

Summary
Fechtner syndrome is an autosomal-dominant variant of Alport syndrome, manifested by nephritis, sensorineural hearing loss, cataract formation, macrothrombocytopenia, and polymorphonuclear inclusion bodies. As opposed to autosomal-recessive and X-linked Alport syndromes, which have been genetically well studied, the genetic basis of Fechtner syndrome remains elusive. We have mapped the disease-causing gene to the long arm of chromosome 22 in an extended Israeli family with Fechtner syndrome plus impaired liver functions and hypercholesterolemia in some individuals. Six markers from chromosome 22q yielded a LOD score >3.00. A maximum two-point LOD score of 7.02 was obtained with the marker D22S283 at a recombination fraction of 0. Recombination analysis placed the disease-causing gene in a 5.5-Mb interval between the markers D22S284 and D22S1167. No collagen genes or genes comprising the basement membrane have been mapped to this region.
Introduction
In 1927, Alport first described the syndrome (MIM 153640) that bears his name (Alport 1927). This syndrome consists of progressive nephropathy, sensorineural hearing loss, and, in many cases, eye abnormalities such as lenticonus or retinal anomalies. Electron-microscopic investigations in the early 1970s identified the thick and irregular glomerular basement membrane as the site of primary renal abnormality (Spear and Slusser 1972; Churg et al. 1973). Spear (1973) later suggested that the association between the three target-organ pathologies stemmed from an abnormality in a structural gene that governs the composition of the basement membrane in the glomerulus, lens capsule, and inner ear. Recent studies have shown that mutations may occur at any of several loci involved in basement-membrane type IV–collagen synthesis.
Both mapping of the Alport loci to specific chromosomes and cloning of the different collagen type IV genes have shown that COL4A1 and COL4A2 are located close together on chromosome 13q33-34 (Boyd et al. 1988). COL4A3 and COL4A4 are located on chromosome 2q35-37 (Mariyama et al. 1992), and COL4A5 and COL4A6 are located on the X chromosome (Hostikka et al. 1990; Myers et al. 1990). Most patients have an X-linked, dominant form of Alport syndrome that arises from a mutation at the COL4A5 locus (Boye et al. 1991; Netzer et al. 1992; Nakazato et al. 1993; Nomura et al. 1993; Tryggvason et al. 1993). In others, mutations and deletions in COL4A5 and COL4A6 cause a combination of Alport syndrome and leiomyomatosis (Antignac et al. 1992; Zhou et al. 1993; Heidet et al. 1995). The autosomal-recessive form results from mutations at the COL4A3 or the COL4A4 locus (Lemmink et al. 1994; Mochizuki et al. 1994). A much less frequent and less characterized condition is the autosomal-dominant form of disease that manifests itself in two forms. (1) Classic Alport syndrome—Jefferson et al. (1997) reported a linkage of a large dominant pedigree to the chromosome 2 COL4A3-COL4A4 locus; no mutations in these genes were found in the affected members of the family. (2) A form that resembles Alport syndrome in its renal, auditory, and ophthalmic manifestations—however, unique hematological features, which include macrothrombocytopenia and typical cytoplasmatic polymorphonuclear inclusion bodies sometimes distinguish it from the classic Alport syndrome. These hematological manifestations resemble those seen in the May-Hegglin anomaly, which also has an autosomal-dominant mode of inheritance (Hegglin 1945). The molecular basis of these syndromes, also named “Fechtner” (Peterson et al. 1985; MIM 153640) or “Epstein” (Epstein et al. 1972; MIM 153650) syndromes, remained elusive. In 1988, Gershoni-Baruch et al. (1988) described a large family in which affected members had a combination of nephropathy, eye abnormalities, high-tone sensorineural hearing loss, impaired liver functions, hypercholesterolemia, macrothrombocytopenia, and polymorphonuclear inclusion bodies. Within this family, the disease segregated in an autosomal-dominant mode. We have recently identified additional family members and have used this family to map the first autosomal-dominant “Alport-like” disease.
