

To the Editor:
Tuberous sclerosis complex (TSC [MIM 191092]), a dominantly inherited disease with a prevalence of 1/6,700 (Sampson et al. 1989; Osborne et al. 1991; Ahlsen et al. 1994) is characterized by hamartomas found in almost every organ system and by malignancy in some organ systems. Two genetic loci have been identified: the TSC1 gene, on chromosome 9q34.3, and TSC2, on chromosome 16p13.3 (The European Chromosome 16 Tuberous Sclerosis Consortium 1993; van Slegtenhorst et al. 1997). A tumor-suppressor function has been suggested for TSC2 by loss of heterozygosity (LOH) in hamartomas (Green et al. 1994; Henske et al. 1995, 1996; Carbonara et al. 1996; Sepp et al. 1996). Thus far, no reports have demonstrated mutations in both alleles of the TSC2 gene in hamartomas in humans. Further evidence of a tumor-suppressor function for TSC2 comes from the Eker rat, a naturally occurring model of tumorigenesis. The Eker rat has been determined to have a germline mutation in the Tsc2 gene (the homolog of the TSC2 gene in humans) (Yeung et al. 1994; Kobayashi et al. 1995; Kubo et al. 1995). LOH and second somatic hit studies of the Tsc2 gene have been reported in 33% of spontaneous or chemically induced Eker rat renal-cell carcinoma (RCC) lines (Kobayashi et al. 1997). The mechanism for cell transformation in humans is expected to be different from that in rats, because RCCs are detectable in 100% of Eker rats by age 1 year, whereas only a small percentage (2.5%) of humans with TSC develop RCCs (Eker et al. 1981; Bjornsson et al. 1996; Cook et al. 1996; Al-Saleem et al. 1998). We collected a variety of hamartomas—angiomyolipomas (AMLs) from three patients, cortical tubers from two patients, and facial angiofibromas (FAs) from one patient—from patients in whom the germline TSC2 mutation had been identified (Au et al. 1998). These tissues were tested for the second hit by Southern blotting, with the TSC2 gene as a probe, and by LOH studies of the TSC2 gene and surrounding markers. The second hit for the TSC2 gene was detected in AMLs from two independent patients and in FAs from another patient, providing direct evidence that mutations in both alleles are required for tumorigenesis in humans affected by TSC (Knudson 1971).
The patients in this study have been described elsewhere (Au et al. 1998). All were diagnosed with TSC according to standard criteria by Roach et al. (1998). Informed consent approved by the institutional review boards at The University of Texas Medical School–Houston, The University of Texas Southwestern Medical School, and the Scottish Rite Hospital in Dallas was obtained. Tissue samples collected included 6 AMLs from the right kidney and 1 AML from the left kidney of patient HOU23-01, 1 AML from patient HOU23-03, 14 AMLs from patient TS94-104, 3 FAs from patient TS93-41, and 1 cortical tuber each from patients TS93-14 and TS94-53.
DNA from the blood lymphocytes of the patients was prepared as previously described. Small samples of tissue were removed from the well-circumscribed tumors at the time of surgery. Fresh tumor tissue weighing ∼0.2 g (or the whole tumor, if it was <0.2 g) was dissected, minced, and rinsed with phosphate-buffered saline to remove red blood cells. The minced tissue was digested with 1 mg proteinase K/ml in 10 ml of lysis buffer, with remaining processing being the same as that for lymphocyte DNA.
Individual TSC2 gene exons containing the germline mutation were amplified by polymerase chain reaction (PCR) from lymphocytic DNA and from the tumor DNA, as described elsewhere (Au et al. 1998). The intragenic polymorphic EcoRV marker in exon 40 was tested in informative cases (Au et al. 1997). Chromosomal markers (D16S525, KG8, and D16S665) flanking the TSC2 gene were tested as described elsewhere (Peral et al. 1994; Shen et al. 1994; Snarey et al. 1994).
