

Summary
Bardet-Biedl syndrome (BBS) is a rare, autosomal recessive disorder; major phenotypic findings include dysmorphic extremities, retinal dystrophy, obesity, male hypogenitalism, and renal anomalies. In the majority of northern European families with BBS, the syndrome is linked to a 26-cM region on chromosome 11q13. However, the finding, so far, of five distinct BBS loci (BBS1, 1q; BBS2, 16q; BBS3, 3p; BBS4, 15q; BBS5, 2q) has hampered the positional cloning of these genes. We use linkage disequilibrium (LD) mapping in an isolated founder population in Newfoundland to significantly reduce the BBS1 critical region. Extensive haplotyping in several unrelated BBS families of English descent revealed that the affected members were homozygous for overlapping portions of a rare, disease-associated ancestral haplotype on chromosome 11q13. The LD data suggest that the BBS1 gene lies in a 1-Mb, sequence-ready region on chromosome 11q13, which should enable its identification.
Introduction
Bardet-Biedl syndrome (BBS; MIM 209900) is a rare, autosomal recessive disorder in which the combination of dysmorphic extremities, retinal dystrophy, obesity, male hypogenitalism, and renal anomalies may be associated, to varying degrees, with mental retardation and/or diabetes mellitus (Green et al. 1989; O'Dea et al. 1996). Of the five genetic subtypes, BBS1 appears to be the most common locus in affected individuals of northern European descent: as many as 50% of pedigrees show linkage to BBS1 (Beales et al. 1997; Bruford et al. 1997; Woods et al. 1999). BBS1 was initially mapped to chromosome 11q13 by combining the positive LOD scores achieved in 17 of 31 North American kindreds as a result of a genome scan (Leppert et al. 1994). The other BBS loci, BBS2 (16q; Kwitek-Black et al. 1993), BBS3 (3p; Sheffield et al. 1994), BBS4 (15q; Carmi et al. 1995) and BBS5 (2q; Young et al. 1999), were located by homozygosity mapping in extended inbred kindreds. In the original study by Leppert et al. (1994), the putative BBS1 gene was tightly linked to two loci on chromosome 11q13: the gene for human muscle glycogen phosphorylase (PYGM) and the anonymous DNA marker D11S913 (AFM164zf12), and was localized to a 26-cM interval between D11S1298 and INT2 (FGF3). A more precise genetic and physical location of BBS1 is required if the BBS1 gene is to be positionally cloned.
Refined mapping of disease genes has recently been accomplished by linkage disequilibrium (LD) mapping in founder populations (reviewed by Jorde 1995 and by Xiong and Guo 1997). When a disease mutation is first introduced into a population, it resides on a single disease haplotype (DH) of linked markers. As a result of meiotic recombination, the length of this DH decreases as a function of genetic distance so that, with successive generations, only the original marker alleles in the vicinity of the disease locus cosegregate on disease chromosomes. Although fine mapping of disease genes was established initially in old populations like the Finnish (Hastbacka et al. 1992), recent success in fine mapping has been accomplished in relatively young populations (Labuda et al. 1996; Groenewald et al. 1998). The island population of Newfoundland, considered a genetic isolate because of the nature of its founding and subsequent isolation, is enriched for BBS, with 10 times the incidence in other white populations of northern European ancestry (Green et al. 1989; O'Dea et al. 1996). A single founder effect has been reported in two recent studies of families from the island with either hereditary nonpolyposis colorectal cancer (HNPCC) or multiple endocrine neoplasia type 1 (MEN1) (Olufemi et al. 1998; Froggatt et al. 1999). In the present study we show that, in five unrelated families from Newfoundland, the family members affected with BBS are homozygous for an ancestral DH, and we use recent and historical recombinations to the ancestral DH to map the BBS1 gene within a 1-Mb region on chromosome 11q13.