Subjects and Methods
Patients and Family Members
The study included 38 individuals, 17 of whom were affected (fig. 1). All patients had macrothrombocytopenia and polymorphonuclear inclusion bodies. They also had various combinations of nephropathy, eye abnormalities, high-tone sensorineural hearing loss, impaired liver functions and hypercholesterolemia. The clinical characteristics of the patients affected by Fechtner syndrome are described in table 1. Patients were recruited in Israel, Belgium, and the United States. In each country, the appropriate institutional review board approved the study, and informed consent was obtained from all participants. Each subject underwent a full physical and ophthalmologic examination, a hearing test, a complete blood count, kidney- and liver-function tests, a lipid profile, and Giemsa staining, under a light microscope, for the study of platelets and polymorphonuclear inclusion bodies.
Figure 1.
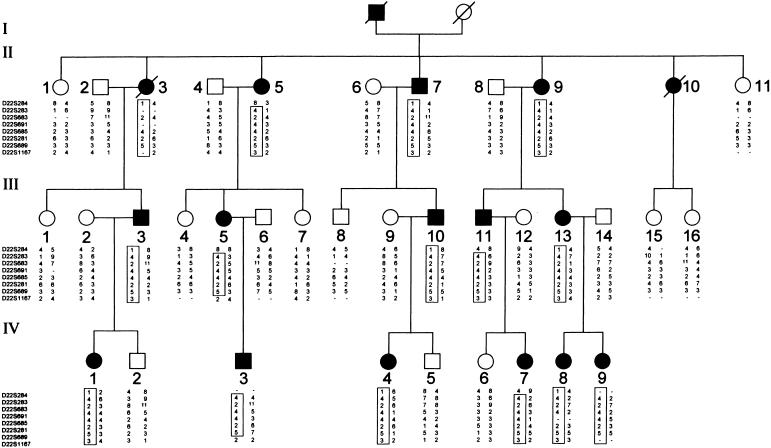
Pedigree and typing for eight chromosome 22 markers. Circles represent females, and squares represent males; unblackened symbols denote unaffected individuals, and blackened symbols denote affected individuals. Critical recombinations in individuals II-5, III-5, III-11, IV-3, and IV-7 define a 4-2-4-4-2-5 haplotype that is coinherited with the disease and that is not shared by the unaffected family members.
Table 1.
Clinical Characteristics of Patients Affected with Fechtner Syndrome[Note]
| Patient | Age(years) | Macrothrombocytopenia+ Inclusion Bodies | Deafness | EyeAbnormalities | Nephropathy | ImpairedLiver Function | Hypercholesterolemia |
| IV1 | 17 | + | − | + | − | + | − |
| IV3 | 15 | + | − | − | − | − | + |
| IV4 | 6 | + | − | − | − | + | + |
| IV7 | 18 | + | − | + | − | − | + |
| IV8 | 18 | + | − | − | − | − | − |
| IV9 | 13 | + | − | − | − | − | − |
| III3 | 50 | + | + | + | + | + | − |
| III5 | 45 | + | + | + | + | + | + |
| III10 | 40 | + | + | − | − | + | + |
| III11 | 43 | + | − | − | + | + | + |
| III13 | 44 | + | + | − | + | + | + |
| II3 | 62 | + | + | + | + | − | − |
| II5 | 74 | + | + | − | + | − | + |
| II7 | 72 | + | + | + | + | − | + |
| II9 | 70 | + | + | + | + | + | + |
| II10 | 55 | + | + | − | + | + | + |
Note.— A plus sign (+) denotes presence; a minus sign (−) denotes absence.