Southern analyses were performed as described elsewhere (Au et al. 1997). The TSC2 gene fragments were identified sequentially, with different segments of the TSC2 cDNA used as probes. One probe, consisting of nucleotides 1–1197 of the TSC2 cDNA, detects two adjacent BamHI fragments of 15 kb (exons 1–8) and 7 kb (exons 9–14) and a 15-kb HindIII fragment (exons 1–11) of the TSC2 gene. Another probe used for verification, consisting of nucleotides 1369–2689 of the TSC2 cDNA, detects two adjacent BamHI fragments—of 7 kb (exons 9–14) and 14 kb (exons 15–24)—and a 15-kb HindIII fragment.
In all cases, the TSC2 allele with the germline mutation was retained in the tumor DNA (table 1). When flanking markers D16S525, KG8, and D16S665 and an intragenic marker, TSC2 exon 40 EcoRV, were used, LOH was not detected in DNA from the one AML from patient HOU23-03, from the three FAs from patient TS93-41, or from the two cortical tubers from patients TS93-14 and TS94-53 (data not shown).
Table 1.
Summary of LOH Studies[Note]
|
LOH Findings
for
Germline Mutation |
|||||||||
| Patient and Tumor Sample | D16S525 | Exon 18 (dl C2070) | Exon 24 (insC 2779) | Exon 30 (dup3611-27) | Exon 36 (dl4770-2) | Exon 37 (A4859T) | Exon 40 (EcoRV) | KG8 | D16S665 |
| HOU23-01: | |||||||||
| 1T | LOH | … | LOH | … | … | … | LOH | NI | LOH |
| rT1 | LOH | … | LOH | … | … | … | LOH | NI | LOH |
| rT2 | LOH | … | LOH | … | … | … | LOH | NI | LOH |
| rT3 | H | … | H | … | … | … | H | NI | H |
| rT4 | H | … | H | … | … | … | H | NI | H |
| rT5 | LOH | … | LOH | … | … | … | LOH | NI | LOH |
| rT6 | LOH | … | LOH | … | … | … | LOH | NI | LOH |
| HOU23-03: | |||||||||
| T | H | … | H | … | … | … | H | NI | H |
| TS94-104: | |||||||||
| T1 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T2 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T3 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T4 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T5 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T6 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T7 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T8 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T9 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T10 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T11 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T12 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T13 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| T14 | NI | LOH | … | … | … | … | NI | LOH | LOH |
| TS93-41: | |||||||||
| FA1 | H | … | … | H | … | … | NI | H | NI |
| FA2 | H | … | … | H | … | … | NI | H | NI |
| FA3 | H | … | … | H | … | … | NI | H | NI |
| TS93-14: | |||||||||
| T | H | … | … | … | H | … | H | H | H |
| TS94-53: | |||||||||
| T | NI | … | … | … | … | H | H | NI | H |
Note.—NI = noninformative; ellipsis (…) = not applicable; and H = maintainance of heterozygosity.
Seven samples of tissue from AMLs were available from patient HOU23-01: six from the right kidney and one from the left kidney. The TSC2 allele carrying the germline mutation was retained in all seven tumors (table 1). Five of the seven AMLs (four from the right and one from the left) showed decreased intensity for one allele with markers D16S525, TSC2 exon 40 EcoRV, and D16S665, indicating LOH for some cells from those samples (fig. 1). The alleles with decreased intensity for the markers were the same in all five tumors (lT, rT1, rT2, rT5, and rT6) and were derived from the chromosome harboring the wild-type TSC2 allele (Rose et al. 1999). We concluded that the second hit in these tumors involved deletion of the complete wild-type TSC2 allele, leaving the nonfunctional germline mutated copy in the tumor. Extent of loss along the chromosome was not determined. In the two AMLs from the right kidney that did not show LOH, Southern blotting with the full-length TSC2 cDNA used as a probe did not reveal aberrant banding patterns (data not shown), implying that large deletion or rearrangement of the wild-type TSC2 allele is not the second hit in these two tumors (rT3 and rT4, from patient HOU23-01).
Figure 1.