Subjects and Methods
Subjects
In a recent population survey, 17 BBS families of English ancestry were haplotyped at the BBS1, BBS2, BBS3, and BBS4 loci. Of these families, three (B8, B10, and B19) were assigned to BBS1 and three (B7, B12, and B15) could not be excluded from this locus because they yielded positive LOD scores and haplotypes consistent with linkage to BBS1 (Woods et al. 1999; table 1). Of these six families, parental consanguinity was documented in family B8 and suspected in families B10, B12, B15, and B19 on the basis of progenitors with the same surname originating from the same community. Extensive genotyping with markers mapping to the BBS1 critical region was performed in the six families, representing 8 BBS patients and 44 first- and second-degree relatives. Informed consent had been obtained previously, and the clinical presentations of the adult patients have been described elsewhere (Green et al. 1989; O'Dea et al. 1996).
Table 1.
DHs on Chromosome 11q13 Segregating in Six BBS Families[Note]
| B7 |
B8 |
B10 |
B12 |
B15 |
B19 |
|||||||
| Marker | p | m | p | m | p | m | p | m | p | m | p | m |
| D11S1298 | 7 | 3 | 10 | 10 | 7 | 10 | 7 | 9 | 11 | 11 | … | … |
| D11S956 | 6 | 8 | 8 | 8 | 10 | 8 | 7 | 5 | 7 | 7 | 11 | 11 |
| D11S480 | 9 | 5 | 6 | 6 | 9 | 6 | 5 | 9 | 5 | 5 | 9 | 9 |
| D11S1883 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 8 | 10 | 10 | 7 | 7 |
| D11S913 | 6 | 7 | 7 | 7 | 6 | 7 | 6 | 5 | 6 | 6 | 6 | 6 |
| FGF3 | 7 | 12 | 5 | 5 | 7 | 5 | 9 | 9 | 9 | 9 | … | … |
Note.— Haplotypes are arranged with the paternal haplotype (p) on the left and the maternal haplotype (m) on the right. For markers D11S913 and FGF3, the phase of the paternal haplotype in B12 is assumed.
Genotyping
DNA was extracted from the lymphocytes of venous blood by a simple salting-out procedure (Miller et al. 1988). Fifty to 100 ng of template DNA in standard 10-μL reactions containing 1.5 pmol of primer, 200 μm dNTPs, 0.125 units of Tf1 DNA polymerase, and a trace amount of γ[32P] end-labeled forward primer was amplified by temperature cycling in a Perkin Elmer 9600 thermocycler. Products were run on 6%–8% polyacrylamide sequencing gels with formamide (Litt et al. 1993) and were subjected to autoradiography. The markers D11S1298, D11S956, D11S480, D11S4205, D11S1883, D11S4945, PYGM, D11S4946, D11S4940, D11S4938, D11S449, D11S4941, D11S913, and FGF3 were typed in key family members. Family B7 was excluded from the study, because several samples failed to amplify.
Family Studies
Haplotypes were constructed, for each family, to give the minimum number of recombinations. DHs were identified from alleles that were transmitted from both unaffected parents to affected offspring. Pairwise linkage analyses between BBS and six markers spanning the BBS1 critical region were performed under an autosomal recessive model with a penetrance of 0.95 and a disease-allele frequency of .004. The disease-allele frequency for BBS was calculated from the Newfoundland population estimate of 1/17,500 (Green et al. 1989), adjusted to reflect an estimated 50% contribution of the BBS1 locus to the overall population frequency. All markers were assumed to have nine alleles of equal frequency in the population. LOD scores were calculated by the MLINK subroutine program of FASTLINK (V3.0P) (Lathrop and Lalouel 1984; Cottingham et al. 1993; Schäffer et al. 1994).
Population Studies
The marker-allele frequencies in disease and normal (nontransmitted) chromosomes of the 10 obligate BBS carriers (parents) were compared. Normal alleles from each parent were used as population controls, to avoid the possibility of inadvertently including BBS1 disease alleles from random carriers in the population. Allelic association was tested by means of Fisher's exact test, with one-sided probability. The DHs were compared between families in the search for (i) common BBS1 haplotypes that would indicate that the parents of two or more families were distantly related and (ii) a common ancestral DH.
