 6,7 | 1,7 |
| Proband | 1,2,3 | 5,9,(9) | 3,4,6 | 2,(2),7 | 1,6 | 2,7 | 5,6,7 | 1,2,7 |
| Family 8: | ||||||||
| Father | 1,4 | 3,7 | 3,4 | 2,6 | 2,9 | 2,6 | 2,3 | 4,7 |
| Motherb | 1,2 | 8,10 | 4,5 | 2,4 | 1,7 | 5,7 | 3,5 | 1,8 |
| Proband | 1,2,4 | 7,8,10 | 3,4,5 | 2,(2),4 | 2,7 | 2,5 | 2,3,5 | 1,7,8 |
| Family 9: | ||||||||
| Father | 3,5 | 8,8 | 3,3 | 2,3 | 1,5 | 5,6 | 2,3 | 5,6 |
| Motherb | 3,4 | 4,5 | 3,4 | 2,4 | 4,5 | 4,8 | 3,7 | 1,7 |
| Proband | 3,4,5 | 4,5,8 | 3,(3),4 | 2,3,4 | 1,4 | 5,8 | 3,(3),7 | 1,5,7 |
| Family 10: | ||||||||
| Father | 2,2 | 7,12 | 4,6 | 2,2 | 1,9 | 5,7 | 3,6 | 2,7 |
| Motherb | 1,5 | 8,10 | 6,6 | 2,2 | 8,10 | 5,7 | 2,7 | 4,8 |
| Proband | 1,2,5 | 7,8,10 | 4,6,(6) | 2,(2),(2) | 9,10 | 7,(7) | 2,3,7 | 2,4,8 |
| Family 11: | ||||||||
| Father | 1,4 | 4,12 | 3,4 | 2,3 | 6,6 | 5,7 | 6,7 | 3,7 |
| Motherb | 1,2 | 6,9 | 3,4 | 2,5 | 2,8 | 2,6 | 6,8 | 3,4 |
| Proband | 1,(1),2 | 4,6,9 | 3,4,(3/4) | 2,3,5 | 6,8 | 6,7 | 6,7,8 | 3,(3),4 |
| Family 12: | ||||||||
| Father | 1,3 | 4,4 | 3,4 | 4,4 | 9,10 | 5,7 | 1,5 | 1,7 |
| Motherb | 1,1 | 1,2 | 4,6 | 2,4 | 2,10 | 5,7 | 1,6 | 5,6 |
| Proband | 1,(1),3 | 1,2,4 | 4,(4),6 | 2,4,(4) | 2,10 | 7,(7) | 1,5,6 | 5,6,7 |
| Family 13: | ||||||||
| Fatherb | 1,2 | 4,4 | 2,6 | 2,2 | 4,5 | 3,6 | 2,5 | 2,9 |
| Mother | 1,1 | 8,8 | 3,4 | 2,2 | 8,8 | 5,6 | 5,8 | 7,7 |
| Proband | 1,(1),2 | 4,(4/8),8 | 2,3,6 | 2,(2),(2) | 4,8 | 3,6 | 2,5,8 | 2,7,9 |
| Family 14: | ||||||||
| Father | 1,2 | 4,8 | 3,6 | 2,2 | 1,6 | 6,6 | 6,8 | 3,6 |
| Motherb | 1,3 | 1,8 | 2,5 | 2,2 | 5,9 | 6,6 | 2,2 | 7,8 |
| Proband | 1,2,3 | 1,8,(8) | 2,3,5 | 2,(2),(2) | 1,5 | 6,(6) | 2,(2),6 | 6,7,8 |
| Family 15: | ||||||||
| Father | 1,3 | 4,7 | 4,6 | 2,4 | 4,6 | 1,6 | 4,6 | 2,3 |
| Motherb | 1,1 | 8,9 | 3,4 | 2,2 | 5,8 | 6,7 | 5,7 | 5,8 |
| Proband | 1,(1),(1) | 7,8,9 | 3,4,6 | 2,(2),4 | 6,8 | 6,(6) | 4,5,7 | 2,5,8 |
| Family 16: | ||||||||
| Father | 2,3 | 8,12 | 4,5 | 2,2 | 5,10 | 5,5 | 2,7 | 5,7 |
| Motherb | 3,5 | 7,11 | 4,4 | 2,2 | 4,8 | 1,5 | 6,9 | 7,7 |
| Proband | 2,3,5 | 7,11,12 | 4,(4),(4) | 2,(2),(2) | 4,10 | 1,5 | 2,6,9 | 7,(7),(7) |
Deduced alleles are shown in parentheses.