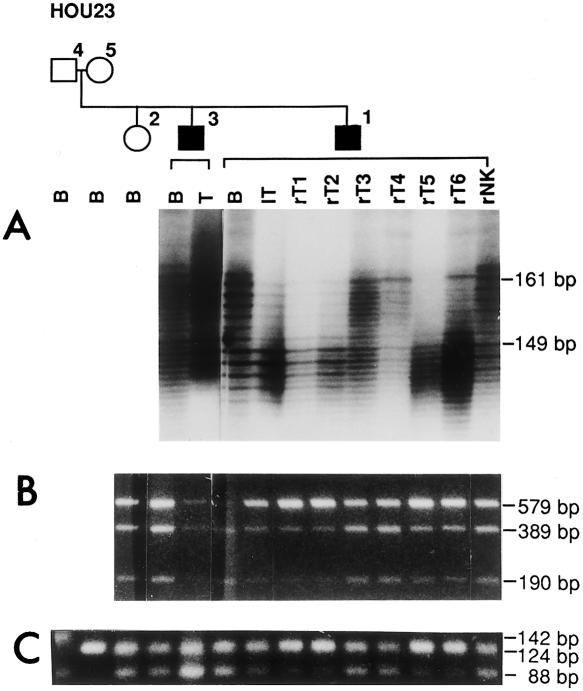
Testing of DNA samples from family HOU23, including members HOU23-04 (unaffected father), HOU23-05 (unaffected mother), HOU23-02 (unaffected child), HOU23-03 (affected child), and HOU23-01 (affected child), as depicted on pedigree shown above gels. The family represents an example of germline mosaicism, details of which are reported elsewhere (Rose et al. 1999). Individual lanes are labeled to indicate tissue from which DNA was extracted: B = blood lymphocytes, T = AML sample from HOU23-03, lT = left kidney AML, rT1–rT6 = six individual right-kidney AML samples, and rNK = right-kidney normal tissue from HOU23-01. Decrease in intensity of bands indicating loss of the wild-type TSC2 allele in some cells of the AMLs from patient HOU23-01 are observed in A–C. A, Marker D16S525 is located ∼95 kb telomeric to the 5′ end of TSC2. Both HOU23-01 and HOU23-03 are heterozygous, with alleles of 149 bp and 161 bp present. The 161-bp allele was significantly reduced in the lT, rT1, rT2, rT5, and rT6 of HOU23-01, whereas the 149-bp and 161-bp alleles were of equal intensity in rT3 or rT4 of HOU23-01 and in the AML (T) from HOU23-03. B, Patients were informative for the EcoRV polymorphism in exon 40 of TSC2, with heterozygotes showing three bands at 579, 389, and 190 bp, respectively. Decrease in the intensity of the wild-type allele was found in the same AML DNAs (lT, rT1, rT2, rT5, and rT6) as in A, indicating LOH for the wild-type allele in some cells from which the DNA was extracted. C, The marker D16S665 is located ∼75 kb from the 3′ end of TSC2. The father (HOU23-04) is heterozygous for the marker, with alleles of 142 and 88 bp, and the mother is homozygous for the 124-bp allele. The results showed the same five AMLs (lT, rT1, rT2, rT5, and rT6) with decreased intensity of the paternal 88-bp allele, while retaining the maternal 124-bp allele. The paternal 142-bp allele is not inherited by any of the offspring in the family.
All 14 AMLs of patient TS94-104 showed loss of the normal copy of exon 18 while retaining the 1-bp smaller mutant copy. The markers KG8 and D16S665 also exhibited LOH in DNA from these tumors (table 1). The somatic events led to a minimal deletion, from exons 18–41 of the patient's wild-type TSC2 allele, in these AMLs. Markers TSC2 exon 40 and D16S525 were not informative for this patient (table 1). Because of the lack of informativeness of markers, we were unable to determine whether the LOH differed among samples—that is, whether they represented one very large (clonal) tumor or multiple tumors.