Results
Preliminary Evidence of a Founder Effect among Families with BBS1
Five of the six families in this study live along the south and southwest coasts of Newfoundland (fig. 1). Although a common ancestor was not identified in ancestral lineages, genetic evidence suggests that the families are interrelated. The affected individuals in family B10 are apparently heterozygous for a copy of the DH identified in family B8 and a portion of the DH transmitted by the father of family B7 (table 1). It was noted that these three families originate from closely-linked communities on the southwest coast of the island (fig. 1). The data also suggest that the father of a south coast family (B12) shares a recent common ancestor with both parents of family B15, the only family not residing on the south coast of the island. Therefore, on the basis of shared DHs and geographical location, preliminary evidence suggested the presence of a founder effect among BBS1 cases in the Newfoundland population.
Figure 1.
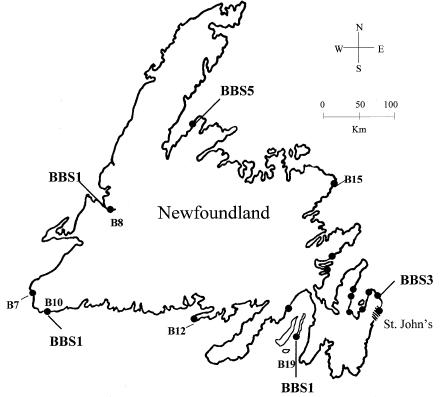
Geographical distribution of 17 Newfoundland families with BBS (blackened circles), indicating linkage with BBS1, BBS3, or BBS5 as determined in previous studies (Woods et al. 1999; Young et al. 1999). The families genotyped in the present study are indicated by family number.
Evidence of Linkage to BBS1
In a previous study, three families (B8, B10, and B19) were assigned to BBS1 (Woods et al. 1999, table 1). Family B10 consists of a large sibship in which two affected individuals share a unique genotype on chromosome 11q13, compared with their nine unaffected sibs (fig. 2). A maximum LOD score of 1.662 at recombination fraction (θ) 0 was obtained with fully informative markers that mapped telomeric to D11S1883. Family B10 was previously excluded from linkage with BBS2, BBS3, BBS4 (Woods et al. 1999), and BBS5 (M. O. Woods, unpublished data) on the basis of established exclusion criteria. For a locus to be excluded, one or both of the following conditions had to be met: (i) an affected individual(s) inherited the same genotype as one or more unaffected sibs, and (ii) two or more affected individuals inherited different genotypes. Family B8 is a consanguineous family in which a single affected offspring was homozygous by descent (HBD) for the entire BBS1 locus (fig. 2). A maximum LOD score of 1.405 (θ=0) was obtained with fully informative markers (e.g., D11S1883). Homozygosity (in the affected member only) was not observed at other BBS loci: BBS2 and BBS3 loci were excluded. Family B19 consists of a large sibship with one affected individual. The affected offspring was homozygous (presumably HBD) for all markers typed within the BBS1 interval, whereas seven unaffected sibs were either heterozygous or homozygous for a normal haplotype. Homozygosity was not observed at other BBS loci, and the BBS3 locus could be excluded (Woods et al. 1999).
Figure 2.

Three families with linkage to BBS1 (B8, B10 and B19) and three unassigned families (B7, B12, and B15) haplotyped for six polymorphic markers spanning the 26-cM BBS1 interval on chromosome 11q13. Only core pedigrees are presented. Blackened symbols indicate individuals with diagnoses of BBS. Boxed haplotypes (solid and dashed lines) indicate DHs. Double marriage lines depict consanguineous unions, either documented (solid line) or suspected (dashed line). “R” indicates that the DH is recombinant.
