

Summary
Duane retraction syndrome (DRS) is a congenital eye-movement disorder characterized by a failure of cranial nerve VI (the abducens nerve) to develop normally, resulting in restriction or absence of abduction, restricted adduction, and narrowing of the palpebral fissure and retraction of the globe on attempted adduction. DRS has a prevalence of ∼0.1% in the general population and accounts for 5% of all strabismus cases. Undiagnosed DRS in children can lead to amblyopia, a permanent uncorrectable loss of vision. A large family with autosomal dominant DRS was examined and tested for genetic linkage. After exclusion of candidate regions previously associated with DRS, a genomewide search with highly polymorphic microsatellite markers was performed, and significant evidence for linkage was obtained at chromosome 2q31 (D2S2314 maximum LOD score 11.73 at maximum recombination fraction .0). Haplotype analysis places the affected gene in a 17.8-cM region between the markers D2S2330 and D2S364. No recombinants were seen with markers between these two loci. The linked region contains the homeobox D gene cluster. Three of the genes within this cluster, known to participate in hindbrain development, were sequenced in affected and control individuals. Coding sequences for these genes were normal or had genetic alterations unlikely to be responsible for the DRS phenotype. Identifying the gene responsible for DRS may lead to an improved understanding of early cranial-nerve development.
Introduction
Duane retraction syndrome (DRS [MIM 126800]) is a congenital disorder of eye movement (Duane 1905), in which patients exhibit partial or complete failure of abduction and partial closure of the eyelids, with retraction of the globe into the orbit on adduction (fig. 1). DRS is a frequent cause of strabismus in children and may result in amblyopia-related visual loss. Although this syndrome can be unilateral and sporadic, many familial cases have been reported; these are usually bilateral and inherited in an autosomal-dominant fashion (Sevel and Kassar 1974).
Figure 1.

Photodocumentation of a patient with DRS, exhibiting incomplete abduction on attempted right or left gaze.
Electromyographic (Huber 1974) data suggest that DRS may result from abnormal development or absence of the abducens nerve (cranial nerve VI). Analysis of autopsy specimens revealed the absence of the abducens nuclei in both unilateral and bilateral cases of DRS (Hotchkiss et al. 1980; Miller et al. 1982). The absence of the left abducens nerve in an individual with unilateral DRS was established by use of magnetic-resonance imaging (Parsa et al. 1998).
DRS can be associated with other nonocular anomalies, such as Klippel-Feil anomaly, Wildervank syndrome, Goldenhar syndrome (Pfaffenbach et al. 1972), Rubinstein-Taybi syndrome (Cruz et al. 1995), and Batten disease (Marshman et al. 1998). Three karyotypic abnormalities, associated with chromosomes 4, 8, and 22, have been found in patients with DRS; however, all three cases exhibited other ocular and nonocular problems. A de novo deletion of a region of chromosome 4 (4q27–31) was observed in a 15-year-old boy with bilateral blepharoptosis, DRS, and “mild learning difficulties” (Chew et al. 1995). An insertion/deletion event resulting in both the deletion of chromosome region 8q12–13 and the insertion of this segment into 6q25 was noted in a patient with DRS, mental retardation, and other anatomic malformations (Calabrese et al. 1998). Cytogenetic analysis of two siblings manifesting DRS, bilateral sensorineural deafness, unilateral renal agenesis, and preauricular skin tags indicated the presence of a supernumerary bisatellited marker chromosome derived from chromosome 22pter-q11 (Cullen et al. 1993).
In the present study, we describe a large four-generation family from Oaxaca, Mexico, with 25 living members affected with DRS transmitted in a fully penetrant autosomal-dominant pattern. Chromosomal loci previously associated with DRS were excluded by genetic linkage analysis (Ott et al. 1999). A genomewide search was undertaken to identify the locus responsible for this form of DRS.