Genotyping
DNA was extracted from whole blood according to a standard phenol-chloroform protocol (Miller et al. 1988). A genomewide search was performed with the CHLC/Weber Human Screening set 8/8RG of polymorphic markers. Markers specifically described in this study include D22S1167, D22S689, D22S281, D22S685, D22S691, D22S683, D22S283, and D22S284 (Genome database). PCR reactions were done in a 15-μl reaction volume containing 150 ng of genomic DNA, 10 pmol of each unlabeled primer, 1.5 mM dNTPs (dCTP depleted), 0.1 μCi dCTP32, 1.5 mM MgCl2, 0.5 U Taq polymerase (Bioline UK), and PCR buffer containing 160 mM (NH4)SO4, 670 mM Tris HCl (pH 8.8), and 0.1% Tween 20. After an initial denaturation for 5 min at 94°C, 35 cycles were performed (94°C for 30 s, 55°C for 30 s, and 72°C for 30 s), followed by a final extension time for 5 min at 72°C. Samples were mixed with 10 μl of loading buffer, denatured at 94°C for 3 min, and electrophoresed on 6% denaturing polyacrylamide gel.
Linkage Analysis
Linkage was calculated with the LINKAGE package of computer programs (version 5.1; Lathrop et al. 1984). Patients with macrothrombocytopenia and polymorphonuclear inclusion bodies, with or without any other manifestations of the syndrome, were considered to be affected. The trait was assumed to be autosomal dominant, with a disease-allele frequency of .001 and penetrance of 1.00. Allele frequencies for the different microsatellite markers were obtained from 38 normal control individuals of Iraqi-Jewish origin (DNA supplied by the Genetic Department of the Sackler School of Medicine, Tel-Aviv University) and from the noncarrier chromosomes in this family.
Results
Eleven of 16 patients evaluated had mild hypercholesterolemia, with a mean value of 298 mg/100 ml, compared with a value of 165 mg/100 ml in the nonaffected members of the family (P<.05). There were typically high levels of LDL cholesterol and normal levels of HDL cholesterol. None of the patients had a nephrotic syndrome that could explain the hypercholesterolemia. Nine of 16 patients evaluated had mildly impaired liver functions: mean aspartate aminotransferase was 80 mg/100 ml (n<40), and mean alanine aminotransferase was 91 mg/100 ml (n<40).
Polymorphic microsatellite repeats from chromosomes 2q35-37 and 13q33-34 enabled us to rule out COL4A1, COL4A2, COL4A3, and COL4A4 as the disease-causing genes in this family. COL4A5 and COL4A6 reside on the X chromosome and therefore were not considered to be candidate genes in this family.
In a subsequent genomewide search using >100 microsatellites, linkage was detected with chromosome 22 markers. A schematic map of the region is shown in figure 2. Six markers from the long arm of chromosome 22 showed an LOD score >3.00. Pairwise LOD scores between Fechtner syndrome and chromosome 22 markers are presented in table 2. A maximum two-point LOD score (Zmax) of 7.02 was obtained with marker D22S283 at a maximum recombination fraction (θ) of 0. LOD-score calculations were performed with allele frequencies derived from 38 normal control individuals of Iraqi origin and from the noncarrier chromosomes of individuals in this family. We repeated the calculations for all markers, using published allele frequencies and equal allele frequencies. In all cases, LOD scores were robust to these changes in allele frequencies. Figure 1 shows typing results for this family, with eight chromosome 22 markers. Two recombinant events in affected family members (II-5 and III-11) established D22S284 as the telomeric boundary of the interval, whereas one such recombination (in individual III-5) defined D22S1167 as the centromeric boundary. No recombination events were detected with six other markers within this interval. Thus, the disease-causing gene maps to an 18-cM interval between markers D22S284 and D22S1167. According to established physical maps, this interval spans 5.5 Mb.
Figure 2.