In three families (B7, B12, and B15), BBS could not be excluded from, or confidently assigned to, the BBS1 locus (Woods et al. 1999). Family B15 consists of a large sibship with one affected and eight unaffected sibs available for genotyping. Homozygosity in the affected individual was observed only at the BBS1 locus, and the BBS3 locus could be excluded. Family B7 has two affected individuals who share a unique genotype on chromosome 11q13, compared with their four unaffected sibs (fig. 2). A maximum LOD score of 1.074 (θ=0) was obtained with fully informative markers, and BBS in this family was excluded from linkage to BBS3, BBS4, and BBS5 (M. O. Woods, unpublished data). Family B12 is relatively uninformative for linkage but was excluded from BBS3 because the only affected individual (PID 14) inherited the same genotype as his unaffected sister. He is homozygous for the BBS1 marker D11S913, but the phase of the paternal chromosomes cannot be determined for the D11S913-FGF3 interval because of an apparent recombination in one of the offspring.
The pairwise LOD scores calculated for the six families at six markers covering the 26-cM BBS1 critical region were summed. Four of six markers examined reached statistical significance when the families were considered together (LOD score >3; table 2). The finding of common DHs among BBS1-assigned families (B8 and B10), between BBS1-assigned and unassigned families (B7 and B10), and among unassigned families with evidence of linkage to BBS1 (B12 and B15) (table 1) corroborates the linkage and haplotype data that these are, in fact, all BBS1 families. Because the exact relationships among the pedigrees are unknown, the summary LOD scores given in table 2 are presumed to be underestimated. Obligate recombinations involving the DHs were detected in several families and used to refine the BBS1 interval. In family B10, an unaffected individual, PID 14, inherited a nonrecombinant DH from his father and a recombinant DH from his mother. The presence of two DHs for the centromeric portion of the BBS1 critical region (D11S1298 to D11S1883) in an unaffected individual suggests that marker D11S1883 is the new centromeric boundary for BBS1 (fig. 2). Similarly, a recombinant paternal haplotype inherited by an unaffected sib in family B8 (PID 12) suggests that BBS1 is located centromeric to FGF3. Intrafamilial recombinations on DHs reduce the BBS1 interval from a 26-cM region to a 15-cM interval between D11S1883 and FGF3 (fig. 3).
Table 2.
Sum of the Pairwise LOD Scores at Chromosome 11q13, in Six Families with BBS
| LOD Score
at θ= |
|||||||
| Marker | .000 | .010 | .050 | .100 | .200 | .300 | .400 |
| D11S1298 | 2.227 | 2.263 | 2.169 | 1.856 | 1.124 | .052 | .142 |
| D11S956 | 3.419 | 3.439 | 3.275 | 2.867 | 1.910 | .972 | .279 |
| D11S480 | 2.809 | 2.819 | 2.623 | 2.202 | 1.288 | .559 | .137 |
| D11S1883 | 3.657 | 3.663 | 3.430 | 2.952 | 1.868 | .894 | .241 |
| D11S913 | 3.019 | 2.923 | 2.546 | 2.093 | 1.264 | .587 | .151 |
| FGF3 | 3.777 | 3.662 | 3.203 | 2.644 | 1.599 | .739 | .190 |
Figure 3.

Refinement of the BBS1 interval by recombinational and LD mapping in Newfoundland families. The markers and their relative positions were selected from the map of the MEN1 region on 11q13 (Manickam et al. 1997; Sixth International SCW 11 Workshop 1998).
LD and Detection of a Founder Haplotype
Extensive genotyping at the BBS1 locus focused on markers within the new BBS1 interval (fig. 3). The distribution of alleles at 14 polymorphic loci in disease and normal chromosomes is shown in table 3. Significant LD between specific marker alleles on DHs was observed across the families. Strong associations were observed between alleles at five consecutive marker loci: D11S4205, D11S1883, D11S4945, PYGM, and D11S4946. Comparison of allele frequencies between disease and normal chromosomes showed that three of these associations were statistically significant. Allele 10 at marker D11S1883 was present on 70% (7/10) of DHs but only 20% (2/10) of normal chromosomes (P<.05). Similarly, allele 8 at the PYGM locus was present on 100% of the DHs and 50% of normal chromosomes (P<.05), and allele 5 at marker D11S4946 was present on 100% of the disease chromosomes and 30% of normal chromosomes (P<.01). Although allele 4 at marker D11S4205 and allele 9 at marker D11S4945 were present on 90% and 100% of disease chromosomes, respectively, these associations were not significant, because both alleles are common in the general population; they are present on 70% of the normal chromosomes (table 3).
