

To the Editor:
Congenital nephrotic syndrome is a clinically and genetically heterogeneous disorder of the glomerular filtration barrier, characterized by massive proteinuria at or shortly after birth. Although nephrotic syndromes encompass a heterogeneous group of disorders, congenital nephrotic syndrome of the Finnish type (NPHS1 [MIM 256300]) is a distinct clinical entity. NPHS1 has an autosomal recessive mode of inheritance and is generally rare—except in Finland, where the incidence is ∼1/10,000 live births (Norio 1966). Nephrin, a putative transmembrane protein belonging to the immunoglobulin family of cell-adhesion molecules, has been identified as the gene mutated in NPHS1, with loss-of-function deletion and missense mutations identified in Finnish and other white patients with NPHS1 (Kestilä et al. 1998; Lenkkeri et al. 1999). Several cases of NPHS1 have also been observed in other populations, with or without direct evidence of Finnish ancestry (Fuchshuber et al. 1996). On the basis of limited haplotype analysis, it has been argued that some cases of non-Finnish NPHS1 may have a common origin in Finland (Männikkö et al. 1996).
We observed a high incidence of NPHS1 among the Old Order Mennonites in Lancaster County, Pennsylvania. In particular, we identified 26 cases of NPHS1, dating from the 1950s, in a very large inbred Mennonite kindred (population). Interestingly, all but one of the cases of NPHS1 in Mennonites occurred in a subgroup known as the “Groffdale Conference” Mennonites. The Groffdale Conference Mennonites formed as a result of a schism in the Weaverland Conference Mennonites of Lancaster County in 1927. The separation between the two groups was based on differing views regarding acculturation to American thought and society. The more conservative group formed the Groffdale Conference, whereas the more progressive members continued in the Weaverland Conference. At present, the populations and birth rates of the two conferences are approximately equal.
We estimated the incidence of NPHS1 among the Groffdale Conference to be .002 or 1/500 live births, on the basis of the complete ascertainment of eight cases among 4,062 births during the period 1985–94. This incidence is 20 times greater than that observed in Finland and predicts that ∼8% of Groffdale Mennonites are carriers of the NPHS1-causing allele. The ancestors of this Old Order group immigrated from Switzerland to Lancaster County during the early 18th century, and no explicit Finnish ancestry is known. In this study, we (1) confirm the role of nephrin in NPHS1, (2) show that a major mutation is shared by families with NPHS1 that are in the Groffdale Conference, and (3) show that this mutation is most likely of recent origin—uncovered by inbreeding and amplified by genetic drift. Our data also suggest that the major Mennonite mutation most likely predated the Weaverland-Groffdale split, since one Weaverland NPHS1 proband is a double heterozygote with one copy of the major nephrin mutation as well as a second novel mutation, possibly contributed through a non-Mennonite lineage.
We initially ascertained four families, in each case through a proband with NPHS1. The four families had 8 obligate carrier parents and 11 unaffected offspring. Only one of the probands from these four families was alive during the study. Subsequent to our analysis of these four families, one additional family—which included a second surviving proband, parents, and paternal grandparents—was ascertained. After our mutation-detection studies were completed, three additional families with NPHS1 agreed to participate.
One of the two living Mennonite probands with NPHS1 (6986) was less-severely affected than the other patients. This patient underwent nephrectomy at age 18 mo, followed by nightly peritoneal dialysis and a renal transplant at age 3.5 years. Prior to nephrectomy, this child had an array of life-threatening problems, including recurrent renal-vein thrombosis, severe hypertension, encephalitis, hypothyroidism, refractory anemia, hypercholesterolemia, edema, ascites, and malnutrition. The second surviving Mennonite child was 30 mo of age at the time of our study. She had minimal medical interventions until age 2 years, at which time she was started on the Finnish protocol of enalapril, thyroid replacement, vitamin D, and erythropoetin (Guez et al. 1998). Within 4 wk of the start of enalapril therapy, her serum albumin increased from 0.6 to 2.1 g/dl and her generalized edema resolved. If she survives until she obtains a weight of 10–12 kg, renal transplantation will be reconsidered.