The three FAs obtained from patient TS93-41 showed no decreased intensity for any of the markers tested; however, junction fragments were detected by Southern blotting. DNA extracted from both FA1 and FA2 of patient TS93-41 revealed 7.5-kb junction fragments detectable by restriction enzyme HindIII (fig. 2A). Junction fragments of ∼11 kb were detected after digestion with BamHI (data not shown). Three additional probes—nucleotides 1–1197 (exons 1–11), nucleotides 1369–2689 (exons 12–23), and a probe containing only exon 3—were tested to define the deletion (data not shown). On the basis of the published restriction map of the TSC2 genomic DNA (The European Chromosome 16 Tuberous Sclerosis Consortium 1993), the missing portion of the gene is roughly 22–25 kb in size and extends from just beyond exon 3 to exon 25 (fig. 2B).
Figure 2.
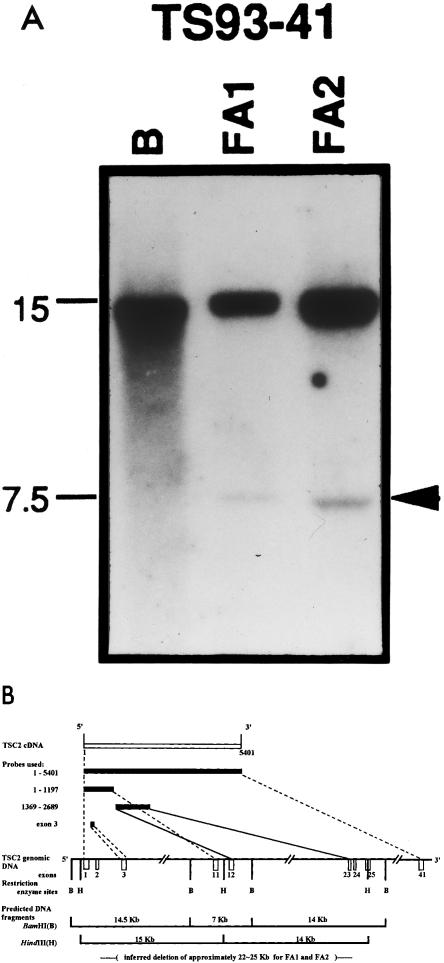
An intragenic deletion of TSC2 is the second (somatic) mutation in two FAs from patient TS93-41. Studies using markers within and flanking TSC2 showed no evidence for LOH in the three FAs obtained. A, Southern analysis of DNA from two FAs (FA1 and FA2), performed by means of a TSC2 cDNA probe spanning nucleotide 1–1197, revealed junction fragments of ∼7.5 kb with HindIII digestion. No junction fragment was detected with the lymphocyte DNA (lane B), even when a much higher (approximately fivefold) concentration of DNA was used. B, Diagram illustrating probes tested, restriction map for BamHI and HindIII of the TSC2 gene, predicted fragment sizes, and approximate size and location of deletion.
Our study confirmed the germline and defined the somatic second hits of the TSC2 gene in AMLs and FAs from three independent patients with TSC. These observations provide direct proof of tumor-suppressor function for the TSC2 gene, in accordance with Knudson's two-hit hypothesis, in the formation of hamartomas in humans. The studies indicate that the second hit in these tumors involved deletion of the complete wild-type TSC2 allele, leaving the nonfunctional germline mutated copy. Another group has recently reported both hits in a malignant islet-cell tumor from a patient with TSC with a germline mutation identified in the TSC2 gene (Verhoef et al. 1999).
An earlier report (Sampson et al. 1997) found a critical role for the PKD1 gene in the etiology of severe renal disease in some patients with TSC. Sampson's patients exhibited a mixed phenotype that included features of TSC and polycystic kidney disease (PKD). Our three patients (HOU23-01, HOU23-03, and TS94-104) had serious renal disease resulting primarily from angiomyolipomas with only minor cysts present; therefore, their symptoms are not consistent with PKD. The three patients had single-base-pair insertion or deletion mutations, all of which are predicted to cause premature termination of tuberin, approximately midway (exons 18 and 24) through the open reading frame of the TSC2 gene. We demonstrate that intragenic TSC2 mutations, without involvement of the PKD1 gene, can result in a life-threatening renal phenotype in patients with TSC.
