

Abstract
Stickler syndrome is a dominantly inherited disorder characterized by arthropathy, midline clefting, hearing loss, midfacial hypoplasia, myopia, and retinal detachment. These features are highly variable both between and within families. Mutations causing the disorder have been found in the COL2A1 and COL11A1 genes. Premature termination codons in COL2A1 that result in haploinsufficiency of type II collagen are a common finding. These produce a characteristic congenital “membranous” anomaly of the vitreous of all affected individuals. Experience has shown that vitreous slit-lamp biomicroscopy can distinguish between patients with COL2A1 mutations and those with dominant negative mutations in COL11A1, who produce a different “beaded” vitreous phenotype. Here we characterize novel dominant negative mutations in COL2A1 that result in Stickler syndrome. Both alter amino acids in the X position of the Gly-X-Y triple-helical region. A recurrent R365C mutation occurred in two unrelated sporadic cases and resulted in the membranous vitreous anomaly associated with haploinsufficiency. In a large family with linkage to COL2A1, with a LOD score of 2.8, a unique L467F mutation produced a novel “afibrillar” vitreous gel devoid of all normal lamella structure. These data extend the mutation spectrum of the COL2A1 gene and help explain the basis for the different vitreous phenotypes seen in Stickler syndrome.
Introduction
Stickler syndrome (hereditary arthro-ophthalmopathy [MIM 108300, MIM 184840, and MIM 120280]) is a dominantly inherited vitreoretinopathy and chondrodysplasia caused by mutations in COL2A1 and COL11A1, the genes for types II and XI collagen (Ahmad et al. 1991; Richards et al. 1996). Systemic features such as arthropathy, midline clefting, hearing loss, and midfacial hypoplasia are highly variable. The incorporation of a premature termination codon into the open reading frame of COL2A1 transcripts, by either frameshift, missplicing, or point mutations is a common feature of the disorder (Ahmad et al. 1991; Brown et al. 1992, 1995; Williams et al. 1996; Richards et al. 2000) and causes haploinsufficiency of type II collagen. In our experience, this results in a characteristic “membranous” congenital anomaly in the vitreous (Scott 1989; Snead et al. 1994; Snead and Yates 1999; Richards et al. 2000; authors' unpublished results). This anomaly is also present in patients with mutations in exon 2, which result in a predominantly ocular disorder (Richards et al. 2000). This feature distinguishes Stickler syndrome due to COL2A1 mutations from a similar disorder caused by mutations in COL11A1, which results in a different vitreous phenotype (Richards et al. 1996; Martin et al. 1999; Snead and Yates 1999).
Types II and XI collagen are members of the family of fibrillar collagens, which also comprises types I, III, and V collagen. These molecules contribute to the strength and shape of the tissues in which they are expressed and are encoded by at least nine different genes (Prockop and Kivirikko 1995). Type I collagen predominates in bone, type II collagen in cartilage and vitreous, and type III collagen in blood vessels. Types V and XI collagen are quantitatively minor collagens that have a role in regulating the diameter of collagen fibrils (Li et al. 1995; Marchant et al. 1996). Increasing the levels of types V and XI with respect to the more abundant collagens results in fibrils with a smaller diameter. Thus, in tissues in which transparency is important, such as cornea and vitreous, greater expression of types V and XI collagen results in thinner collagen fibrils. All fibrillar collagens have a long uninterrupted repetitive amino acid sequence of glycine-X-Y, which forms the extended collagen triple helix. It is obligatory that glycine occupies every third position within the helix, since other amino acids disrupt the correct conformation of the helix. Defects in the genes for fibrillar collagens are known to cause a variety of inherited disorders (Prockop and Kivirikko 1995; Vikkula et al. 1995; Nicholls et al. 1996; Richards et al. 1996; Michalickova et al. 1998)—osteogenesis imperfecta (COL1A1 and COL1A2), some forms of chondrodysplasias, including Stickler syndrome (COL2A1 and COL11A1), otospondylomegaepiphyseal dysplasia (COL11A2), and some types of Ehlers-Danlos syndrome (COL1A1, COL1A2, COL3A1, COL5A1, and COL5A2). There are many examples of mutations that result in the substitution of a helical glycine residue by a bulkier amino acid, causing the aforementioned disorders (Kuivaniemi et al. 1997). Such mutations in the abundant collagens have been shown to reduce secretion of the mutant molecules and to lead to intracellular degradation (Prockop and Kivirikko 1995). Similar mutations in COL11A1 probably have the same effect on type XI collagen, resulting in the irregularly thickened fiber bundles seen clinically in the vitreous of patients who have such alterations.