Table 3.
LD at the BBS1 Locus on Chromosome 11q13 among Five Newfoundland Kindreds[Note]
|
DH for Kindred |
Normal Haplotype
for Kindred |
|||||||||||||||||||
| B8 |
B10 |
B12 |
B15 |
B19 |
B8 |
B10 |
B12 |
B15 |
B19 |
|||||||||||
| Locus | p | m | p | m | p | m | p | m | p | m | p | m | p | m | p | m | p | m | p | m |
| 1298 | 10 | 10 | 7 | 10 | 7 | 9 | 11 | 11 | … | … | 5 | 7 | 10 | 7 | 4 | 7 | 8 | 13 | … | … |
| 956 | 8 | 8 | 10 | 8 | 7 | 5 | 7 | 7 | 11 | 11 | 10 | 3 | 11 | 7 | 1 | 2 | 5 | 6 | 11 | 7 |
| 480 | 6 |
6 |
9 | 6 |
5 |
9 |
5 |
5 |
9 |
9 |
9 | 5 |
5 |
5 | 6 |
5 |
8 | 5 |
4 |
5 |
| 4205 | 4 | 4 | 3 |
4 | 4 | 4 | 4 | 4 | 4 |
4 |
3 | 4 |
4 |
5 | 4 |
4 | 3 | 4 |
4 | 4 |
| 1883 | 10 | 10 | 10 | 10 | 10 | 8a | 10 | 10 | 7 |
7 |
9 |
12 |
12 |
9 |
13 |
10 |
12 | 11 | 10 | 7 |
| 4945 | 9 | 9 | 9 | 9 | 9 | 9 | 9 | 9 | 9 | 9 | 9 | 9 | 9 | 9 | 9 |
4 | 10 | 10 |
9 | 9 |
| PYGM | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
8 | 6a | 8 |
2 | 4 | 4 | 8 | 8 |
4 |
| 4946 | 5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
4 | 5 |
5 |
4 | 4 | 1 | 1 | 5 |
3 | 3 |
| 4940 | 5 | 5 | 7 | 5 | 7 | 5 | 7 | 7 | 3 | 3 | 5 | 5 | 5 | 5 | 5 | 3 | 5 | 7 | 7 | 5 |
| 4938 | 5 | 5 | 3 | 5 | 5 | 3 | 5 | 5 | 5 | 5 | 9 | 3 | 5 | 3 | 5 | 9 | 5 | 5 | … | 9 |
| 449 | 7 | 7 | 3 | 7 | 3 | 6 | 5 | 5 | 3 | 3 | 5 | 3 | 4 | 3 | 4 | 2 | 5 | 5 | 3 | 4 |
| 4941 | 9 | 9 | 11 | 9 | 2 | 2 | 4 | 4 | 4 | 4 | 8 | 3 | 6 | 11 | 2 | 4 | 4 | 6 | 4 | 4 |
| 913 | 7 | 7 | 6 | 7 | 6 | 5 | 6 | 6 | 6 | 6 | 7 | 6 | 5 | 5 | 5 | 7 | 6 | 6 | 7 | 6 |
| FGF3 | 5 | 5 | 7 | 5 | 9 | 9 | 9 | 9 | … | … | 4 | 5 | 5 | 9 | 5 | 5 | 12 | 5 | … | … |
Note.— Haplotypes are arranged with paternal haplotype (p) on the left and the maternal haplotype (m) on the right. Specific alleles associated with disease chromosomes are boxed.
Allele is not associated with disease chromosome.