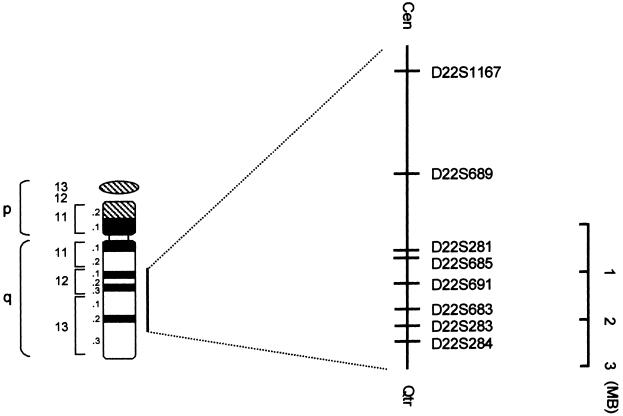
Schematic map of the interval containing the disease gene on chromosome 22. The markers are illustrated on the right.
Table 2.
Two-Point LOD Scores between Fechtner Syndrome and Chromosome 22 Markers
|
LOD
Score at θ
= |
|||||||||
| Marker | .00 | .05 | .10 | .15 | .20 | .25 | .30 | θmax | Zmax |
| D22S284 | −∞ | −7.65 | −4.63 | −2.99 | −1.93 | −1.20 | −.69 | .48 | .01 |
| D22S283 | 7.02 | 6.44 | 5.84 | 5.20 | 4.53 | 3.81 | 3.06 | .00 | 7.02 |
| D22S691 | 6.11 | 5.74 | 5.29 | 4.77 | 4.19 | 3.56 | 2.87 | .00 | 6.11 |
| D22S685 | 4.82 | 4.42 | 3.99 | 3.55 | 3.07 | 2.57 | 2.03 | .00 | 4.82 |
| D22S683 | 6.00 | 5.50 | 4.97 | 4.42 | 3.84 | 3.23 | 2.59 | .00 | 6.00 |
| D22S281 | 6.92 | 6.37 | 5.78 | 5.16 | 4.50 | 3.80 | 3.05 | .00 | 6.92 |
| D22S689 | 6.17 | 5.64 | 5.08 | 4.50 | 3.89 | 3.25 | 2.58 | .00 | 6.17 |
| D22S1167 | −∞ | 3.14 | 3.04 | 2.79 | 2.47 | 2.09 | 1.67 | .05 | 3.14 |
Discussion
We have demonstrated that, in the family described above, the disease-causing gene maps to chromosome 22q. Six markers on the long arm of chromosome 22 gave an LOD score >3.00. Haplotype analysis placed the disease gene in a 5.5-Mb interval between D22S284 and D22S1167.
During the last 3 decades, a number of families sharing Alport-like features and an autosomal-dominant mode of inheritance have been reported. A comparison between these families, the X-linked and recessive forms of Alport syndrome, is illustrated in table 3. Peterson et al. (1985) reported a four-generation family in which eight members had various combinations of nephritis, sensorineural hearing loss, congenital cataracts, macrothrombocytopenia, and cytoplasmatic polymorphonuclear inclusion bodies. They named the syndrome “Fechtner” after the index family. The inclusion bodies were similar to those seen, under a light microscope, in the May-Hegglin anomaly; however, under electron microscopy, they were found to contain spread filaments, ribosomes, and some segments of rough and smooth endoplasmatic reticulum. Gershoni-Baruch et al. (1988) reported a second family, with 16 affected individuals. These individuals resembled those described by Peterson et al., but, in addition, they had impaired liver functions and hypercholesterolemia. Rocca et al. (1993) described a third family with very similar symptoms. Epstein syndrome is yet another closely related condition, which manifests differently from the conditions of the families described above by the lack of polymorphonuclear inclusion bodies (Epstein et al. 1972; Eckstein et al. 1975; Bernheim et al. 1976; Parsa et al. 1976; Clare et al. 1979). No mutations in COL4A5 were found in two Japanese patients with this syndrome (Kawai et al. 1995). Sebastian platelet syndrome shares the hematological manifestations of Fechtner syndrome but lacks its renal, auditory, and ophthalmologic findings (Greinacher et al. 1990). It is currently unknown, for most of the autosomal-dominant families presented in table 2, whether the disease-causing gene maps to chromosomes 2, 13, or 22 or to other, yet-undetermined loci.