Carrier parent.
To affirm both biparental inheritance of chromosome 11 and a 3:1 MI malsegregation mechanism, markers from chromosome 11 were also tested. In each of the 16 families tested, the proband was deduced to be trisomic for chromosome 11 markers distal to the t(11;22) breakpoint as a result of 3:1 meiosis I malsegregation (fig 1B and table 1). Only the presence of three different alleles for a marker was considered to be evidence for trisomy, and, in each case, at least one of the two markers distal to the breakpoint was informative. For D11S614 and D11S1356, each of the 16 probands had received two of his or her three alleles from the translocation-carrier parent and had received one from the noncarrier parent. Marker D11S1317, which is proximal to the t(11;22) breakpoint, was informative for 13 of the 16 families (fig. 1B), by virtue of biparental inheritance. Each proband had received from each parent one allele of this marker. This suggested that the proband had received one normal chromosome 11 from each parent and had received the der(22) from the carrier parent. This result is also indicative of a 3:1 meiosis I malsegregation in the carrier parent. Taken together, the results from analysis of chromosome 11 markers and chromosome 22 markers rule out alternative mechanisms and strongly support a 3:1 meiosis I malsegregation in the translocation-carrier parent as the predominant mechanism that leads to the supernumerary-der(22) syndrome.
Discussion
The unbalanced karyotype associated with the supernumerary-der(22) syndrome has been generally accepted to be the result of 3:1 meiotic malsegregation in carriers of the constitutional t(11;22)(q23;q11.2) translocation, the only known recurrent non-Robertsonian translocation in humans (Fraccaro et al. 1980; Zackai and Emanuel 1980). Although numerous t(11;22) families have been ascertained, de novo 11;22 translocations have been detected in a limited number of cases, suggesting a low mutation frequency for the rearrangement (Fraccaro et al. 1980; Zackai and Emanuel 1980; Dawson et al. 1996). We have detected no additional de novo events in the families in the present study, perhaps because access to antecedent generations was limited. Nonetheless, the families reported in this study, as well as those previously reported, demonstrate extremely wide geographical, ethnic, and racial distribution. Furthermore, different heteromorphic cytogenetic variants of the chromosomes 22, as well as varied haplotypes for the polymorphic alleles for DNA markers, both for chromosome 11 and for chromosome 22 (table 1), would tend to exclude relationship between the families. Taken together, these observations appear to argue against a common ancestral origin for the t(11;22).
There are several reported cases of supernumerary-der(22) syndrome that cannot be accounted for by 3:1 malsegregation in the translocation carrier. In most of these cases the unbalanced-translocation progeny have inherited the balanced translocation as well as the supernumerary der(22), and their resultant karyotype is 47,XX/XY,t(11;22)(q23;q11.2),+der(22)t(11;22)(q23;q11.2) (Lockwood et al. 1989; Abeliovich and Carmi 1990; Lurie and Podleschuk 1992; Simi et al. 1992). In these cases the malsegregation has been suggested to be the result of alternate segregation at meiosis I, followed by either meiosis II or postzygotic nondisjunction of the der(22). In one case, it was determined that the unbalanced karyotype most likely resulted from adjacent-1 segregation in a de novo paternally derived translocation, resulting in a gamete that had a normal chromosome 11 and the der(22) but no normal chromosome 22. The embryo was rescued by complementation with a maternal gamete disomic for chromosome 22 (Dawson et al. 1996).