Two of the AML DNAs (rT3 and rT4) from HOU23-01 did not reveal an aberrant banding pattern on Southern blots (not shown), and no LOH of the tested markers was evident; therefore, deletion of the entire wild-type TSC2 allele was not the second event leading to formation of these two tumors. More subtle, undefined mutations are likely the cause of the loss of function of the second allele in these tumors.
Previous attempts to search for LOH of markers within and around the TSC2 gene in facial angiofibromas have been unsuccessful (Henske et al. 1996). Our success in detecting junction fragments relied on the relative abundance of DNA we could extract from the two FAs available and on the use of the two smaller TSC2 cDNA probes (1–1197 and 1369–2689) to detect the specific TSC2 genomic fragments in Southern analyses. The relatively small amount of DNA represented by the junction fragments in these FAs suggests that the tumor cells were in low abundance. A slight reduction of one of the heterozygous alleles of these markers would be difficult to detect. Previous failure to demonstrate LOH in TSC brain lesions could reflect the same difficulties observed in FAs. Perhaps tumorigenesis for FAs and brain tumors does not require the extensive LOH seen in AMLs.
Acknowledgments
The work was supported by National Institutes of Health grant R29NS32300-05 and by a National Tuberous Sclerosis Association Senior Investigator Award (97-4) to H.N.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for X75621)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for TSC [MIM 191092]) [PubMed]
References
- Ahlsen G, Gilberg IC, Lindblom R, Gilberg C (1994) Tuberous sclerosis in western Sweden. Arch Neurol 51:76–81 [DOI] [PubMed]
- Al-Saleem T, Wessner LL, Scheithauer BW, Patterson K, Roach ES, Dreyer SJ, Fujikawa K, et al (1998) Malignant tumors of the kidney, brain, and soft tissues in children and young adults with the tuberous sclerosis complex. Cancer 83:2208–2216 [PubMed]
- Au K-S, Rodriguez JA, Finch JL, Volcik KA, Roach ES, Delgado MR, Rodriguez E Jr, et al (1998) Germline mutations of the TSC2 gene in 90 tuberous sclerosis patients. Am J Hum Genet 62:286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au K-S, Rodriguez JA, Rodriguez E Jr, Dobyns WB, Delgado MR, Northrup H (1997) Mutations and polymorphisms in the tuberous sclerosis complex gene on chromosome 16. Hum Mutat 9:23–29 [DOI] [PubMed]
- Bjornsson J, Short MP, Kwaitkowski DJ, Henske EP (1996) Tuberous sclerosis-associated renal cell carcinoma: clinical, pathological, and genetic features. Am J Pathol 149:1201–1208 [PMC free article] [PubMed]
- Carbonara C, Loga L, Grosso E, Mazzuco G, Borrone C, Garre ML, Brisigotti M, et al (1996) Apparent preferential loss of heterozygosity at TSC2 over TSC1 chromosomal regions in tuberous sclerosis hamartomas. Genes Chromosomes Cancer 15:18–25 [DOI] [PubMed]
- Cook JA, Oliver K, Mueller RF, Sampson J (1996) A cross sectional study of renal involvement in tuberous sclerosis. J Med Genet 33:480–484 [DOI] [PMC free article] [PubMed]
- Eker R, Mossige J, Johannessen JV, Aars H (1981) Hereditary renal adenomas and adenocarcinomas in rats. Diagn Histopathol 4:99–110 [PubMed]
- European Chromosome 16 Tuberous Sclerosis Consortium, The (1993) Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75:1305–1315 [DOI] [PubMed]
- Green AJ, Smith M, Yates JRW (1994) Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet 6:193–196 [DOI] [PubMed]