For genetic analysis, we obtained, with informed consent, either peripheral blood samples or buccal swabs from members of the eight families. Genomic DNA was extracted according to standard procedures. No pathology samples were available from the deceased affected individuals, to perform additional analyses. Microsatellite-marker alleles were amplified and genotyped via standard PCR methods (Puffenberger et al. 1994). Allele sizes for all markers (except D19S608, D19S609, and D19S610) were determined in comparison with CEPH individual 1347-02; allele numbers for D19S608, D19S609, and D19S610 were assigned arbitrarily. To determine the genomic structure of nephrin, we used the “BLAST 2-sequences” option of the program BLAST (Tatusova et al. 1999), to align nephrin cDNA sequence and overlapping genomic sequence from cosmid R33502 (GenBank AFO35835 and AC002133). The gene consists of 29 exons and spans ∼26 kb; primer sequences, from the program Oligo, version 4.0, were designed for PCR amplification of all 29 exons in 19 reactions (table 1). For mutation analysis, nephrin exons were PCR amplified from one patient (3898), the patient's father (3833), and a CEPH control DNA. After PCR amplification, products were sequenced on an ABI377 sequencer (PE Biosystems), with the use of standard dye-terminator chemistry.
Table 1.
Oligonucleotide Primers for Amplification of NPHS1 Exons
| Exon(s) | Primer Sequences | Annealing Temperature (oC) | Product Size (bp) |
| 1, 2 | Forward: AGACGCAAGGTGGCTGGCAGCG | 60 | 474 |
| Reverse: TTCCGCTGGTGGCTGAGGGTC | |||
| 3, 4 | Forward: AGCGGAGCTGCGGCCCTGACT | 63 | 520 |
| Reverse: CCTTCCCACTCCAGAGGCTTCA | |||
| 5 | Forward: TCGCACCCACCCTGAGGACTTC | 60 | 252 |
| Reverse: ATGCTTGCATCCCTGGGGTCTG | |||
| 6 | Forward: CCCCACCTCTTCTCCCTGACT | 60 | 246 |
| Reverse: ATCCCCCCACACCCCCCAGTG | |||
| 7, 8 | Forward: GGAAAGAGTGGATGGGCTACT | 60 | 486 |
| Reverse: CTCTGAGGCACAGACCGACAG | |||
| 9 | Forward: TCTGGGCTGGTCTGTGAGAAA | 55 | 264 |
| Reverse: TCAGCCCCCTCCATGCTCAGA | |||
| 10, 11 | Forward: GTGCTGCAGTCCCCACGTCTGReverse: CATTCCTGGCCACCCCCATAG | 60 | 435 |
| 12 | Forward: TGCTGATGAGAGTGCTTCTCC | 60 | 467 |
| Reverse: CACCCCAGGCTCCGCCCAGTC | |||
| 13 | Forward: CTGGGCGGAGCCTGGGGTGCA | 60 | 308 |
| Reverse: CAGAGGCTGGAGAGGCACTAG | |||
| 14 | Forward: ATCCCTCCCCTCTCTGGTCTG | 60 | 356 |
| Reverse: AAGGTAAGACCCAAGGAGTAG | |||
| 15, 16 | Forward: AACCTTAAACCCCGTCGTGAC | 60 | 543 |
| Reverse: CAATGAGGAGACTCCACAATG | |||
| 17 | Forward: AACCTTCCTGGAACCCCTAAG | 57 | 300 |
| Reverse: CACTCCCAAGGAACTCACAGT | |||
| 18, 19 | Forward: GTGATGGATCTGGGGCTAGAC | 59 | 558 |
| Reverse: AGGGACTCAGGGAGGGGAAGT | |||
| 20 | Forward: GATAGATAGGCAGACGGTTAC | 55 | 382 |
| Reverse: AACTCCATCCTCACACATACA | |||
| 21, 22 | Forward: TCTTCTGGAATTCTTTTGTAT | 55a | 474 |
| Reverse: TACACATCCTCTGAGGAATAC | |||
| 23 | Forward: ACCAGTTCCCATGAATCTAATA | 55 | 262 |
| Reverse: GTCGTTAAGCAGCTGTGACTAC | |||
| 24–26 | Forward: CAGCCTGTTGTCTGGGATTC | 60 | 610 |
| Reverse: GCTTCAGTCGCCGTCGGTGC | |||
| 27, 28 | Forward: TCCGGGCACAGTGGGGTCAGAG | 59 | 454 |
| Reverse: GCAGCTAGCTGGCCCTAACTAA | |||
| 29 | Forward: TCCATGGTTCTAACTCACTT | 60 | 580 |
| Reverse: GCACTTAGGGACCCTCAGAATAG |
Requires 10% DMSO.