Well-characterized mutations altering X- or Y-position amino acids are much rarer, but the substitution of cysteine into the X or Y position of the type II collagen helix has been reported. This amino acid is usually absent from the helical region of all fibrillar collagens, except type III, in which it occurs at the extreme carboxy-terminal end of the molecule.
The phenotypes caused by different mutations in COL2A1 range from severe achondrogenesis II through spondyloepiphyseal dysplasia (SED) congenita (SEDC), Kniest dysplasia to the milder Stickler syndrome and precocious arthritis. The most severe forms result from glycine substitutions and other, more disruptive mutations. Three different cysteine substitutions result in a range of phenotypes—namely, familial osteoarthritis, mild SED, or SEDC (Ala-Kokko et al. 1990; Chan et al. 1993; Williams et al. 1993), whereas a fourth produces a Stickler-like syndrome (Ballo et al. 1998). The substitution of a cysteine into the α1(I) and α2(XI) collagen chains produces relatively mild phenotypes, compared with glycine substitutions in the same α chains (McGuirt et al. 1999; Nuytinck et al. 2000).
In this report we characterize a fifth example of a cysteine substitution in type II collagen and show that it results in the same vitreous phenotype as that caused by nonsense mutations. In contrast, a unique L467F mutation in the X position of the type II collagen helix results in a novel vitreous phenotype that is also different from that caused by mutations in COL11A1. These data extend the mutation spectrum of the COL2A1 gene and help to explain the basis for the different vitreous phenotypes seen in Stickler syndrome.
Patients, Material, and Methods
Clinical Examination
Patients and pedigrees were identified from the Vitreous Clinic database at Addenbrooke’s Hospital. We obtained informed written consent in all cases and prior ethical approval (LRC92/019) for the study. The diagnostic criteria used for Stickler syndrome were as published elsewhere (Snead and Yates 1999)—(1) congenital vitreous anomaly and (2) any three of the following features:
-
a.
myopia with onset at age <6 years;
-
b.
rhegmatogenous retinal detachment or paravascular pigmented lattice retinopathy;
-
c.
joint hypermobility with abnormal Beighton score (with or without radiological evidence of joint degeneration);
-
d.
audiometric confirmation of sensorineural hearing defect;
-
e.
midline clefting.
Physical Examination
All patients were examined subjectively and objectively using methods described elsewhere (Richards et al. 2000). In brief, a subjective assessment was made of the nonquantifiable parameters retrognathia and anteversion of nares. All other parameters were measured objectively, according to a standardized method (Hall et al. 1989). Anteroposterior and lateral facial photographs at a standardized scale of 1:8 and photographs of both hands at a scale of 1:10 were taken using a Nikon FM2 camera with Micro Nikon 105-mm medical lens and Kodachrome 64 film at F16. A 1-cm grid was printed and then photographed at a scale of 1:8, to match the facial films, and at a scale of 1:10, to match the hand views. Clinical measurements of outer canthal distance, inner canthal distance, philtrum length, and middle-finger length were also recorded at the time of initial examination. These were used subsequently to test the accuracy of the method of photographic calibration. Control measurements of inner and outer canthal distance, interpupillary distance, and philtrum length in 20 unaffected siblings and 60 age-matched controls (recruited from the general ophthalmic clinic) were also recorded.