To determine whether 3:1 meiotic malsegregation is the predominant mechanism leading to viable +der(22) offspring, we have analyzed 16 t(11;22) families, using markers from both chromosome 11 and chromosome 22, to determine the parental origin of normal chromosome 11, chromosome 22, and the der(22). Analysis of chromosome 22 markers demonstrated that the unbalanced-translocation proband had inherited one normal chromosome 22 from each parent, as well as the der(22) from the translocation-carrier parent. This result strongly suggests a 3:1 meiosis I malsegregation in the translocation-carrier parent. However, the result would also be the same if a 2:2 adjacent-2 malsegregant gamete containing the normal chromosome 22 and the der(22) and missing a chromosome 11 were rescued by fertilization with a gamete disomic for chromosome 11. This situation would be similar to one case that has been reported elsewhere (Dawson et al. 1996). To rule out this possibility, chromosome 11 markers were tested, and it was demonstrated that, in each case, the proband demonstrated biparental inheritance of his or her normal chromosomes 11. Taken together, these data rule out alternative mechanisms and demonstrate that, in the 16 families analyzed, the supernumerary der(22) resulted from a 3:1 meiosis I malsegregation in the translocation carrier. In 1 of the 16 families analyzed, the father was the carrier parent, and the mechanism that produced the unbalanced-translocation offspring was still 3:1 meiosis I malsegregation. It is interesting to note that, in four of the five reported cases of variant segregations, the translocation was paternal in origin. Thus, the existing data would suggest that, for paternal translocations, all modes of segregation are possible. Additional t(11;22) families with paternal carriers will need to be analyzed before it can be determined whether parental origin of the translocation plays a significant role in the mechanism of malsegregation.
It has been shown that balanced-translocation carriers produce normal as well as unbalanced gametes with all possible 2:2 (alternate, adjacent-1 and adjacent-2) and 3:1 segregations in approximately equal frequencies (Martin 1984; Soler et al. 1993). Therefore, it is theoretically possible that unbalanced-translocation offspring of t(11;22) carriers could result from multiple different segregation patterns. However, there have been very few reported live-born cases that have resulted from such alternative mechanisms. We propose that the majority of patients with supernumerary-der(22) syndrome present as the result of 3:1 meiosis I malsegregation, because this may be the segregation most likely to result in a viable pregnancy. This hypothesis is supported by the observation that there is an increased frequency of spontaneous abortion in the t(11;22)-carrier population (Zackai and Emanuel 1980). The spontaneous abortions are presumed to be loss of pregnancies carrying other unbalanced chromosomal complements.
The 22q11.2 region, between markers D22S788 and ZNF74, where the constitutional t(11;22) breakpoint is located is also involved in numerous other rearrangements. There are multiple translocations whose breakpoints appear to cluster in this region including a balanced t(20;22) (de la Chapelle et al. 1981), three unbalanced translocations—t(12;22), t(4;22), and t(17;22) (Li et al. 1995)—and a balanced t(1;22) (Rhodes et al. 1997). We have found that, although the majority (90%) of the 22q11 deletions associated with DGS/VCFS are 3 Mb in size, there is a small subset (7%) of patients with a smaller, 1.5-Mb deletion (Carlson et al. 1997; Emanuel et al. 1998, and in press) and that both the larger and smaller deletions share their proximal breakpoints. Interestingly, one of the distal breakpoints of the smaller deletions also localizes to the region between D22S788 and ZNF74 (Emanuel et al. 1998, and in press; Funke et al. 1999).
Large blocks (∼200 kb) of duplicated DNA sequences have been identified in multiple 22q11 regions that may be involved in the different rearrangements of chromosome 22, leading to disorders such as CES and DGS/VCFS (Halford et al. 1993; Collins et al. 1997; Emanuel et al. 1998; McTaggart et al. 1998; Edelmann et al. 1999; Saitta et al. 1999). Some sequence components that lie within the region between D22S788 and ZNF74 are also found in other copies of the duplication (Funke et al. 1999; Saitta et al. 1999; also see University of Oklahoma Advanced Center for Genome Technology), suggesting that all or part of the duplication may be present within this interval. The presence of duplicated sequences, as well as the underrepresentation of this region in the available genomic libraries, continues to impede the identification of the t(11;22) breakpoint on chromosome 22. Furthermore, extreme caution must be exercised in the assignment of clones to the t(11;22) breakpoint region, since clones corresponding to other copies of the duplication often appear to map to this region on the basis of marker content and metaphase FISH analysis. This is a result of the high level of sequence identity that occurs over ∼200 kb of genomic DNA. In our experience, establishing that there is an overlap between clones containing duplicated sequences is best accomplished by sequence analysis (T. H. Shaikh, M. L. Budarf, and B. S. Emanuel, unpublished results). Only clones that share 100% sequence identity with each other should be considered as truly overlapping.