We expected that any NPHS1 candidate locus will have a unique haplotype in Mennonites, which is homozygous in affected individuals, heterozygous in the 8 obligate heterozygote parents, and not homozygous in the 11 unaffected siblings (fig. 1 shows all individuals but the unaffected siblings). Using 10 microsatellite markers, Männikkö et al. (1995) mapped NPHS1 to chromosome 19q13.1 by linkage and linkage-disequilibrium analysis in Finnish patients. We genotyped these same markers in the 20 available individuals and identified a common haplotype (table 2) for five markers in the 3.5-cM interval D19S609–D19S220, satisfying the above criteria and implicating nephrin in Mennonite NPHS1. It is unclear whether any of the Finnish NPHS1 haplotypes are in common with Mennonite NPHS1 haplotypes, since Kestilä et al. (1998) have not published actual allele sizes at the marker loci studied.
Figure 1.
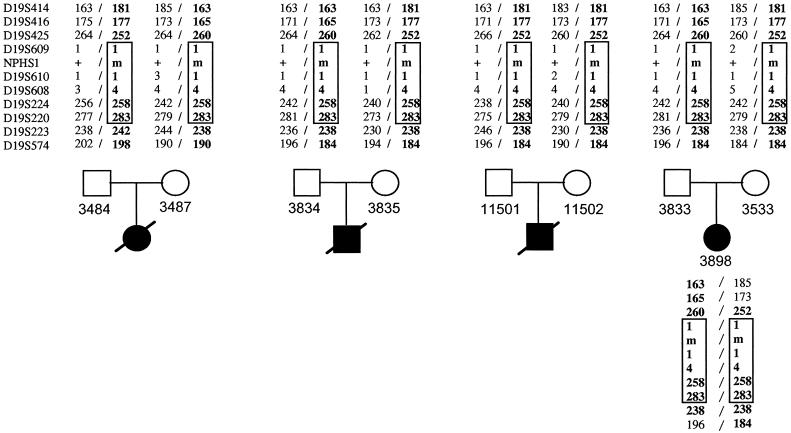
Haplotype analysis of four Mennonite nuclear families with NPHS1. Chromosome 19q13 haplotypes, of the markers shown in table 2, of NPHS1-transmitting parents and homozygosity for a haplotype in proband 3898 are shown. None of the 11 unaffected siblings from the four families were homozygous for the identified haplotype (data not shown). The common mutation, 1481delC, segregates with the haplotype designated in boldface.
Table 2.
Mennonite NPHS1 Mutant-Allele–Carrying Haplotypes
| Markera | Intermarker Distance (cM)b | Haplotypec | |||
| D19S414 | 4.9 | 181 | 163 | 163 | 181 |
| D19S416 | .6 | 177 | 165 | 165 | 177 |
| D19S425 | 1.0 | 252 | 260 |
260 |
252 |
| D19S609 | 1 | 1 | 1 | 1 | |
| D19S610 | 3.0 | 1 | 1 | 1 | 1 |
| D19S608 | 4 | 4 | 4 | 4 | |
| D19S224 | .5 | 258 | 258 | 258 | 258 |
| D19S220 | 2.2 | 283 | 283 | 283 | 283 |
| D19S223 | 5.6 | 238 | 238 | 238 |
242 |
| D19S574 | 184 |
184 |
190 | 198 | |
Markers in the segment common to all haplotypes and that implicate nephrin in NPHS1 span a genetic distance of ⩾3.5 cM and are underlined.