Subjects and Methods
Clinical Evaluations
After informed consent was obtained, ophthalmic, neurological, and surgical histories were studied. All living family members (with the exception of individuals II-4, II-8, and III-17) were examined, and peripheral blood was drawn for direct genomic DNA isolation or transformation of B-lymphoblasts. All subjects were tested for visual acuity, ocular ductions and versions, ocular alignment by prism-cover testing, fixation preference, and globe retraction. Determination of the Duane phenotype was made by one examiner (M.S.B.), on the basis of limitation of abduction in one or both eyes, incomitant strabismus, and lid fissure narrowing on adduction of affected eyes. For a more detailed description of the clinical diagnosis performed, see Chung et al. (in press). Patients with a history of strabismus surgery who did not meet the above diagnostic criteria were considered to be of indeterminate status and were considered unknown in subsequent linkage analysis. Studies were performed in accordance with a protocol approved by the Committee on Clinical Investigations of Childrens Hospital Los Angeles
Genotyping and Linkage Analysis
Genotyping was performed by use of 396 fluorescent dye–labeled dinucleotide-repeat markers (Prism Linkage Mapping Set version 2; PE Biosystems). The PCR conditions employed are given in detail at the PE Biosystems database. The PCR products were electrophoresed in 48-lane, 5% denaturing polyacrylamide gels in ABI 377 sequencers. The GENESCAN and GENOTYPER software packages (PE Biosystems) were used to generate genotypes. Parametric two-point and multipoint LOD-score analyses were performed by use of FASTLINK (Lathrop et al. 1984). The genetic model assumed dominant inheritance with penetrance equal to .99. On the basis of the prevalence of DRS in the population (DeRespinis et al. 1993), a phenocopy rate of .001 was assumed. In regions showing significant evidence of linkage, lower penetrances, of .9 and .8, were also tested. Only one disease liability was considered. For the genome scan, allele frequencies were estimated on the basis of data on the founders of the pedigree.
Fine Mapping and Haplotype Analysis
After evidence for linkage was obtained at 2q31, panels of additional dye-labeled markers were designed. Haplotype analysis, using D2S2333, D2S2216, D2S160, D2S347, D2S112, D2S2313, D2S142, D2S2330, D2S335, D2S364, D2S117, D2S325, D2S2382, D2S126, D2S396, D2S206 and D2S125 (ABI-PRISM Linkage Mapping Set) and D2S333, D2S1238, D2S2314, D2S1244, D2S1245, homeobox D (HOXD), and D2S138, which span the interval between D2S2330 and D2S364 (Genetic Location Database), was performed to identify the markers flanking the disease region. Genotyping was performed as described above, and haplotypes were constructed by use of GENEHUNTER 2.0 (Kruglyak et al. 1996).
Sequence Analysis of Candidate Homeobox Genes
PCR primers flanking each of the coding exons of the human genes HOX D3 (GenBank accession number, D11117) and D4 (GenBank accession number, X17360) and D1 (cloned during this study) were used to amplify and sequence these genetic segments in affected and control subjects. PCR reactions contained 50–100 ng of genomic DNA, 20 pmol of each primer, 0.5 U of Pfu polymerase (Stratagene), 4 mM of each dNTP and 5 μl of a 10× cloned Pfu buffer in a total reaction volume of 50 μl. PCR reactions were optimized on a Robocylcer Gradient 96 (Stratagene); conditions typically were 96°C for 5 min, followed by 30 cycles of 95°C for 1 min, 56°C–68°C for 1 min, and 72°C for 1 min. For difficult templates, such as exon 1 of D1, 4% dimethyl sulfoxide was added to the PCR reaction mix before amplification. PCR products were electrophoresed through 1.5% agarose and were purified by use of a QIAquick gel extraction kit (Qiagen) prior to direct cycle sequencing. Sequencing was performed on an automated ABI 310 sequencer by use of dye-terminator chemistry and the manufacturer's protocols (Applied Biosystems). The structure and sequence of the human HOX D1 gene was determined (data not shown). A human bacterial artificial chromosome (BAC) library was screened by PCR, by use of primers designed to the second exon and 3′ UTR of the HOX D3 gene, to isolate a clone containing the adjacent HOX D1 (situated 3′ to HOX D3). On the basis of the mouse homologue, primers were designed to amplify HOX D1 coding segments from the positive BAC, to obtain the corresponding human sequence. The products were sequenced, and the remainder of the HOX D1 coding region was obtained by sequencing the BAC directly. HOX D1 exons were amplified and sequenced as described above.