Table 3.
Clinical Manifestations of the Various Alport-Like Syndromes
| Syndrome and Inheritance | Mutated Gene/Locus | Macrothrombocytopenia | Inclusion Bodies | Nephropathy | Deafness | EyeAbnormalities | Impaired Liver Function | Hypercholesterolemia |
| Alport | ||||||||
| X-linked | COL4A5, COL4A6 | − | − | + | + | + | − | − |
| Autosomal recessive | COL4A3, COL4A4 | − | − | + | + | + | − | − |
| Autosomal dominant | COL4A3(?), COL4A4(?) | − | − | + | + | + | − | − |
| May-Hegglin | ||||||||
| Autosomal dominant | ? | + | +a | − | − | − | − | − |
| Epstein | ||||||||
| Autosomal dominant | ? | + | − | + | + | + | − | − |
| Fechtnerb | ||||||||
| Autosomal dominant | Chromosome 22q11-13 | + | +a | + | + | + | + | + |
| Sebastian | ||||||||
| Autosomal dominant | ? | + | +a | − | − | − | − | − |
The three families with Fechtner syndrome that have been described so far differ slightly from each other, as described in the text.
The ultrastructure of the inclusion bodies in Fechtner syndrome resemble those in Sebastian syndrome and are different from those seen in the May-Hegglin anomaly.
An update of the data presented by Gershoni-Baruch et al. (1988) shows more convincingly that there is a progressive deterioration of all tissues involved in the disease. The children usually present with hematological manifestations, whereas the adults show the full-blown phenotype by the 5th decade.
Other congenital disorders may be associated with macrothrombocytopenia or inclusion bodies in the neutrophils. The gene coding for glycoprotein 1b-β, localized in chromosome 22q11.2 (a few million bases from the area containing the disease-causing gene in our family), was recently found to be deleted in another macrothrombocytopenic state, Bernard-Soulier syndrome (Budarf et al. 1995; MIM 231200). The gene for Chediak-Higashi syndrome immune deficiency, characterized by inclusion bodies made of giant lysosomes in the granulocytes in addition to partial oculocutaneous albinism, was localized to 1q42-43 (Barrat et al. 1996). It may be hypothesized that the Fechtner gene is similar or homologous to one of these genes.
The 5.5-Mb interval on chromosome 22q, to which the disease-causing gene maps, contains 91 known genes, none of which encode collagen. Moreover, none of them encode a protein that is part of a basement membrane, such as laminin, entactin, heparin sulfate proteoglycans, or other glycoproteins, that may be expressed in a wide variety of tissues and that could account for the diverse symptomatology and laboratory abnormalities seen in Fechtner syndrome. This region, however, also contains 60 expressed-sequence tags, whose function and tissue expression are unknown. Although Fechtner syndrome is very rare, the wide variety of tissues involved in this disease suggests that the cloning of this gene may have a bearing on the understanding of other genetic and nongenetic diseases in which these specific tissues are involved.