Between the marker shown and that immediately below (except for “3.0,” which denotes the distance spanned by the four markers underlined).
The boxed portion indicates a common haplotype segment shared by all parents of the affected individuals whom we studied. The number of copies is four, two, one, and one, respectively.
Nucleotide sequencing of all nephrin exons (table 1) from one proband revealed the deletion of one nucleotide within exon 12 (1481delC), which segregated with the NPHS1 haplotypes identified. In addition to the families shown in figure 1, six parents from three additional Mennonite sibships with NPHS1 were genotyped for the 1481delC mutation; all were heterozygous. This deletion mutation is novel (Kestilä et al. 1998) and leads to a frameshift and premature truncation of the protein, to 547 residues. Nephrin is a 1,241-residue protein predicted to be a cell-adhesion receptor and/or a signaling protein. Its predicted structure includes eight Ig-like domains and one fibronectin-like domain, adjacent to a transmembrane region. The truncation caused by the Mennonite mutation occurs well before the putative transmembrane domain (residues 1059–1068); therefore, it follows that 1481delC is very likely a null allele and that homozygotes have no functional protein.
Subsequent to the identification of 1481delC, we ascertained the only other living Mennonite proband (6986 [fig. 2]) and his immediate family. Interestingly, this patient was found to be heterozygous for 1481delC and inherited the allele maternally. After nephrin sequence analysis of this patient and his father, we detected a second frameshift mutation, 3250delG, for which both were heterozygous. Genealogical analysis revealed that proband 6986 has a partially non-Mennonite lineage; his maternal great-grandmother married into the Mennonite kindred (fig. 2). We were able to trace the second mutation, 3250delG, to the patient's paternal grandfather by DNA analysis. Since the proband's non-Mennonite great-grandmother is deceased, we could not establish whether she contributed this mutation or whether the occurrence of 3250delG is a new mutation in the proband's grandfather. Nonetheless, we identified a second unique mutation, which also results in premature truncation of nephrin protein, resulting in a 1,141-residue protein. According to the predicted structure of the protein, it is likely that the 1,141-residue protein inserts into the membrane but lacks the complete cytosolic domain. Thus, some residual nephrin function may exist and may explain the less-severe phenotype of this patient.
Figure 2.
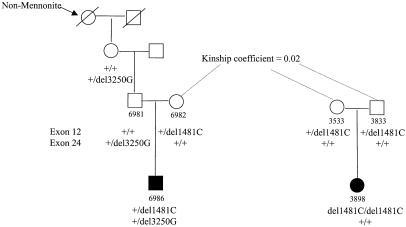
Genealogy of two nephrin-deletion mutations. The single Mennonite proband who is doubly heterozygous and who inherited the common Mennonite mutation and a second mutation from a non-Mennonite ancestor is shown. The kinship coefficient of the ancestors shown clarifies that this proband is closely related to other Mennonites with NPHS1.
Our results confirm the role of nephrin in NPHS1 and allelic heterogeneity within the Mennonites. NPHS1 is very rare throughout the world, and its low incidence is likely under mutation-selection balance; for a recessive lethal mutation, the incidence would equal its de novo mutation rate per generation, which has a mean value of ∼10−6 (Cavalli-Sforza and Bodmer 1971). The ∼1/10,000 incidence in Finland is two orders of magnitude larger and is likely to have increased by genetic drift and founder effects during the past 100 generations (2,000 years) of Finnish history. Moreover, the occurrence of at least three distinct mutations, each on a specific marker haplotype with estimated frequencies of 0.78%, 0.16%, and 0.06% (Kestilä et al. 1998), suggests that the age of the more frequent mutation is not recent. This is corroborated by the observation of linkage disequilibrium, over a small distance (∼150 kb), for the major Finnish NPHS1 mutation (Kestilä et al. 1998). As we have shown here, these effects are further amplified in the Old Order Mennonites, which have a much more recent history and a much smaller effective population size.