A single ancestral DH was readily identified in the vicinity of marker loci with strong allelic associations. All disease chromosomes segregating in BBS1 families contained the -9-8-5- subhaplotype at D11S4945, PYGM, and D11S4946, respectively. The affected members in families B10 and B12 that initially appeared to be heterozygous at the BBS1 locus (table 1) are, in fact, HBD for a region centered around the PYGM locus (table 3). As well, the DHs that appeared unique on the basis of six polymorphic markers (e.g., family B19, table 1) are indistinguishable from all other DHs identified in the population. In contrast, the -9-8-5- subhaplotype was found only on a single normal haplotype (family B8). This suggest that, although rare, the -9-8-5- subhaplotype is not exclusive to BBS1-carrying chromosomes in the Newfoundland population. The longer - 4-10-9-8-5- haplotype (encompassing the -9-8-5- subhaplotype and the centromeric markers D11S4205 and D11S1883) was identified on 60% of the DHs but none of the normal chromosomes. This longer haplotype may represent a larger portion of the ancestral DH. In this case, DHs discordant for allele 10 at D11S1883 (families B12 and B19) may have resulted from either (i) historical recombinations to the ancestral DH between D11S1883 and D11S4945 that were not informative at D11S4205, a notably uninformative marker in the population, or (ii) de novo mutations at the more polymorphic D11S1883 locus. In any case, the evidence suggests that the -9-8-5- subhaplotype represents either the remnants of a founder BBS1 chromosome imported from England or the background haplotype that sustained a BBS1 mutation de novo in the germline of a single English settler.
Location of BBS1
LD mapping supports a position for the BBS1 gene within a 1-cM region between markers D11S1883 and D11S4940, surrounding the PYGM locus. The D11S1883 boundary is also supported on the basis of intrafamilial recombination (fig. 3). This 1-cM genetic interval represents a physical distance of ∼1 Mb (Manickam et al. 1997) within a region of the genome that is gene rich and, fortuitously, sequence ready as a result of physical mapping efforts to clone the Best vitelliform macular dystrophy (VMD-2) and multiple endocrine neoplasia (MEN1) genes that map to this region (Cooper et al. 1997; Guru et al. 1997; Manickam et al. 1997).
Discussion
The island of Newfoundland is a sparsely populated region of Canada in which 50% of the population of 560,000 reside in small coastal communities. The colonization of the island occurred primarily by a natural increase from northern European settlers of predominantly English and Irish extraction who arrived before 1835. Most founders originated from the West Country of England and from southeast Ireland (Mannion 1977). Mating segregation between Irish Catholics and English Protestants, low immigration, and geographical isolation of communities have resulted in genetic isolation of the population. In a review of the historical development of genetic isolation in three Newfoundland outports, Bear et al. (1988) observed that only 1%–8% of breeding parents were immigrants to the area and 60% of births had been to parents originating from the same small community.
The scattered distribution of families with BBS and the recent identification of at least three distinct genetic BBS subtypes in Newfoundland (Woods et al. 1999) is not consistent with the expectation of a single cluster of families with a recessive disease in a young founder population. However, we noted that five of the six families with evidence of linkage to BBS1 reside on the south and southwest coasts of the island, a region that was settled predominantly by the spread of settlers by sea in an east-to-west direction (Mannion 1977). The genotyping data also suggest that these families have complex relationships with each other in that large regions (up to 26 cM) on disease-associated haplotypes were shared among kindreds. In the absence of mutation analysis, the identification of a single founder effect would require that all patients were homozygous for specific alleles at marker loci tightly linked to the BBS1 gene. We have shown that all BBS1 patients identified in the Newfoundland population are homozygous for a relatively rare haplotype spanning a 1-cM region centered around the PYGM gene on chromosome 11q13. The relatively high incidence of BBS and the scattered distribution of affected families in the Newfoundland population are attributable to a combination of locus heterogeneity and at least one founder effect.
