

To the Editor:
Several mitochondrial mutations have been identified that exhibit matrilineal transmission of deafness either in isolation or with other features such as diabetes. The A1555G substitution in the 12S rRNA gene is known to be associated with nonsyndromic deafness, with or without aminoglycoside exposure, in several different populations (Higashi 1989; Hu et al. 1991; Jaber et al. 1992; Prezant et al. 1993; Pandya et al. 1997; Estivill et al. 1998). Hamaski et al. (1997) demonstrated specific binding of aminoglycoside antibiotics to the proposed 1555A→G transition in a mitochondrial 12S rRNA construct, providing evidence in support of the aminoglycoside-ototoxicity hypothesis suggested by Prezant et al. (1993). However, the explanation for tissue specificity and for the pathogenesis in individuals without exposure to aminoglycosides remains unclear. An additional mutation in a nuclear gene has been postulated as one possible cause for the latter, but efforts to identify such a modifier gene have not been successful (Guan et al. 1996; Bykhovskaya et al. 1998). Another mitochondrial mutation, A7445G, was initially identified, in a Scottish pedigree, as a cause of sensorineural hearing loss with incomplete penetrance (Reid et al. 1994; Vernham et al. 1994). In two subsequent pedigrees, from New Zealand and Japan, a relatively mild palmoplantar keratoderma (MIM 148350) was also noted in most affected individuals (Sevior et al. 1998). The A→G substitution at position 7445 changes the final residue of the cytochrome oxidase (COI) stop codon on the H strand and is also located immediately adjacent to the 3′ end of tRNA ser(UCN), whose precursor is encoded on the L strand. The A7445G mutation was recently shown to retard the normal processing of the tRNA precursor, resulting in a 70% reduction in tRNA ser(UCN) production and in a 45% decrease in protein synthesis (Guan et al. 1998). A simultaneous decrease in mRNA for the NADH (nicotinamide adenine dinucleotide [reduced]) dehydrogenase (complex I) ND6 subunit may reflect an upstream effect of the delayed processing of the polycistronic RNA2 molecule. One or more of these three defects could contribute to the pathogenesis of deafness (Guan et al. 1998). In previous work we documented the A1555G mutation in three Mongolian families with matrilineal deafness (Pandya et al. 1997). To estimate the prevalence of both these mitochondrial mutations in the deaf population in Mongolia, we screened 480 deaf students and family members ascertained through the only residential School for Deaf and Blind in Mongolia, which is located in Ulaanbaatar, as well as 389 Mongolian controls with normal hearing. Our sample is estimated to include >70% of the student-age deaf population of Mongolia. We identified 37 deaf students with the A1555G mutation and 9 with the 7445 mutation, by screening for loss of a restriction site for the Alw26I and the XbaI restriction enzymes, respectively. Direct sequencing of DNA from the nine individuals positive for the loss of an XbaI restriction site revealed heterogeneity with novel mutations at one of three adjacent base pairs—7443, 7444, or 7445. Six students had coexistent A1555G and G7444A substitutions, which has not previously been reported.
After informed consent was provided, the clinical history was obtained and an examination was performed on each subject by one of the investigators, with special emphasis on identification of environmental causes of deafness, such as trauma, meningitis, otitis media, or pharmacologic ototoxicity, as well as syndromic forms of genetic deafness. An audiogram was obtained on most deaf individuals, with air conduction being tested at 250, 500, 1,000, 2,000, 3,000, 4,000, 6,000, and 8,000 Hz. For initial screening, DNA samples were tested for the presence of A1555G and A7445G mitochondrial mutations, by PCR amplification and subsequent digestion with Alw26I and XbaI restriction enzymes, as described elsewhere (Prezant et al. 1993; Reid et al. 1994; Pandya et al. 1997). The primer pairs used for PCR amplification were as follows: for the 1555 region, nucleotide (nt) 1261–1282 of the Cambridge sequence (Anderson et al. 1981) was used for the forward primer, and nt 2866–2843 was used for the reverse primer. For the 7445 region, nt 7178–7198 and nt 7840–7821 were used for the forward and reverse primers, respectively. The amplification products were resolved on a 2% NuSieve gel, were stained with ethidium bromide, and were photographed. On the basis of the restriction analysis, 31 individuals were found to be positive for the A1555G mutation alone, 3 were positive for the 7445 mutation alone, and 6 were positive for both (fig. 1).
Figure 1.