The 11q23 region where the constitutional t(11;22) breakpoints cluster has not been mapped as extensively as 22q11. Because the phenotype of balanced-t(11;22) carriers is normal, there has been limited interest in the 11q23 region involved in the constitutional-t(11;22) breakpoint. In contrast, the areas immediately flanking this region, on either side, have been studied extensively. Multiple tumor-associated rearrangements of chromosome 11 map to the region, in 11q22-11q23, that is just proximal to APOA1 (Arai et al. 1996, and references within). On the telomeric side, distal to APOA1, is the region containing the MLL gene, which is most frequently rearranged in hematopoietic malignant disorders. More distally located are both the ES/NE-associated t(11;22) breakpoint (Budarf et al. 1989) and the JS breakpoints in 11q23.3-11q24.2 (Tunacliffe et al. 1993, 1999). Taken together, these data suggest that the constitutional-t(11;22) breakpoints cluster in a region that is within an unstable segment of the long arm of chromosome 11. Despite these observations, neither FISH nor molecular analysis of the chromosome 11 t(11;22) breakpoint spanning BAC 442e11 suggests the presence of chromosome 11–specific duplications.
There are a number of disorders that are caused by chromosomal rearrangements that are believed to be due to homologous recombination between large blocks of duplicated sequences. These genomic disorders include Charcot-Marie Tooth disease type 1A (CMT1A [MIM 118220]) and hereditary neuropathy with liability to pressure palsies, on 17p11.2 (Chance et al. 1994); Smith-Magenis syndrome (SMS [MIM 182290]), on 17p11.2 (Chen et al. 1997); Prader-Willi syndrome (PWS [MIM 176270]) and Angelman syndrome (AS [MIM 105830]), on 15q11-q13 (Christian et al. 1999); and Williams-Beuren syndrome (WBS [MIM 194050]), on 7q11.23 (Perez Jurado et al. 1998). The presence of duplicated sequences in the region where the t(11;22) breakpoints cluster on chromosome 22 suggests their involvement in the translocation. However, to suggest a mechanism involving homologous recombination would require the presence of homologous sequences on chromosome 11. Our data from interphase and metaphase FISH with the breakpoint-spanning BAC 442e11 do not suggest any significant sequence homology between chromosome 11 and chromosome 22. Therefore, it seems unlikely that chromosome 11 has blocks of duplicated sequence that are identical or similar to those seen on chromosome 22. It is possible that, on both chromosome 11 and chromosome 22, there may be small stretches of homologous sequences or site-specific recombination signals, undetectable by FISH, that may play a role in the t(11;22) translocation. Sequence analysis of the regions that contain the t(11;22) breakpoint region should allow us to answer questions about the mechanism involved in the etiology of the constitutional t(11;22). Another factor that may play a role in the generation of the constitutional t(11;22) is the spatial organization of chromosomes 11 and 22 in meiotic and mitotic interphase nuclei. It is possible that 11q23 and 22q11 are in close proximity to one another during interphase, facilitating exchange between these two chromosomes. FISH with probes from both chromosomes can be used to examine the relative position of 11q23 and 22q11 during meiotic and mitotic interphase. This should provide insight into the role that chromatin organization plays in facilitating the constitutional t(11;22).
Acknowledgments
We would like to thank Bea Sellinger, Holly Mensch, and Yvonne Tatsumura for excellent technical assistance. The chromosome-specific gene library LL22NC03 used here was constructed at the Biomedical Sciences Division, Lawrence Livermore National Laboratory, under the auspices of the National Laboratory Gene Library Project sponsored by the U.S. Department of Energy. This work was supported by grants CA39926 and DC02027 (to M.L.B. and B.S.E.) and by funds from the Letitia Scott and Charles E. H. Upham endowed chairs from The Children's Hospital of Philadelphia (support to B.S.E.)