- Henske EP, Neumann HPH, Scheithauer BW, Herbst EW, Short MP, Kwaitkowski DJ (1995) Loss of heterozygosity in the tuberous sclerosis (TSC2) region of chromosome band 16p13.3 occurs in sporadic as well as TSC-associated renal angiomyolipomas. Genes Chromosomes Cancer 13:295–298 [DOI] [PubMed]
- Henske EP, Scheithauer BW, Short MP, Wollmann R, Nahmias J, Hornigold N, van Slegtenhorst M, et al (1996) Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet 59:400–406 [PMC free article] [PubMed]
- Knudson AG (1971) Mutations and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820–823 [DOI] [PMC free article] [PubMed]
- Kobayashi T, Hirayama Y, Kobayashi E, Kubo Y, Hino O (1995) A germline insertion in the tuberous sclerosis (Tsc2) gene gives rise to the Eker rat model of dominantly inherited cancer. Nat Genet 9:70–74 [DOI] [PubMed]
- Kobayashi T, Urakami S, Hirayama Y, Yamamoto T, Nishizawa M, Takahara T, Kubo Y, et al (1997) Intragenic Tsc2 somatic mutations as Knudson's second hit in spontaneous and chemically induced renal carcinomas in the Eker rat model. Jpn J Cancer Res 88:254–261 [DOI] [PMC free article] [PubMed]
- Kubo Y, Klimek F, Kikuchi Y, Bannasch P, Hino O (1995) Early detection of Knudson's two-hits in preneoplastic renal cells of the Eker rat model by the laser microdissection procedure. Cancer Res 55:989–990 [PubMed]
- Osborne JP, Fryer A, Webb D (1991) Epidemiology of tuberous sclerosis. Ann NY Acad Sci 615:125–127 [DOI] [PubMed]
- Peral B, Ward CJ, San Millan JL, Thomas S, Stallings RL, Moreno F, Harris PC (1994) Evidence of linkage disequilibrium in the Spanish polycystic kidney disease population. Am J Hum Genet 54:899–908 [PMC free article] [PubMed]
- Roach ES, Gomez MR, Northrup H (1998) Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 13:624–628 [DOI] [PubMed]
- Rose VM, Au K-S, Pollom G, Roach ES, Prashner HR, Northrup H (1999) Germ-line mosaicism in tuberous sclerosis: how common? Am J Hum Genet 64:986–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JR, Maheshwar MM, Aspinwall R, Thompson P, Cheadle JP, Ravine D, Roy S, et al (1997) Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet 61:843–851 [DOI] [PMC free article] [PubMed]
- Sampson JR, Scahill SJ, Stephenson JB, Mann L, Connor JM (1989) Genetic aspects of tuberous sclerosis in the west of Scotland. J Med Genet 26:28–32 [DOI] [PMC free article] [PubMed]
- Sepp T, Yates JRW, Green AJ (1996) Loss of heterozygosity in tuberous sclerosis hamartomas. J Med Genet 33:962–964 [DOI] [PMC free article] [PubMed]
- Shen Y, Holman K, Doggett NA, Callen DF, Sutherland GR, Richards RI (1994) Dinucleotide repeat polymorphisms at the D16S525, D16S359, D16S531, and D16S522 loci. Hum Mol Genet 3:210 [DOI] [PubMed]
- Snarey A, Thomas S, Schneider MC, Pound SE, Barton N, Wright AF, Somlo S, et al (1994) Linkage disequilibrium in the region of the autosomal dominant polycystic kidney disease gene (PKDI). Am J Hum Genet 55:365–371 [PMC free article] [PubMed]
- van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Jansen B, Verhoef S, Lindhout D, et al (1997) Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277:805–808 [DOI] [PubMed]
- Verhoef S, van Diemen-Steenvoorde R, Akkersdijk WL, Bax NM, Ariyurek Y, Hermans CJ, van Nieuwenhuizen O, et al (1999) Malignant pancreatic tumour within the spectrum of tuberous sclerosis complex in childhood. Eur J Pediatr 158:284–287 [DOI] [PubMed]
- Yeung RS, Xiao GH, Jin F, Lee W-C, Teata JR, Knudson AG (1994) Predisposition to renal carcinomas in the Eker rat is determined by germline mutation of the tuberous sclerosis 2 (Tsc2) gene. Proc Natl Acad Sci USA 91:11413–11416 [DOI] [PMC free article] [PubMed]