Results
The family consists of 118 members, spanning four generations, of which 24 individuals, after clinical examination, were definitively diagnosed with DRS (fig. 2). All living affected individuals have limited abduction, with narrowing of the palpebral fissure and retraction of the globe on adduction. Some individuals showed limited adduction (Chung et al., in press). Individual III-16 was orthotropic; however, she had a history of strabismus surgery for esotropia noted at an early age and so was considered to be “indeterminate” for the purposes of analysis (fig. 2). No apparent nonocular associated dysmorphism was observed (Chung et al., in press). Before a genomewide search was performed, candidate chromosome loci were tested for linkage. Twenty-four microsatellite markers were typed for chromosomes 4, 8, and 22, and all are associated with LOD scores <.5 (Ott et al. 1999).
Figure 2.
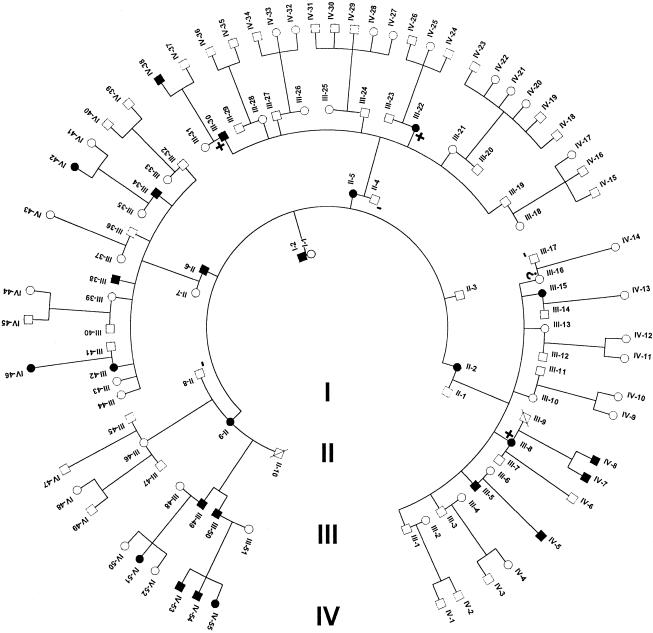
Pedigree of the large DRS family used for linkage analysis. All individuals, with the exception of the deceased and individuals (denoted by a minus sign [−]), were examined for DRS. DNA was prepared from subjects and was genotyped for linkage analysis. Individuals denoted with a plus sign (+) were used to confine the critical disease region (see fig. 5). Individual III-16 was phenotypically classified as “indeterminate.”
Linkage and Haplotype Analysis
The genomewide screen with highly polymorphic markers spaced at ∼10-cM intervals revealed linkage only at 2q31. Maximum two-point LOD scores of 6.9 (θ=.1), 4.9 (θ=0), and 6.3 (θ=.1) were seen at markers D2S2330, D2S335, and D2S364, respectively. A maximum two-point LOD score of 11.73 (θ=0) was subsequently observed with marker D2S2314, and a maximum multipoint LOD score of 12.69 centering on marker D2S2314 was obtained. Microsatellite markers yielding significant LOD scores are presented in table 1, and the multipoint linkage analyses are presented in figure 3.
Table 1.
Two-Point LOD Scores
|
Two-Point
LOD Score at
θ =a |
||||||
| Chromosome2 Marker | .0 | .05 | .1 | .2 | .3 | .4 |
| D2S112 | −22.75 | −4.29 | −1.55 | .5 | .94 | .58 |
| D2S2313 | −29.31 | −5.94 | −2.75 | −.11 | .62 | .45 |
| D2S142 | −7.58 | −1.24 | −.12 | .68 | .79 | .54 |
| D2S2330 | −3.53 | 6.79 | 6.87 | 5.92 | 4.32 | 2.25 |
| D2S333 | 9.05 | 8.37 | 7.63 | 6.00 | 4.15 | 2.06 |
| D2S335 | 4.9 | 4.53 | 4.14 | 3.27 | 2.26 | 1.13 |
| D2S1238 | 7.54 | 6.95 | 6.34 | 5.0 | 3.48 | 1.74 |
| D2S2314 | 11.73 | 10.87 | 9.96 | 7.92 | 5.58 | 2.88 |
| D2S1244 | 10.82 | 10.04 | 9.20 | 7.31 | 5.15 | 2.64 |
| D2S1245 | 6.3 | 5.83 | 5.32 | 4.19 | 2.9 | 1.4 |
| HOXD13 | 1.38 | 1.17 | .96 | .55 | .24 | .03 |
| D2S138 | 11.11 | 10.29 | 9.40 | 4.19 | 2.90 | 1.40 |
| D2S364 | −8.79 | 6.19 | 6.33 | 5.50 | 4.04 | 2.11 |
| D2S117 | −16.77 | 2.11 | 3.03 | 3.12 | 2.31 | 1.08 |
| D2S325 | −19.05 | 1.40 | 2.79 | 3.31 | 2.72 | 1.5 |
Calculated under the assumption of an autosomal-dominant, fully penetrant genetic model (penetrance .99) with a phenocopy rate of .001.