Acknowledgments
We are grateful to Rabbi Elimelech Firer, for helping us obtain the blood samples from overseas members of the family, and to Mrs. Anna Gorshnik, for excellent technical assistance.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Genome Database, http://gdbwww.gdb.org (for markers for microsatellite studies)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for Alport syndrome [MIM 153640], Fechtner syndrome [MIM 153640], Epstein syndrome [MIM153650], and Bernard-Soulier syndrome [MIM 231200.0001])
References
- Alport AC (1927) Hereditary familial congenital hemorrhagic nephritis. Br J Med 1:504–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antignac C, Zhou J, Sanak M, Cochat P, Roussel B, Deschenes G, Gros F, et al (1992) Alport syndrome and diffuse leiomyomatosis: deletions in the 5′ end of the COL4A5 gene. Kidney Int 42:1178–1183 [DOI] [PubMed]
- Barrat FJ, Auloge L, Pastural E, Dufourcq Lagelouse R, Vilmer E, Cant AJ, Weissenbach J, et al (1996) Genetic and physical mapping of the Chediak-Higashi syndrome on chromosome 1q42-43. Am J Hum Genet 59:625–632 [PMC free article] [PubMed]
- Bernheim J, Dechavanne M, Byron PA, Lagards M, Colon S, Pozet N, Traeger J (1976) Thrombocytopenia, macrothrombopathia, nephritis and deafness. Am J Med 61:145–150 [DOI] [PubMed]
- Boyd CD, Toth-Fejel S, Dadi IK, Litt M, Condon MR, Kolbe M (1988) The genes coding for human pro alpha 1(IV) and pro alpha 2(IV) collagen are both located at the end of the long arm of chromosome 13. Am J Hum Genet 42:309–314 [PMC free article] [PubMed]
- Boye E, Vetrie D, Flinter F, Buckle B, Pihlajaniemi T, Hamalainen ER, Myers JC, et al (1991) Major rearrangements in alpha 5(IV) collagen gene in three patients with Alport syndrome. Genomics 11:1125–1132 [DOI] [PubMed]
- Budarf ML, Konkle B, Ludlow LB, Michaud D, Li M, Yamashiro DJ, McDonald-Mcginn D, et al (1995) Identification of a patient with Bernard-Soulier syndrome and a deletion in the DiGeorge/velo-cardio-facial chromosomal region in 22q11.2. Hum Mol Genet 4:763–766 [DOI] [PubMed]
- Churg J, Sherman RL (1973) Pathology of hereditary nephritis. Arch Pathol 95:374–379 [PubMed]
- Clare NM, Montiel MM, Lifschitz MD, Bannayan GA (1979) Alport's syndrome associated with macrothrombatic thrombocytopenia. Am J Clin Pathol 72:111–117 [DOI] [PubMed]
- Eckstein JD, Filip DJ, Watts JC (1975) Hereditary thrombocytopenia, deafness, and renal disease. Ann Intern Med 82:639–645 [DOI] [PubMed]
- Epstein CJ, Shaud MA, Piel CF, Goodman JR, Bernfield MR, Kushner JH, Ablin AR (1972) Hereditary macrothrombocytopenia, nephritis and deafness. Am J Med 52:299–310 [DOI] [PubMed]
- Gershoni-Baruch R, Baruch Y, Viener A, Lichtig C (1988) Fechtner syndrome: clinical and genetic aspects. Am J Med Genet 31:357–367 [DOI] [PubMed]
- Greinacher A, Nieuwenhuis HK, White JC (1990) Sebastian platelet syndrome: a new variant of hereditary macrothrombocytopenia with leukocyte inclusions. Blut 61:282–288 [DOI] [PubMed]
- Hegglin R (1945) Gleichzeitige konstitutionelle Veranderungen an Neutrophilen und Thrombozyten. Helv Med Acta 12:439–440 [PubMed] [Google Scholar]
- Heidet L, Dahan K, Zhou J, Xu Z, Cochat P, Gould JDM, Leppig KA, et al (1995) Deletions of both alfa5(VI) and alfa6(IV) collagen genes in Alport syndrome and in Alport syndrome associated with smooth muscle tumors. Hum Mol Genet 4:99–108 [DOI] [PubMed]