The power of LD mapping is well illustrated from this study: a total of six affected individuals in five families were used to fine map the BBS1 gene to within 1 Mb by this method. Even families not informative for linkage (e.g., B12) provided valuable information on historical recombinations. In contrast, two recent studies reporting on a total of 47 families with BBS failed to identify informative recombinations at the BBS1 locus (Beales et al. 1997; Bruford et al. 1997). The localization of BBS1 within a 1-Mb interval between D11S1883 and D11S4940 is small enough to promote the positional cloning of the BBS1 gene. The Newfoundland population may also be useful for the analysis of other genetic diseases, especially those that have a relatively high incidence in the population: nonsyndromic deafness, various forms of retinal dystrophy, neuronal ceroid lipofuscinosis, and neural tube defects (J. Green, unpublished data).
Acknowledgments
We thank the families for their participation in this study and Lynette Penney for technical assistance. We thank Settara Chandrasekharappa for providing primers and Ban Younghusband for reviewing the manuscript. Financial support for this work came from the Kidney Foundation of Canada and the Medical Research Council of Canada. T.L.Y. and M.O.W. are recipients of Graduate Fellowships from the Faculty of Medicine at Memorial University. J.S.G was a recipient of a grant from the Canadian Genetic Diseases Network.
Electronic-Database Information
Accession number and URLs for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for BBS [MIM 209900])
- Sixth International SCW 11 Workshop (Nice, France, 1998), http://www.genetics.wustl.edu/gerhard/SCW11/SCW11.html
References
- Beales PL, Warner AM, Hitman GA, Thakker R, Flinter FA (1997) Bardet-Biedl syndrome: a molecular and phenotypic study of 18 families. J Med Genet 34:92–98 [DOI] [PMC free article] [PubMed]
- Bear JC, Nemec TF, Kennedy JC, Marshall WH, Power AA, Kolonel VM, Burke GB (1988) Inbreeding in outport Newfoundland. Am J Med Genet 29:649–660 [DOI] [PubMed]
- Bruford EA, Riise R, Teague PW, Porter K, Thomson KL, Moore AT, Jay M, et al (1997) Linkage mapping in 29 Bardet-Biedl syndrome families confirms loci in chromosomal regions 11q13, 15q22.3-q23, and 16q21. Genomics 41:93–99 [DOI] [PubMed]
- Carmi R, Rokhlina T, Kwitek-Black AE, Elbedour K, Nishimura D, Stone EM, Sheffield VC (1995) Use of a DNA pooling strategy to identify a human obesity syndrome locus on chromosome 15. Hum Mol Genet 4:9–13 [DOI] [PubMed]
- Cooper PR, Nowak NJ, Higgins MJ, Simpson SA, Marquardt A, Stoehr H, Weber BHF, et al (1997) A sequence-ready high resolution physical map of the Best macular dystrophy gene region in 11q13. Genomics 41:185–192 [DOI] [PubMed]
- Cottingham RW Jr, Idury RM, Schäffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed]
- Froggatt NJ, Green J, Brassett C, Evans DGR, Bishop DT, Kolodner R, Maher ER (1999) A common MSH2 mutation in English and North American HNPCC families: origin, phenotypic expression, and sex specific differences in colorectal cancer. J Med Genet 36:97–102 [PMC free article] [PubMed]
- Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, et al (1989) The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med 321:1002–1009 [DOI] [PubMed]
- Groenewald JZ, Liebenberg J, Groenewald IM, Warnich L (1998) Linkage disequilibrium analysis in a recently founded population: evaluation of the variegate porphyria in South African Afrikaners. Am J Hum Genet 62:1254–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guru SC, Olufemi SE, Manickam P, Cummings C, Gieser LM, Pike BL, Bittner ML, et al (1997) A 2.8-Mb clone contig of the multiple endocrine neoplasia type I (MEN1) region at 11q13. Genomics 42:436–445 [DOI] [PubMed]