Figure 3.
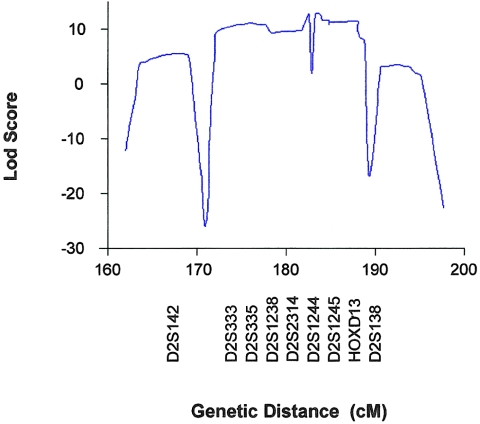
Graph of multipoint LOD scores, between DRS and chromosome 2 markers D2S142, D2S333, D2S335, D2S1238, D2S2314, D2S1244, D2S1245, HOXD13, and D2S138, for the large Mexican family.
Haplotype analysis has refined the region to a 17.8-cM interval situated between D2S2330 and D2S364 (fig. 4). GENEHUNTER predicted a common haplotype, comprising the markers D2S333, D2S335, D2S1238, D2S2314, D2S1244, D2S1245, HOXD13, and D2S138, which segregate with all individuals diagnosed with the DRS phenotype. The proximal flanking marker D2S2330 and the distal flanking marker D2S364 are defined by recombination events within individuals III-30 and III-8/III-22, respectively (fig. 5). Individual III-16, who was originally classified as “indeterminate” because of a history of strabismus surgery, also carried the common “disease-associated” haplotype, indicating that this person is affected. Thus, although only 24 individuals were clinically diagnosed with DRS, we feel both that it is likely that individual III-16 is affected and that her strabismus surgery corrected a DRS-related esotropia.
Figure 4.
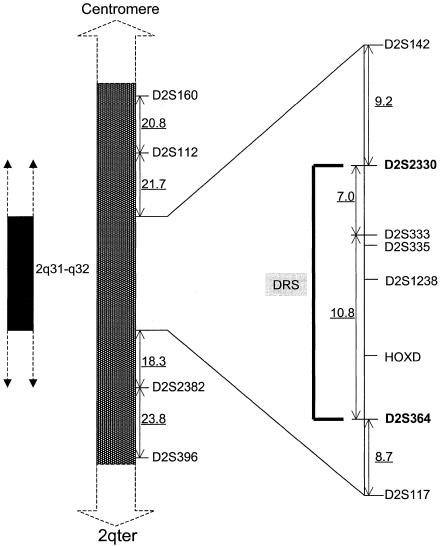
Schematic representation of the genetic map of the region containing the DRS locus. The flanking proximal and distal markers, separated by 17.8 cM, are shown in boldface. The distances (in cM) between the markers D2S160, D2S112, D2S142, D2S2330, D2S333, D2S364, D2S117, D2S382, and D2S396 are underlined. The order of and the genetic distances between markers were obtained from the PE Biosystems ABI PRISM mapping Web page.
Figure 5.