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- NCBI GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for searches of the BLAST Database and for APOAI cDNA [accession number J00098], PAC 201m18 [accession number AC000097], and BAC 422e11 [BAC b1030 in ; accession number AC007707])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for AS [MIM 105830], CES [MIM 115470], CMT1A [MIM 118220], ES/NE [MIM 133450], JS [MIM 147791], PWS [MIM 176270], SMS [MIM 182290], DGS [MIM 188400], VCFS [MIM 192430], and WBS [MIM 194050])
- University of Oklahoma Advanced Center for Genome Technology http://dna1.chem.ou.edu/index.html (for maps of 22q11 and sequences of clones from 22q11.2)
References
- Abeliovich D, Carmi R (1990) The translocation 11q;22q: a novel unbalanced karyotype. Am J Med Genet. 37:288 [DOI] [PubMed] [Google Scholar]
- Arai Y, Hosoda F, Nakayama K, Ohki M (1996) A yeast artificial chromosome contig and NotI restriction map that spans the tumor suppressor gene(s) locus, 11q22.2 -q23.3. Genomics 35:196–206 [DOI] [PubMed]
- Aurias A, Rimbaut C, Buffe D, Zucker JM, Mazabraud A (1984) Translocation involving chromosome 22 in Ewing's sarcoma: a cytogenetic study of four fresh tumors. Cancer Genet Cytogenet 12(1): 21–25 [DOI] [PubMed]
- Budarf ML, Emanuel BS (1997) Progress in autosomal segmental aneusomy syndromes (SASs): single or multi-locus disorders? Hum Mol Genet 6:1657–1665 [DOI] [PubMed]
- Budarf M, Sellinger B, Griffin C, Emanuel BS (1989) Comparative mapping of the constitutional and tumor-associated 11;22 translocations. Am J Hum Genet 45:128–139 [PMC free article] [PubMed]
- Carlson C, Sirotkin H, Pandita R, Goldberg R, Mckie J, Wadey R, Patanjali SR, et al (1997) Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet 61:620–629 [DOI] [PMC free article] [PubMed]
- Chance PF, Abbas N, Lensch MW, Pentao L, Rao BB, Patel PI, Lupski JR (1994) Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Mol Genet 3:223–228 [DOI] [PubMed]
- Chen KS, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault AC, Lee CC, et al (1997) Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet 17:154–163 [DOI] [PubMed]
- Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH (1999) Large genomic dulicons map to sites of instability in Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet 8:1025–1037 [DOI] [PubMed]
- Collins JE, Mungall AJ, Badcock KL, Fay JM, Dunham I (1997) The organization of gamma-glutamyltransferase genes and other low copy repeats in human chromosome 22q11. Genome Res 7:522–531 [DOI] [PubMed]
- Dawson AJ, Mears AJ, Chudley AE, Bech-Hansen T, McDermid H (1996) der(22)t(11;22) resulting from a paternal de novo translocation, adjacent-1 segregation, and maternal heterodisomy of chromosome 22. J Med Genet 33:952- 956 [DOI] [PMC free article] [PubMed]
- de la Chapelle A, Herva R, Koivisto M, Aula P (1981) A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet 57:253–256 [DOI] [PubMed]
- Driscoll DA, Emanuel BS, Mitchell LE, Budarf ML (1997) PCR assay for screening patients at risk for 22q11.2 deletion. Genet Testing 1(2): 109–113 [DOI] [PubMed]
- Edelmann L, Pandita RK, Morrow BE (1999) Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet 64:1076–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel BS, Budarf ML, Scambler PJ (1999) The genetic basis of conotruncal cardiac defects: the chromosome 22q11.2 deletion. In: Harvey R, Rosenthal N (eds) Heart development. Academic Press, New York, pp 463–478 [Google Scholar]