By dual projection of identically calibrated projectors, the matched scales were superimposed over each facial view in turn, so that the quantifiable facial measurements could be made. This was repeated using the matched scale for the hand views. All measurements were made according to criteria published elsewhere (Hall et al. 1989).
Clinical Features
Pedigree MS12
The proband was referred to the Vitreoretinal Service at age 4 years, having been found to be myopic at age 1 year. Cycloplegic refraction revealed extreme myopia in both eyes: OD −23.50/-2.00×90=6/9; OS -23.50DS=6/36. There was a moderate left esotropia for near and distance vision. Anterior-segment examination was unremarkable, with normal anterior-chamber drainage-angle development and clear crystalline lenses. Vitreous examination confirmed the presence of the type 1 membranous vitreous anomaly, set well back toward the posterior segment of both eyes. Both retinae were attached, with no peripheral lattice retinopathy. The optic disks were normal, with no signs of “myopic” peripapillary chorioretinal atrophy. At age 4 years, the patient underwent bilateral contiguous 360° prophylactic retinal cryotherapy. The patient is now age 17 years, and vision has remained stable in both eyes. Results of cardiovascular examination and echocardiography were normal. Stature is normal. Audiometric testing at age 7 years confirmed mild, bilateral sensorineural deafness, which has remained stable. Joint hypermobility has been present since early childhood. Formal testing confirmed a Beighton (1993) score of 6/9. On radiological screening, there was minimal evidence of spondyloepiphyseal dysplasia or degenerative joint disease. The patient exhibited the classical facial features of Stickler syndrome, with midfacial hypoplasia and a short and underdeveloped nasal bridge with mildly anteverted nares (fig. 1A–C). There was no evidence of midline clefting.
Figure 1 .

Clinical phenotypes. A–C, MS12, age 17 years, showing mild nasal-root hypolasia and moderate midfacial hypoplasia and slender digits. D–F, MS16, age 14 years. Note anteverted nares and nasal-root hyoplasia and joint laxity. G–I, III-7, MS25, age 29 years. Note well-developed nasal bridge and root, moderate midfacial hypoplasia, and slender digits.
Pedigree MS16
The proband was adopted by foster parents, and a full family history is unavailable. She was born at 38 wk gestation, weighing 2.94 kg. There was a paternal history of congenital heart disease, and heart failure was diagnosed at age 3 d, with pulmonary-valve stenosis and a patent ventriculoseptal defect. At age 2 years, the patient underwent surgery to repair the ventriculoseptal defect and to relieve the valvular stenosis. At age 5 years, the patient underwent a major craniofacial reconstructive procedure to correct mid- and upper-facial hypoplasia and nasal hypoplasia. Three years later, she developed a secondary hydrocephalus requiring a ventriculoperitoneal shunt. The shunt was revised and replaced in 1999.
Myopia had been diagnosed at age 6 years, and the patient suffered a giant-tear retinal detachment in her right eye that was not successfully repaired. The following year, the right eye was enucleated. A year later, the patient was referred to the Vitreoretinal Service at Addenbrooke’s Hospital, with a tentative diagnosis of Crouzon syndrome. Examination of the left eye under cycloplegic refraction confirmed extreme myopia: OS -32.50/-2.00×90. Anterior-segment examination showed a wedge-shaped cortical lens opacity (Scott 1989; Seery et al. 1990). Assessment of the craniofacial phenotype was difficult because of previous craniofacial surgery and hydrocephalus. However, the extreme myopia, sectorial cortical lens opacity, previous giant retinal tear, joint laxity, mixed conductive and sensorineural deafness, and midline cleft were all suggestive of Stickler syndrome rather than Marshall syndrome (MIM 154780) (table 1). This was further supported by the presence of the type 1 membranous vitreous anomaly (STL1 [MIM 108300] [Snead et al. 1994]), which is not a feature of Marshall syndrome. The patient underwent contiguous 360° retinal cryotherapy for multiple 360° retinal tears in the left eye. Vision remains stable, and the left retina remains attached after 13 years' follow-up.