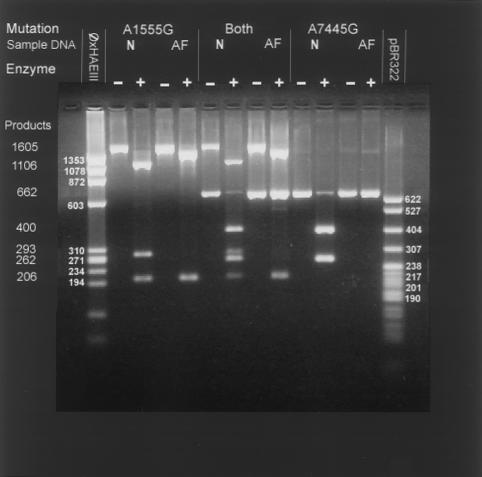
Representative gel with restriction enzyme digestion pattern of individuals with the A1555G mutation alone (lanes 4 and 5), with the 7445 mutation alone (lanes 12 and 13), and with both mutations (lanes 8 and 9). The band pattern for individuals with normal hearing is depicted for each category with and without digestion with the appropriate restriction enzyme. The A1555G mutation abolishes a site for the Alw26I enzyme. The undigested PCR product of 1605 bp is digested in normal individuals to yield bands of 1106, 293, and 206 bp. In affected individuals, lack of digestion results in a larger band at 1399 bp and in the lower band at 206 bp. The PCR product obtained with primers for the nt7445 mutation is 662 bp. In unaffected individuals, digestion with the restriction enzyme XbaI results in two bands—400 and 262 bp in size. The presence of a mutation at this nt abolishes the recognition sequence for this enzyme with absence of digestion after incubation with the enzyme.
To confirm the nature of the mutations, sequencing of DNA samples was performed by means of the Sequenase PCR-product sequencing kit from Amersham Life Science. Approximately 200 ng of the PCR product was incubated with 10 U of exonuclease 1 and 2 U of alkaline phosphatase, at 37°C for 30 min (Hanke and Wink 1994), followed by heat inactivation at 80°C for 15 min. The appropriate forward or reverse primer was added to an aliquot of the enzyme-treated PCR product, followed by the labeling solution mix made up of 40 mM Tris-Hcl (pH 7.5), 20 mM MgCl2, 50 mM NaCl, 5 mM DTT, 0.375 μM 7-deaza dNTP = s, 10 μCi [35S-α] dATP, and 3.2 U Sequenase. A 4-μl aliquot of this mixture was transferred to a tube containing 2.5 μl of the ddNTPs and incubated at 37°C for 10 min. The reaction was stopped with 4 μl of stop dye, and the samples were denatured and immediately loaded onto a 6% 1 × GTG acrylamide sequencing gel. After electrophoresis, the gel was dried and placed on an x-ray plate overnight, before the film was developed. Primers used to sequence the 1555 region were as described elsewhere (Pandya et al. 1997). The sequencing primers for the 7445 region were as follows: forward (nt 7321–7341) and reverse (nt 7623–7600). Ten of the 31 samples that were positive for the loss of a restriction site at 1555 were sequenced, and all had the A1555G substitution, as expected (data not shown). In contrast, direct sequencing of DNA from the nine individuals with loss of the XbaI restriction site showed mutational heterogeneity, as seen in figure 2. Two of the three individuals with the isolated loss of the XbaI restriction site had an A7445C substitution, whereas the third had an A7443G substitution. All six students with the double mutation showed a G7444A substitution. This mutation has been reported elsewhere in association, with Leber hereditary optic neuropathy (LHON) in whites, but it has been considered to be a secondary change that may increase the penetrance of the primary mutation but that is insufficient in itself to cause LHON (Brown et al. 1995). The G7444A substitution has not been reported in association with deafness, however. All the mutations appeared homoplasmic in every affected individual who tested positive, within the limits of detection by PCR-RFLP analysis and direct sequencing of PCR products. No subject with loss of either restriction site was detected among the 389 control samples from hearing Mongolians. Overall, our results indicate an incidence of 7.7%, 0.33%, 0.33%, and 1.33% for the A1555G, A7443G, A7445C, and A1555G + A7444G changes, respectively, in our population of deaf students.
Figure 2.