- Emanuel BS, Budarf ML, Shaikh T, Driscoll DA (1998) Blocks of duplicated sequence define the endpoints of DGS/VCFS 22q11.2 deletions. Am J Hum Genet Suppl 63:A11 [Google Scholar]
- Emanuel BS, Goldmuntz E, Budarf ML, Shaikh T, McGrath J, McDonald-McGinn D, Zackai EH, et al. Blocks of duplicated sequence define the endpoints of DGS/VCFS 22q11.2 deletions. In: Clark E, Nakazawa M, Takao A (eds) Etiology and morphogenesis of congenital heart disease. Futura Publishing, New York (in press) [Google Scholar]
- Fraccaro M, Lindsten J, Ford CE, Iselius L (1980) The 11q;22q translocation: a European collaborative analysis of 43 cases. Hum Genet 56:21–51 [DOI] [PubMed]
- Funke B, Edelmann L, McCain N, Pandita RK, Ferreira J, Merscher S, Zohouri M, et al (1999) der(22) syndrome and velo-cardio-facial syndrome/DiGeorge syndrome share a 1.5-Mb region of overlap on chromosome 22q11. Am J Hum Genet 64:747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyapay G, Morissette J, Vignal A, Dib C, Fizames C, Millasseau P, Marc S, et al (1994) The 1993–94 Généthon human genetic linkage map. Nat Genet 7:246–339 [DOI] [PubMed]
- Halford S, Lindsay E, Natudu M, Carey AH, Baldini A, Scambler PJ (1993) Low-copy- number repeat sequences flank the DiGeorge/velo-cardio-facial syndrome loci at 22q11. Hum Mol Genet 2:191–196 [DOI] [PubMed]
- Holmes SE, Riazi MA, Gong W, McDermid HE, Sellinger BT, Hua A, Chen F, et al (1997) Disruption of the clathrin heavy chain-like gene (CLTCL) associated with features of DGS/VCFS: a balanced (21;22)(p12;q11) translocation. Hum Mol Genet 6:357–367 [DOI] [PubMed]
- Kaplan JC, Aurias A, Julier C, Prieur M (1987) Human chromosome 22. J Med Genet 24:65–78 [DOI] [PMC free article] [PubMed]
- Li M, Budarf ML, Chien P, Barnoski BL, Emanuel BS, Driscoll DA (1995) Clustering of DiGeorge/velocardiofacial-associated translocations suggestive of a translocation “hot spot.” Am J Hum Genet Suppl 57:A119 [Google Scholar]
- Lichter P, Cremer T, Borden J, Manuelidis L, Ward DC (1988) Delineation of individual human chromosomes in metaphase and interphase cells by in situ suppression hybridization using recombinant DNA libraries. Hum Genet 80:224–234 [DOI] [PubMed]
- Lin AE, Bernar J, Chin A, Sparkes R, Emanuel BS, Zackai EH (1986) Congenital heart disease in supernumerary der(22), t(11;22) syndrome. Clin Genet 29:269–275 [DOI] [PubMed]
- Lindenbaum RH, Bobrow M (1975) Reciprocal translocations in man: 3:1 meiotic disjunction resulting in 47- or 45-chromosome offspring. J Med Genet 12:29–43 [DOI] [PMC free article] [PubMed]
- Lockwood DH, Farrier A, Hecht F, Allanson J (1989) Not all chromosome imbalance resulting from the 11q;22q translocation is due to 3:1 segregation in first meiosis. Hum Genet 83:287–288 [DOI] [PubMed]
- Lurie IW, Podleschuk LV (1992) 11q;22q Translocation: third case of imbalance not due to 3:1 nondisjunction in first meiosis. Am J Med Genet 42:216 [DOI] [PubMed]
- Marineau C, Aubry M, Julien JP, Rouleau GA (1992) Dinucleotide repeat polymorphism at the D22S264 locus. Nucleic Acids Res 20:1430 [PMC free article] [PubMed]
- Martin RH (1984) Analysis of human sperm chromosome complements from a male heterozygous for a reciprocal translocation t(11;22)(q23;q11). Clin Genet 25:357–361 [DOI] [PubMed]
- McDermid HE, Duncan AMV, Brasch KR, Holden JJA, Magenis E, Sheehy R, Burn J, et al (1986) Characterization of the supernumerary chromosome in cat eye syndrome. Science 232:646–648 [DOI] [PubMed]