- Hastbacka J, de la Chapelle A, Kaitila I, Sistonen P, Weaver A, Lander E (1992) Linkage disequilibrium mapping in isolated founder populations: diastrophic dysplasia in Finland. Nat Genet 2:204–211 [DOI] [PubMed]
- Jorde LB (1995) Linkage disequilibrium as a gene-mapping tool. Am J Hum Genet 56:11–14 [PMC free article] [PubMed]
- Kwitek-Black AE, Carmi R, Duyk GM, Buetow KH, Elbedour K, Parvari R, Yandava C, et al (1993) Linkage of Bardet-Biedl syndrome to chromosome 16q and evidence for non-allelic genetic heterogeneity. Nat Genet 5:392–396 [DOI] [PubMed]
- Labuda M, Labuda D, Korab-Laskowska M, Cole DEC, Zietkiewicz E, Weissenbach J, Popowska E (1996) Linkage disequilibrium analysis in young populations: pseudo-vitamin D–deficiency rickets and the founder effect in French Canadians. Am J Hum Genet 59:633–643 [PMC free article] [PubMed]
- Lathrop GM, Lalouel JM (1984) Easy calculations of LOD scores and genetic risks on small computers. Am J Hum Genet 36:460–465 [PMC free article] [PubMed]
- Leppert M, Baird L, Anderson KL, Otterud B, Lupski JR, Lewis RA (1994) Bardet-Biedl syndrome is linked to DNA markers on chromosome 11q and is genetically heterogeneous. Nat Genet 7:108–112 [DOI] [PubMed]
- Litt M, Hauge X, Sharma V (1993) Shadow bands seen when typing polymorphic dinucleotide repeats: some causes and cures. BioTechniques 15:280–284 [PubMed]
- Manion JJ (ed) (1977) The peopling of Newfoundland: essays in historical geography. Institute of Social and Economic Research, Memorial University of Newfoundland, St. John's, Newfoundland [Google Scholar]
- Manickam P, Guru SC, Debelenko LV, Agarwal SK, Olufemi S-E, Weisemann JM, Boguski MS, et al (1997) Eighteen new polymorphic markers in the multiple endocrine neoplasia type 1 (MEN1) region. Hum Genet 101:102–108 [DOI] [PubMed]
- Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215 [DOI] [PMC free article] [PubMed]
- O'Dea D, Parfrey P, Harnett JD, Hefferton D, Cramer BC, Green J (1996) The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kid Dis 27:776–783 [DOI] [PubMed]
- Olufemi SE, Green JS, Manickam P, Guru SC, Agarwal SK, Kester MB, Dong Q, Burns AL et al. (1998) Common ancestral mutation in the MEN1 gene is likely responsible for the prolactinoma variant of MEN1 (MEN1Burin) in four kindreds from Newfoundland. Hum Mutat 11:264–269 [DOI] [PubMed]
- Schäffer AA, Gupta SK, Shriram K, Cottingham RW Jr (1994) Avoiding recomputation in linkage analysis. Hum Hered 44:225–237 [DOI] [PubMed]
- Sheffield VS, Carmi R, Kwitek-Black A, Rokhlina T, Nishimura D, Duyk GM, Elbedour K, et al (1994) Identification of a Bardet-Biedl syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mapping. Hum Mol Genet 3:1331–1335 [DOI] [PubMed]
- Woods MO, Young TL, Parfrey PS, Hefferton D, Green JS, Davidson WS (1999) Genetic heterogeneity of Bardet-Biedl syndrome in a distinct Canadian population: evidence for a fifth locus. Genomics 55:2–9 [DOI] [PubMed]
- Xiong M, Guo, S-W (1997) Fine-scale genetic mapping based on linkage disequilibrium: theory and practice. Am J Hum Genet 60:1513–1531 [DOI] [PMC free article] [PubMed]
- Young TL, Penney L, Woods MO, Parfrey PS, Green JS, Hefferton D, Davidson WS (1999) A fifth locus for Bardet-Biedl syndrome maps to 2q31. Am J Hum Genet 64:900–904 [DOI] [PMC free article] [PubMed] [Google Scholar]