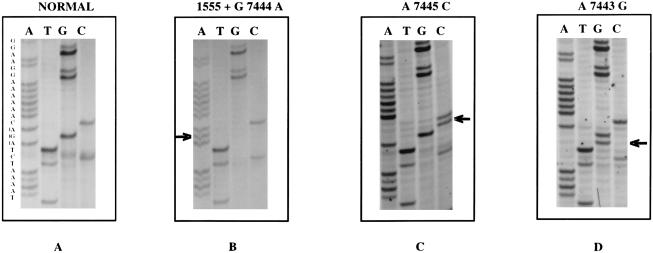
Sense strand sequence of the 3′ region of the tRNA ser(UCN) in a normal individual (A) and in three deaf students (B-D) from Mongolia. B, A deaf student with the double mutation showing the AGA→AAA substitution at nt7444. C, A deaf student with isolated nt 7445 mutation, with substitution of AGA→AGC. D, A deaf student with isolated nt 7443 change, with AGA→GGA substitution.
The six individuals with the G7444A + A1555G double mutation included five with matrilineal transmission of hearing loss and one with no family history of deafness. A definite history of aminoglycoside exposure could be elicited in only two individuals, but all six had severe to profound bilateral sensorineural hearing loss detected at birth or during early infancy. Both individuals with the isolated A7445C mutation had onset of deafness after an illness at age 1–4 years, and the one individual with the A7443G mutation had been deaf since infancy. These findings differ from those reported for other families with the A7445G mutation, in that the Scottish family exhibited incomplete penetrance (Reid et al. 1994) and the New Zealand family had a late onset (in the second decade) of hearing loss, with subsequent progression to profound deafness and palmoplantar keratoderma (Sevior et al. 1998). The variability in phenotypic expression noted in these reports was attributed to differences in the background mitochondrial haplotypes of the two pedigrees, thereby implying an interaction between the A7445G mutation and a second mitochondrial change. Although a history of aminoglycoside use was present in two of the six individuals, it did not appear to be a prerequisite, in view of the fact that some affected individuals had hearing loss detected soon after birth. The phenotype of deafness in individuals with isolated A1555G mutation is quite variable, with some reported patients developing late-onset hearing loss in the absence of aminoglycoside exposure (Estivill et al. 1998). These observations raise the possibility that the coexistence of the two mutations may influence the age at onset and severity of hearing loss, even in the absence of aminoglycoside exposure. The sample size is too small to make any definitive conclusions, and further observations would be required to establish digenic epistasis as a likely explanation for the apparent phenotypic heterogeneity in these families.
The A7445G mutation is located in the final residue of the COI gene on the H strand. It alters the normal stop codon, AGA, to another stop codon, AGG, and should therefore be a silent change. However, this substitution has been shown to dramatically alter the processing of the tRNA ser(UCN) precursor on the L strand, as noted previously. It also inhibits the maturation of other, more distal genes on the L strand, by destabilizing the major mitochondrial polycistronic transcription product, RNA 2 (Guan et al. 1998). All three newly described substitutions—A7445C, G7444A, and A7443G—also abolish the XbaI cleavage site at nt 7445. Since all are also associated with deafness, it seems plausible that they may share with A7445G a common pathogenic mechanism, possibly involving interference with the activity of the endogenous 3′ nuclease, which is required for normal processing of the tRNA ser(UCN) precursor. If so, we would predict that these three mutations would act similarly to A7445G and would alter tRNA ser(UCN) and upstream NADH dehydrogenase (complex I) ND6 subunit–message maturation, as well as protein synthesis. An RNASE P–like enzyme and a tRNA 3′ endonuclease have been implicated as the key enzymes in the precise cleavage at the 5′ and 3′ ends of mitochondrial precursor tRNAs (Rossmanith et al. 1995). Although these enzymes have been isolated and partially purified (Rossmanith 1997; Rossmanith and Karwan 1998), their number, molecular structure, and substrate recognition sequences have not yet been well characterized. On the H strand, however, the three new mutations we have described would all result in a read-through of the normal stop codon of the COI message, leading to the addition of three amino acids to the protein. The predicted tripeptides additions are SerGlnLys, LysGlnLys, and ArgGlnLys for the A7445C, G7444A, and A7443G substitutions, respectively. These changes are not lethal but could certainly contribute to the observed phenotype by altering the activity of COI either directly or by modifying the assembly of cytochrome oxidase (complex IV) holoenzyme. The resulting deficit in mitochondrial energy transduction may in turn contribute to defective development and function of the auditory sensory tissues. Hence, the potential involvement of the identified mutations in the synthesis and integrity of COI and in mitochondrial RNA processing provide two likely targets for further analysis of the molecular basis of the phenotype of deafness.
Acknowledgments
The authors would like to thank Wanda Hunt and Razieh Javaheri for their technical assistance. This research was supported in part by NIH grants KO8 HD-01172-01A1 and DC02530-01A2 to A.P. and W.E.N.