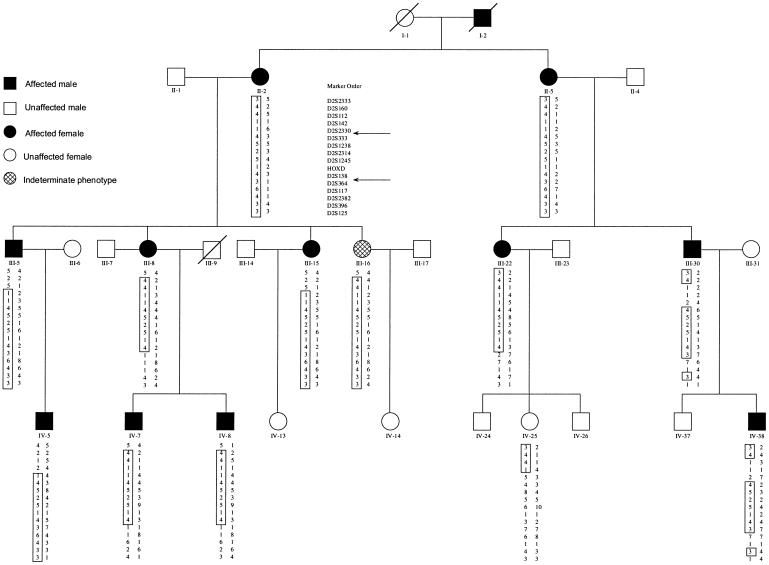
Haplotypes for chromosome 2 markers, for a subset of the large Mexican family with DRS. The order of the DNA markers is shown. The disease-associated alleles are boxed. The critical interval lies between markers D2S2330 and D2S364 and is indicated by the arrows. Two other markers, D2S335 and D2S1244 (not shown), are also within this critical interval.
Sequence Analysis of Candidate Homeobox Genes
Sequencing of the coding regions of the HOX D1, D3, and D4 genes from patients with DRS in this particular family presented no changes that were indicative of a functional disruption of an expressed protein. A homozygous nucleotide change, G→C, was observed in exon 1 of the D3 gene (position 1745; GenBank accession number D11117) which would result in an amino change of a cysteine (codon TGT) to a serine (codon TCT), in both affected and normal individuals (data not shown). This TCT codon is also seen within an alternative HOX D3 sequence submitted to the database (EMBL accession number Y09980), suggesting that this cysteine-to-serine substitution is a polymorphism that occurs without consequence in the general population.
A heterozygous genotype (T/C) in exon 2 of D3 at position 4270 (GenBank accession number D11117), segregated with the affected individuals, whereas the homozygous genotype, C/C, was present in all the unaffected individuals (data not shown). Although this results in a change of a codon from CTG to TTG (codon 335), both of these codons code for the amino acid leucine; thus, the primary protein sequence of HOX D3 is the same on both of the chromosomes from the affected individuals.
Discussion
This study provides strong evidence for linkage of an autosomal-dominant form of DRS to 2q31. Chromosomal localization of DRS is the initial step toward the isolation and functional characterization of the gene/protein responsible for this phenotype and should increase our understanding of cranial-nerve development. It should be noted that DRS may be genetically heterogeneous. As mentioned earlier, there is evidence that suggests that additional loci associated with DRS map to chromosomes 4, 8, and 22.
Electromyographic data (Sato 1960; Huber 1974) and postmortem histopathologic data attribute the DRS phenotype to the abnormal development of the abducens nerve nuclei. The association of other congenital anatomic malformations with DRS has led some to suggest that the dysgenesis event occurs sometime between the 4th and 10th wk of gestation (Cross and Pfaffenbach 1972). Functional characterization of the DRS gene will help us understand the physiology and mechanism of cranial-nerve embryogenesis.
There are a number of candidate genes, known to be expressed during embryogenesis, within this disease interval, including the genes within the HOXD cluster. HOX genes are developmental control genes that regulate morphogenesis and cell differentiation in animals, and the expression of HOX genes can be detected as early as gastrulation in mammals (Mark et al. 1997).