- McTaggart KE, Budarf ML, Driscoll DA, Emanuel BS, Ferreira P, McDermid HE (1998) Cat eye syndrome chromosome breakpoint clustering: identification of two intervals also associated with 22q11 deletion syndrome breakpoints. Cytogenet Cell Genet 81:222–228 [DOI] [PubMed]
- Mears AJ, Dunca AMV, Budarf ML, Emanuel BS, Sellinger B, Siegel-Bartelt J, Greenberg CR, et al (1994) Molecular characterization of the marker chromosome associated with cat eye syndrome. Am J Hum Genet 55:134–142 [PMC free article] [PubMed]
- Morrow B, Goldberg R, Carlson C, Dasgupta R, Sirotkin H, Collins J, Dunham I, et al (1995) Molecular definition of the 22q11 deletions in velo-cardio-facial syndrome. Am J Hum Genet 56:1391–1403 [PMC free article] [PubMed]
- Morton NE (1991) Parameters of the human genome. Proc Natl Acad Sci USA 88:7474–7476 [DOI] [PMC free article] [PubMed]
- Perez Jurado LA, Wang YK, Peoples R, Coloma A, Cruces J, Francke U (1998) A duplicated gene in the breakpoint regions of the 7q11.23 Williams-Beuren syndrome deletion encodes the initiator binding protein TFII-I and BAP-135, a phosphorylation target of BTK. Hum Mol Genet 7:325–334 [DOI] [PubMed]
- Philippsen P, Stotz A, Scherf C (1991) DNA of Saccharomyces cerevisiae. Methods Enzymol 194:169–172 [DOI] [PubMed]
- Quackenbush J, Davies C, Bailis JM, Khristich JV, Diggle K, Marchuck Y, Tobin J, et al (1995) An STS content map of human chromosome 11: localization of 910 YAC clones and 109 islands. Genomics 29:512–525 [DOI] [PubMed]
- Rhodes CH, Call KM, Budarf ML, Barnoski BL, Bell CJ, Emanuel BS, Bigner SH, et al (1997) Molecular studies of an ependymoma-associated constitutional t(1;22)(p22;q11.2). Cytogenet Cell Genet 78:247–252 [DOI] [PubMed]
- Saitta SC, McGrath JM, Mensch H, Shaikh TH, Zackai EH, Emanuel BS (1999) A 22q11.2 deletion that excludes UFD1L and CDC45L in a patient with conotruncal and craniofacial defects. Am J Hum Genet 1999:562–566 [DOI] [PMC free article] [PubMed]
- Shaikh TH, Driscoll D, Budarf M, Emanuel BS (1997) Molecular evidence for 3:1 meiotic (MI) malsegregation in the supernumerary der(22) syndrome. Am J Hum Genet Suppl 61:A50 [Google Scholar]
- Simi P, Ceccarelli M, Barachini A, Floridia G, Zuffardi O (1992) The unbalanced offspring of the male carriers of the 11q;22q translocation: nondisjunction at meiosis II in a balanced spermatocyte. Hum Genet 88:482–483 [DOI] [PubMed]
- Soler A, Carrio A, Perez-Vidal MT, Borrell A, Fortuny A (1993) Unusual segregation for 11q;22q parental translocation in a triplet pregnancy: prenatal diagnosis in chorionic villi and amniotic fluid. Prenat Diagn 13:137–141 [DOI] [PubMed]
- Trask BJ, Massa H, Kenwrick S, Gitschier J (1991) Mapping of human chromosome Xq28 by two-color fluorescence in situ hybridization of DNA sequences to interphase cell nuclei. Am J Hum Genet 48:1–15 [PMC free article] [PubMed]
- Tunacliffe A, Jones C, Le Paslier D, Todd R, Cherif D, Birdsall M, Devenish L, et al (1999) Localization of Jacobsen syndrome breakpoints on a 40-Mb physical map of distal chromosome 11q. Genome Res 9:44–52 [PMC free article] [PubMed]
- Tunnacliffe A, Perry H, Radice P, Budarf ML, Emanuel BS (1993) A panel of sequence tagged sites for chromosome band 11q23. Genomics 17:744–747 [DOI] [PubMed]
- Turc-Carel C, Philip I, Berger MP, Philip T, Lenoir GM (1984) Chromosome study of Ewing's sarcoma (ES) cell lines: consistency of a reciprocal translocation t(11;22)(q24;q12). Cancer Genet Cytogenet 12(1): 1–19 [DOI] [PubMed]
- Zackai EH, Emanuel BS (1980) Site-specific reciprocal translocation, t(11;22)(q23;q11), in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet 7:507–521 [DOI] [PubMed]