Electronic-Database Information
The accession number and URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.nih.gov/Omim (for keratoderma, palmoplantar, with deafness [MIM 148350])
References
- Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, Coulson AR, Drouin J, Eperon IC, et al (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465 [DOI] [PubMed]
- Brown MD, Torroni A, Reckord CL, Wallace DC (1995) Phylogenetic analysis of Leber's hereditary optic neuropathy mitochondrial DNA's indicates multiple independent occurrences of the common mutations. Hum Mutat 6:311–325 [DOI] [PubMed]
- Bykhovskaya Y, Shohat M, Ehrenman K, Johnson D, Hamon M, Cantor RM, Aouizerat B, et al (1998) Evidence for complex nuclear inheritance in a pedigree with nonsyndromic deafness due to a homoplasmic mitochondrial mutation. Am J Med Genet 77:421–426 [DOI] [PubMed]
- Estivill X, Govea N, Barceló A, Perelló E, Badenas C, Romero E, Moral L, et al (1998) Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment with aminoglycosides. Am J Hum Genet 62:27–35 [DOI] [PMC free article] [PubMed]
- Guan M, Enriquez JA, Fischel-Ghodsian N, Puranam RS, Lin CP, Maw MA, Attardi G (1998) The deafness-associated mitochondrial DNA mutation at position 7445, which affects tRNAser(UCN)precursor processing, has long-range effects on NADH dehydrogenase subunit ND6 gene expression. Mol Cell Biol 18:5868–5879 [DOI] [PMC free article] [PubMed]
- Guan M, Fischel-Ghodsian N, Attardi G (1996) Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum Mol Genet 5:963–971 [DOI] [PubMed]
- Hamasaki K, Rando RR (1997) Specific binding of aminoglycosides to a human rRNA construct based on a DNA polymorphism which causes aminoglycoside-induced deafness. Biochemistry 36:12323–12328 [DOI] [PubMed]
- Hanke M, Wink M (1994) Direct DNA sequencing of PCR-amplified vector inserts following enzymatic degradation of primer and dNTPs. Biotechniques 17:858–860 [PubMed]
- Higashi K (1989) Unique inheritance of streptomycin-induced deafness. Clin Genet 35:433–436 [PubMed]
- Hu DN, Qiu WQ, Wu BT, Fang LZ, Zhou F, Gu YP, Zhang QH, et al (1991) Genetic aspects of antibiotic induced deafness: mitochondrial inheritance. J Med Genet 28:79–83 [DOI] [PMC free article] [PubMed]
- Jaber L, Shohat M, Bu X, Fischel-Ghodsian N, Yang HY, Wang SJ, Rotter JI, et al (1992) Sensorineural deafness inherited as a tissue specific mitochondrial disorder. J Med Genet 29:86–90 [DOI] [PMC free article] [PubMed]
- Pandya A, Xia X, Radnaabazar J, Batsuuri J, Dangaansuren B, Fischel-Ghodsian N, Nance WE (1997) Mutation in the mitochondrial 12S rRNA gene in two families from Mongolia with matrilineal aminoglycoside ototoxicity. J Med Genet 34:169–172 [DOI] [PMC free article] [PubMed]
- Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, et al (1993) Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and nonsyndromic deafness. Nat Genet 4:289–294 [DOI] [PubMed]
- Reid FM, Vernham GA, Jacobs HT (1994) A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Hum Mutat 3:243–247 [DOI] [PubMed]
- Rossmanith W (1997) Processing of human mitochondrial tRNA(Ser(AGY))GCU: a novel pathway in tRNA biosynthesis. J Mol Biol 265:365–371 [DOI] [PubMed]
- Rossmanith W, Karwan RM (1998) Characterization of human mitochondrial RNase P: novel aspects in tRNA processing. Biochem Biophys Res Commun 247:234–241 [DOI] [PubMed]
- Rossmanith W, Tullo A, Potuschak T, Karwan R, Sbisà E (1995) Human mitochondrial tRNA processing. J Biol Chem 270:12885–12891 [DOI] [PubMed]
- Sevior KB, Hatamochi A, Stewart IA, Bykhovskaya Y, Allen-Powell DR, Fischel-Ghodsian N, Maw MA (1998) Mitochondrial A7445G mutation in two pedigrees with palmoplantar keratoderma and deafness. Am J Med Genet 75:179–185 [PubMed]
- Vernham GA, Reid FM, Rundle PA, Jacobs HT (1994) Bilateral sensorineural hearing loss in members of a maternal lineage with a mitochondrial point mutation. Clin Otolaryngol 19:314–319 [DOI] [PubMed]