In humans there are four HOX gene clusters—HOXA, -B, -C, and -D—situated on chromosomes 7, 17, 12, and 2, respectively. The spatial pattern of HOX-gene expression in the developing embryo is directly related to the chromosomal order of the genes within the cluster. Genes at the 5′-most end of the HOX cluster have the most posterior (caudal) boundary of expression, and each successive gene 3′ is progressively expressed in more-anterior (rostral) regions (Dubuole and Dollé 1989; Giampaolo et al. 1989; Graham et al. 1989; Guant 1991). The 3′-most genes within the HOXD cluster are D1, D3, and D4. Embryonic-gene-expression studies and gene knockout data in mouse homologues of the human D1, D3, and D4 and their paralogue genes ( A1, B1, A3, B3, A4, B4, and C4) indicate that these genes are necessary for the normal development of the head, the hindbrain, and associated structures (Frohman et al. 1990; Hunt et al. 1991; Frohman and Martin 1992; Carpenter et al. 1993; Dollé et al. 1993; Bedford et al. 1995; Manley and Capecchi 1997). Interestingly, disruption of the mouse Hoxb1 gene resulted in failure of the motor nucleus of the facial (VIIth) nerve to develop (Goddard et al. 1996). Thus, genes that are expressed early in embryogenesis and that are involved in the development of the hindbrain are strong candidates for DRS.
The fact that the HOX D1, D3, and D4 genes fit the above criteria prompted us to amplify and sequence these genes from individuals in the family studied. Although no coding-sequence mutations likely to cause the disease phenotype were found within the HOX genes analyzed in this study, we are continuing our efforts to search for causative mutations outside the coding sequence of these genes.
The expression of HOX genes is known to be regulated by retinoids: vitamin A derivatives, usually all-trans- and 9-cis-retinoic acid (RA [Simeone et al. 1991; Moroni et al. 1993]). Enhancers containing RA response elements (RARE), which are responsible for the RA-regulated expression, are found within 3′ regions of the mouse Hoxa1 and Hoxb1 genes (Langston et al. 1997) and within both the 3′ and 5′ regions of the mouse Hoxd4 gene (Zhang et al. 1997). A conserved RARE was found associated with the chick and mouse and Hoxb1 genes that acts to restrict expression to a specific segment (rhombomere 4) of the normal developing hindbrain (Studer et al. 1994). It is interesting to note that in vivo disruption of the mouse Hoxa1 3′ RARE results in cranial-nerve and hindbrain abnormalities similar to the phenotypes of the Hoxa1 knockouts (Dupe et al. 1997). Abnormal development of the VIIth–XIth cranial nerves can occur as a result of disruption of the 3′ RARE of the Hoxa1 and Hoxb1 genes (Gavalas et al. 1998). It is possible that disruption of regulatory elements associated with the human HOX genes within the D cluster may result in DRS. With this in mind we are characterizing the 3′ and 5′ regions of the HOX D1, D3, and D4 genes in affected individuals of this large family.
As far as we are aware, this is the first linkage data for a family with an autosomal dominant form of DRS without other associated dysmorphisms. Characterization of other families with DRS may permit refinement of the critical region. Many mouse homeobox genes have been targeted for disruption of normal function; howeve, none to date exhibit a phenotype that is reminiscent of DRS. Considering the phenotype of the mouse Hoxb1 knockout, which fails to develop a normal VIIth-nerve motor nucleus (Goddard et al. 1996), it would be of great interest to know the phenotype of a Hoxd1-deficient mouse. Isolation and functional analysis of the gene responsible for DRS will give us a new insight into the intricate processes involved in the early development of the hindbrain and cranial nerves. Assignment of this new locus for nonsyndromic DRS is a significant advance toward identification of the genetic basis of this condition.
Acknowledgments
We are very grateful to all the family members who willingly participated in this study. We thank Dr. Y. Taniguchi for his kind donation of the D3/D4 cosmid clone. This work was supported by the Knights Templar Eye Foundation (support to B.A.) and the Clayton Foundation for Research (support to J.T.S.).