In this study, we have estimated the Mennonite NPHS1 incidence at ∼1/500 live births, or an overall 20-fold increase compared with the Finnish incidence. This large increase is undoubtedly from genetic drift and founder effects (Chakravarti and Chakraborty 1978), consistent with the finding of a major common mutation (1481delC) in a population with an estimated effective number of ∼200 (A. Chakravarti and E. G. Puffenberger, unpublished data). We argue that the 1481delC mutation is likely of very recent origin. Table 1 shows that the core NPHS1 haplotype, on which the 1481delC mutation is found, is shared by all eight mutant chromosomes and is ⩾3.5 cM; however, haplotypes 1 and 4 (five copies total) and haplotypes 2 and 3 (three copies total) each share the longer D19S414–D19S220 haplotype, of 10 cM. Therefore, the average time to origin of the common mutation in the Mennonites is ∼1/0.1 morgans, or ∼10 generations. It is possible either that this was a rare mutation in the European Mennonite groups and that it expanded in the United States or that the mutation is of recent U.S. origin.
As mentioned above, the distribution of NPHS1 is not random within the Old Order Mennonite community and is largely restricted to the Groffdale Conference. The Groffdale and Weaverland Conferences share very recent ancestry, yet the distribution and allelic spectrum of NPHS1 mutations are not equal within these two groups. At present, only 1 case of NPHS1 has been observed in the Weaverland Mennonites, whereas 25 cases have been identified in the Groffdale Mennonites since the 1950s. We have found that the single Weaverland Conference patient with NPHS1 (6986) is a compound heterozygote for the major Mennonite mutation, 1481delC (same core haplotype as in table 2), and a second mutation, 3250delG. Our analysis shows that 1481delC is the only mutation segregating in seven nuclear families from the Groffdale Conference (fig. 2). It is interesting that the origin of 3250delG is non-Mennonite, since it is inherited through the non-Mennonite ancestors of proband 6986. These data emphasize the rarity of NPHS1 mutations in most populations, identified here among non-Mennonites only when combined with a common Mennonite mutation. Moreover, the concentration of 1481delC in the Groffdale Mennonites suggests that NPHS1 occurs at an incidence much higher than 1/500. The incidence within each conference cannot as yet be determined with any greater precision.
The accumulation of NPHS1 in the Groffdale Conference and the rarity of 1481delC in the Weaverland Conference can be explained in multiple ways. First, a subfounder effect might explain the high incidence within the Groffdale group. However, genealogical and mutational analyses do not support this hypothesis, since there is no common ancestor for all carriers of NPHS1 who were born after the 1927 schism and 1481delC is present in the Weaverland Mennonite population. Second, 1481delC may have arisen in the Groffdale Mennonites and may have been reintroduced into the Weaverland Conference. Since marriage between the groups is extremely rare, it is unlikely, although not impossible, that the mutation was contributed to the Weaverland Conference after the 1927 split. An alternative and simpler explanation is that 1481delC arose prior to the split and that it has increased in frequency through genetic drift. The formation of the Groffdale Conference by family groups wishing to preserve a simpler lifestyle may have led, by chance, to a relative increase in the mutant-allele frequency in one conference compared with the other.