- Hostikka SL, Eddy RL, Byers MG, Hoyhtya M, Shows TB, Tryggvason K (1990) Identification of a distinct type IV collagen alpha chain with restricted kidney distribution and assignment of its gene to the locus of X chromosome-linked Alport syndrome. Proc Natl Acad Sci USA 87:1606–1610 [DOI] [PMC free article] [PubMed]
- Jefferson JA, Lemmink HH, Hughes AE, Hill CM, Smeets HJ, Doherty CC, Maxwell AP (1997) Autosomal dominant Alport syndrome linked to type IV collagen alpha 3 and alpha 4 genes (COL4A3 and COL4A4). Nephrol Dial Transplant 12:1595–1599 [DOI] [PubMed]
- Kawai S, Nomura S, Harno T, Harno K, Fukushima T, Osawa G (1995) The COL4A5 gene in Japanese Alport syndrome patients: spectrum of mutations of all exons. Kidney Int 49:814–822 [DOI] [PubMed]
- Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446 [DOI] [PMC free article] [PubMed]
- Lemmink HH, Mochizuki T, Van Den Heuvel LPWJ, Schroder CH, Barrientos A, Monnens LAH, van Oost BA, et al (1994) Mutations in the type IV collagen alfa3 (COL4A3) gene in autosomal recessive Alport syndrome. Hum Mol Genet 3:1269–1273 [DOI] [PubMed]
- Mariyama M, Zheng K, Yang-Feng TL, Reeders ST (1992) Colocalization of the genes for the alpha 3(IV) and alpha 4(IV) chains of type IV collagen to chromosome 2 bands q35-q37. Genomics 13:809–813 [DOI] [PubMed]
- Miller SA, Dykes DD, Plesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215 [DOI] [PMC free article] [PubMed]
- Mochizuki T, Lemmink HH, Mariyama M, Antignac C, Gubler MC, Pirson Y, Verellen-Dumoulin C, et al (1994) Identification of mutations in the alfa3(IV) and alfa4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 8:77–82 [DOI] [PubMed]
- Myers JC, Jones TA, Pohjolainen ER, Kadri AS, Goddard AD, Sheer D, Solomon E, et al (1990) Molecular cloning of the alpha 5 (IV) collagen and assignment of the gene to the region of the X chromosome containing the Alport syndrome locus. Am J Hum Genet 46:1024–1033 [PMC free article] [PubMed]
- Nakazato H, Hattori S, Matsuura T, Koitabashi Y, Endo F, Matsuda I (1993) Identification of a single base insertion in the COL4A5 gene in Alport syndrome. Kidney Int 44:1091–1096 [DOI] [PubMed]
- Netzer KO, Renders L, Zhou J, Pullig O, Tryggvason K, Weber M (1992) Deletions of the COL4A5 gene in patients with Alport syndrome. Kidney Int 42:1336–1344 [DOI] [PubMed]
- Nomura S, Osawa G, Sai T, Harano T, Harano K (1993) A splicing mutation in the alpha 5(IV) collagen gene of a family with Alport's syndrome. Kidney Int 43: 1116–1124 [DOI] [PubMed]
- Parsa KP, Lee DBN, Zamboni L, Glassock RJ (1976) Hereditary nephritis, deafness and abnormal thrombopoiesis: study of a new kindred. Am J Med 60:665–672 [DOI] [PubMed]
- Peterson LC, Rao KV, Crosson JT, White JG (1985) Fechtner syndrome: a variant of Alport's syndrome with leukocyte inclusions and macrothrombocytopenia. Blood 65:397–406 [PubMed]
- Rocca B, Laghi F, Zini G, Maggiano N, Landolfi R (1993) Fechtner syndrome: report of a third family and literature review. Br J Haematol 85:423–426 [DOI] [PubMed]
- Spear GS (1973) Alport syndrome: a consideration of pathogenesis. Clin Nephrol 1:336–337 [PubMed]
- Spear GS, Slusser RJ (1972) Alport's syndrome: emphasizing electron microscopic studies of the glomerulus. Am J Pathol 69:213–222 [PMC free article] [PubMed]
- Tryggvason K, Zhou J, Hostikka SL, Shows TB (1993) Molecular genetics of Alport syndrome. Kidney Int 43:38–44 [DOI] [PubMed]
- Zhou J, Mochizuki T, Smeets H, Antignac C, Laurila P, DE Paepe A, Tryggvason K, et al (1993) Deletions of the paired alfa5(IV) and alfa6(IV) collagen genes in inherited smooth muscle tumors. Science 261:1167–1169 [DOI] [PubMed]