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/GenBank (for gene locations)
- Genetic Location Database, http://cedar.genetics.soton.ac.uk/ (for markers used for haplotype analysis)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim (for Duane retraction syndrome [MIM 126800])
- PE Biosystems, http://www.pebio.com/ab/apply/dr/dra1a3.html (for markers used for linkage analysis), http://www2.perkin-elmer.com/ab/apply/dr/lmsv2/chrom2.html (for ABI PRISM mapping)
References
- Bedford M, Arman E, Orr-Urtreger A, Lonai P (1995) Analysis of the Hoxd-3 gene: structure and localization of its sense and natural antisense transcripts. DNA Cell Biol 14(4): 295–304 [DOI] [PubMed]
- Calabrese GSL, Morizio E, Franchi PG, Pompetti F, Mingarelli R, Marsilio T, Rocchi M, et al (1998) Detection of an insertion deletion of region 8q13-q21.2 in a patient with Duane syndrome: implications for mapping and cloning a Duane gene. Eur J Hum Genet 6(3): 187–193 [DOI] [PubMed]
- Carpenter EM, Goddard JM, Chisaka O, Manley NR, Capecchi MR (1993) Loss of Hox-A1 (Hox-1.6) function results in the reorganization of the murine hindbrain. Development 118:1063–1075 [DOI] [PubMed]
- Chew CK, Foster P, Hurst JA, Salmon JF (1995) Duane's retraction syndrome associated with chromosome 4q27-31 segment deletion. Am J Ophthalmol 119(6): 807–809 [DOI] [PubMed]
- Chung M, Stout JT, Borchert MS. Clinical diversity of hereditary duane's retraction syndrome. Ophthalmology (in press) [DOI] [PubMed] [Google Scholar]
- Cross HE, Pfaffenbach DD (1972) Duane's retraction syndrome and associated congenital malformations. Am J Ophthalmol 73:442–450 [DOI] [PubMed]
- Cruz OA, Mason DM, Eswara MS, Lueder GT (1995) Duane retraction syndrome associated with Rubinstein-Taybi syndrome. Ophthalmic Genet 16(4): 171–175 [DOI] [PubMed]
- Cullen P, Rodgers C, Callen DF, Connolly VM, Eyre H, Fells P, Gordon H, et al (1993) Association of familial Duane anomaly and urogenital abnormalities with a bisatellited marker derived from chromosome 22. Am J Med Genet 47:925–930 [DOI] [PubMed]
- DeRespinis PA, Caputo AR, Wagner RS, Guo S (1993) Review: Duane's Retraction Syndrome. Surv of Ophthalmol 38(3): 257–288 [DOI] [PubMed]
- Dollé P, Lufkin T, Krumlauf R, Mark M, Duboule D, Chambon P (1993) Local alterations of Krox-20 and Hox gene expression in the hindbrain suggest lack of rhombomeres 4 and 5 in homozygote null Hoxa-1 (Hox-1.6) mutant embryos. Proc Natl Acad Sci USA 90:7666–7670 [DOI] [PMC free article] [PubMed]
- Duane A (1905) Congenital deficiency of abduction associated with impairment of adduction, retraction movement, contraction of the palpebral fissure and oblique movements of eye. Arch Ophthalmol 34:133–159 [DOI] [PubMed]
- Dubuole D, Dollé P (1989) The structural and functional organization of the murine HOX gene family resembles that of Drosophila homeotic genes. EMBO J 8:1497–1505 [DOI] [PMC free article] [PubMed]
- Dupe V, Davenne M, Brocard J, Dollé P, Mark M, Dierich A, Chambon P, et al (1997) In vivo functional analysis of the Hoxa-1 3′ retinoic acid response element (3′RARE). Development 124:399–410 [DOI] [PubMed]
- Frohman MA, Boyle M, Martin G (1990) Isolation of the mouse Hox-2.9 gene: analysis of embryonic expression suggests that positional information along the anterior-posterior axis is specified by mesoderm. Development 110:589–607 [DOI] [PubMed]
- Frohman MA, Martin G (1992) Isolation and analysis of embryonic expression of Hox-4.9, a member of the murine labial-like gene family. Mech Dev 38:55–67 [DOI] [PubMed]
- Gavalas A, Studer M, Lumsden A, Rijli FM, Krumlauf R, Chambon P (1998) Hoxa1 and Hoxb1 synergize in patterning the hindbrain, cranial nerves and second pharyngeal arch. Development 126:1123–1136 [DOI] [PubMed]