It is surprising that a recent mutation has achieved such a high frequency in a short time. This is likely a result of the intense inbreeding that occurs within the Old Order Mennonites in each generation—not from high rates of social consanguinity but from the limited ancestral pool and the sharing of multiple common ancestors between any two individuals. We have established a genealogical database of >7,500 individuals from the Old Order Mennonites that allows direct calculation of kinship coefficients between any two individuals, equivalently the inbreeding coefficient for their offspring. We studied 12 pairs of parents with complete genealogies and with an offspring affected with NPHS1; the average kinship coefficient was .020 (range .010–.037), corresponding to a relationship between first cousins once removed and second cousins. For the four pairs of parents that we report here, the average kinship coefficient was .017 (range .014–.019). These rates are relevant for most individuals in the last several generations. Consequently, any de novo mutation occurring in the recent past has a high chance of becoming homozygous, through inbreeding, in the Mennonites. This is in contrast to outbred populations, in which, with random mating, rare mutants become homozygous only rarely.
Acknowledgments
We are very indebted to the members of the numerous families with nephrotic syndrome, for sharing information and donating tissue samples; without these families this work would have been impossible. We gratefully acknowledge the superb assistance of Jennifer Scott, for family studies; Kimberly Bentley, Andrew Wong, and Gilbert Wong, for DNA sequencing and technical assistance; and Carl Kashuk, for informatics. This work was supported by National Institutes of Health grant HD28088 to A.C.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- BLAST, http://www.ncbi.nlm.nih.gov/BLAST (for alignment of sequences)
- GenBank, http://www.ncbi.nlm.nih.gov/Web/Search (for nephrin cDNA [accession number AF035835] and for cosmid R33502 [accession number AC002133])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for NPHS1 [MIM 256300])
References
- Cavalli-Sforza LL, Bodmer WF (1971) The genetics of human populations. WH Freeman, San Francisco [Google Scholar]
- Chakravarti A, Chakraborty R (1978) Elevated frequency of Tay-Sachs disease among Ashkenazic Jews unlikely by genetic drift alone. Am J Hum Genet 30:256–261 [PMC free article] [PubMed]
- Fuchshuber A, Niaudet P, Gribouval O, Jean G, Gubler M, Broyer M, Antignac C (1996) Congenital nephrotic syndrome of the Finnish type: linkage to the locus in a non-Finnish population. Pediatr Nephrol 10:135–138 [DOI] [PubMed]
- Guez S, Giani M, Melzi ML, Antignac C, Assael BM (1998) Adequate clinical control of congenital nephrotic syndrome by enalapril. Pediatr Nephrol 12:130–132 [DOI] [PubMed]
- Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, et al (1998) Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1:575–582 [DOI] [PubMed]
- Lenkkeri U, Männikkö M, McCready P, Lamerdin J, Gribouval O, Niaudet PM, Antignac C, et al (1999) Structure of the gene for congenital nephrotic syndrome of the Finnish type (NPHS1) and characterization of mutations. Am J Hum Genet 64:51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Männikkö M, Kestilä M, Holmberg C, Norio R, Ryynänen M, Olsen A, Peltonen L, et al (1995) Fine mapping and haplotype analysis of the locus for congenital nephrotic syndrome on chromosome 19q13.1. Am J Hum Genet 57:1377–1383 [PMC free article] [PubMed]
- Männikkö M, Lenkkeri U, Kashtan CE, Kestilä M, Holmberg C, Tryggvason K (1996) Haplotype analysis of congenital nephrotic syndrome of the Finnish type in non-Finnish families. J Am Soc Nephrol 7:2700–2703 [DOI] [PubMed]
- Norio R (1966) Heredity of the congenital nephrotic syndrome. Ann Paediatr Fenn 12 Suppl 27:1–94 [PubMed] [Google Scholar]
- Puffenberger EG, Kauffman ER, Bolk S, Matise TC, Washington SS, Angrist M, Weissenbach J, et al (1994) Identity-by-descent and association mapping of a recessive gene for Hirschsprung disease on human chromosome 13q22. Hum Mol Genet 3:1217–1225 [DOI] [PubMed]
- Tatusova TA, Madden TL (1999) Blast 2 sequences: a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett 174:247–250 [DOI] [PubMed]