- Giampaolo A, Acampora D, Zappavigna V, Pannese M, D'Esposito M, Care A, Faiella A, et al (1989) Differential expression of human HOX-2 genes along the anterior-posterior axis in embryonic central nervous system. Nature 40:191–197 [DOI] [PubMed]
- Goddard JM, Rossei M, Manley NR, Capecchi MR (1996) Mice with targeted disruption of Hoxb-1 fail to form the motor nucleus of the VIIth nerve. Development 122:3217–3228 [DOI] [PubMed]
- Graham A, Papalopulu N, Krumlauf R (1989) The murine and Drosophila homeobox gene complexes have common features of organization and expression. Cell 57(3): 367–378 [DOI] [PubMed]
- Guant SJ (1991) Expression patterns of mouse Hox genes: clues to understanding of developmental and evolutionary strategies. Bioessays 13:505–513 [DOI] [PubMed]
- Hotchkiss MG, Miller NR, Clark AW, Green WR (1980) Bilateral Duane's retraction syndrome: a clinical-pathologic case report. Arch Ophthalmol 98:870–874 [DOI] [PubMed]
- Huber A (1974) Electrophysiology of the retraction syndromes. Br J Ophthalmol 58:293–300 [DOI] [PMC free article] [PubMed]
- Hunt P, Gulisano M, Cook M, Sham M-H, Faiella A, Wilkinson D, Boncinelli E, et al (1991) A distinct Hox code for the branchial region of the vertebrate head. Nature 353:861–864 [DOI] [PubMed]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed]
- Langston AW, Thompson JR, Gudas LJ (1997) Retinoic acid-responsive enhancers located 3′ of the Hox A and Hox B homeobox gene clusters: functional analysis. J Biol Chem 272:2167–2175 [DOI] [PubMed]
- Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446 [DOI] [PMC free article] [PubMed]
- Manley NR, Capecchi MR (1997) Hox group 3 paralogous genes act synergistically in the formation of somitic and neural crest-derived structures. Dev Biol 192:274–288 [DOI] [PubMed]
- Mark M, Rijli FM, Chambon P (1997) Homeobox genes in embryogenesis and pathogenesis. Pediatr Res 42(4): 421–429 [DOI] [PubMed]
- Marshman WE, Lee JP, Jones B, Schalit G, Holder GE (1998) Duane's retraction syndrome and juvenile Batten's disease: a new association? Aust N Z J Ophthalmol 26(3): 251–254 [DOI] [PubMed]
- Miller NR, Kiel SM, Green WR, Clark AW (1982) Unilateral Duane's retraction syndrome (type 1). Arch Ophthalmol 100:1468–1472 [DOI] [PubMed]
- Moroni M, Viganó MA, Mavilio F (1993) Regulation of the human HOXD4 gene by retinoids. Mech Dev 44:139–154 [DOI] [PubMed]
- Ott S, Borchert M, Chung M, Appukuttan B, Wang X, Weinberg K, Stout JT (1999) Exclusion of candidate genetic loci for Duane's retraction syndrome. Am J Ophthalmol 127:358–360 [DOI] [PubMed]
- Parsa CF, Grant E, Dillon WP Jr, du Lac S, Hoyt WF (1998) Absence of the abducens nerve in Duane syndrome verified by magnetic resonance imaging. Am J Ophthalmol 125(3): 399–401 [DOI] [PubMed]
- Pfaffenbach DD, Cross HE, Kearns TP (1972) Congenital anomalies in Duane's retraction syndrome. Arch Ophthalmol 88:635–639 [DOI] [PubMed]
- Sato S (1960) Electromyographic study on retraction syndrome. Jpn J Ophthalmol 4:57–66 [Google Scholar]
- Sevel D, Kassar BS (1974) Bilateral Duane syndrome: occurrence in three successive generations. Arch Ophthalmol 91:492–494 [DOI] [PubMed]
- Simeone A, Acampora D, Nigro V, Faiella A, D'Esposito M, Stornaiuolo, Mavilio F, et al (1991) Differential regulation by retinoic acid of the homeobox genes of the four HOX loci in human embryonal carcincoma cells. Mech Dev 33:215–228 [DOI] [PubMed]
- Studer M, Popperl H, Marshall H, Kuroiwa A, Krumlauf R (1994) Role of a conserved retinoic acid response element in rhombomere restriction of Hoxb-1. Science 265:1728–1732 [DOI] [PubMed]
- Zhang F, Popperl H, Morrison A, Kovacs EN, Prideaux V, Schwarz L, Krumlauf R, et al (1997) Elements both 5′ and 3′ to the murine Hoxd4 gene establish anterior borders of expression in mesoderm and neuroectoderm. Mech Dev 67(1): 49–58 [DOI] [PubMed]