Table 1.
Phenotypes
|
Ocular Phenotype |
Articular Phenotype |
AuralPhenotype |
Orofacial Phenotypea |
|||||
| Familyand Subject | Myopiab | RhegmatogenousRetinalDetachmentc | JointLaxityd | RadiologicalJointAbnormality | HearingLosse | MidfacialHypoplasia | AbnormalNasalDevelopment | MidlineCleftingf |
| MS12: | ||||||||
| II-1 | +++ | NA | 3 | 0 | + (S) | ++ | + | 0 |
| MS16: | ||||||||
| II-1 | +++ | 1 | 3 | + | ++ (M) | + | ++ | ++ |
| MS25: | ||||||||
| II-1 | ++ | 2 | 0 | 0 | + (S) | 0 | 0 | 0 |
| II-3 | +++ | NA | 0 | + | 0 | + | 0 | 0 |
| II-5 | +++ | 2 | 0 | 0 | + (S) | 0 | 0 | 0 |
| III-1 | +++ | 2 | 0 | 0 | 0 | 0 | + | 0 |
| III-3 | +++ | 1 | 0 | 0 | 0 | 0 | 0 | ++ |
| III-5 | +++ | NA | 2 | 0 | + (S) | 0 | 0 | ++ |
| III-7 | +++ | 0 | 0 | 0 | 0 | ++ | 0 | 0 |
| III-10 | +++ | NA | 0 | 0 | 0 | + | 0 | 0 |
| IV-2 | +++ | NA | 2 | 0 | 0 | 0 | + | 0 |
Assessed according to age/sex-matched control (Hall et al. 1989)
++ = −5 to −10D and +++ = >−10D.
0 = no retinal detachment; 1 = single eye; 2 = both eyes; NA = not applicable because of prophylactic treatment.
Beighton score = 1, 2, or ⩾3 more than that in age/sex-matched controls (Beighton 1993).
+ = Mild (30 dB); ++ = moderate (30–60 dB); and +++ severe = (>60 dB), compared with age/sex matched controls (data not shown). S = sensorineural; M = mixed.
+ = Bifid uvula; ++ = high-arch palate; +++ = cleft-palate repair.
The patient has a history of serous otitis media, with multiple grommet procedures in the past. In addition, audiometric testing confirmed mild, bilateral sensorineural deafness that has since remained stable. Joint hypermobility had been noted since early childhood. Formal testing revealed a Beighton score of 7/9. The patient is now age 20 years and is under orthopedic review because of premature arthritis that principally affects the ankles and knees. The patient exhibits the typical facial features of Stickler syndrome, with midfacial hypoplasia and a short and underdeveloped nasal bridge with anteverted nares. There is a high arch to the palate, and the ears are “low set” (fig. 1D–F).
Pedigree MS25
The proband from pedigree MS25 was identified using the standardized diagnostic protocol given above. However, although in this pedigree all affected individuals exhibited the major criterion of abnormal vitreous architecture, systemic involvement was found to be highly variable and generally mild. The clinical phenotype is illustrated by individual III-7 (fig. 1G–I). Strikingly, no individual in this pedigree exhibited the type 1 membranous vitreous anomaly. Instead, an “afibrillar” vitreous gel devoid of all normal lamella structure filled the entire posterior segment. No individual exhibited the “beaded” vitreous phenotype associated with mutations in the COL11A1 gene (Richards et al. 1996; Martin et al. 1999; authors' unpublished data). The three distinctive vitreous phenotypes, together with a normal one, for comparison, are shown in figure 2A–D.
Figure 2 .
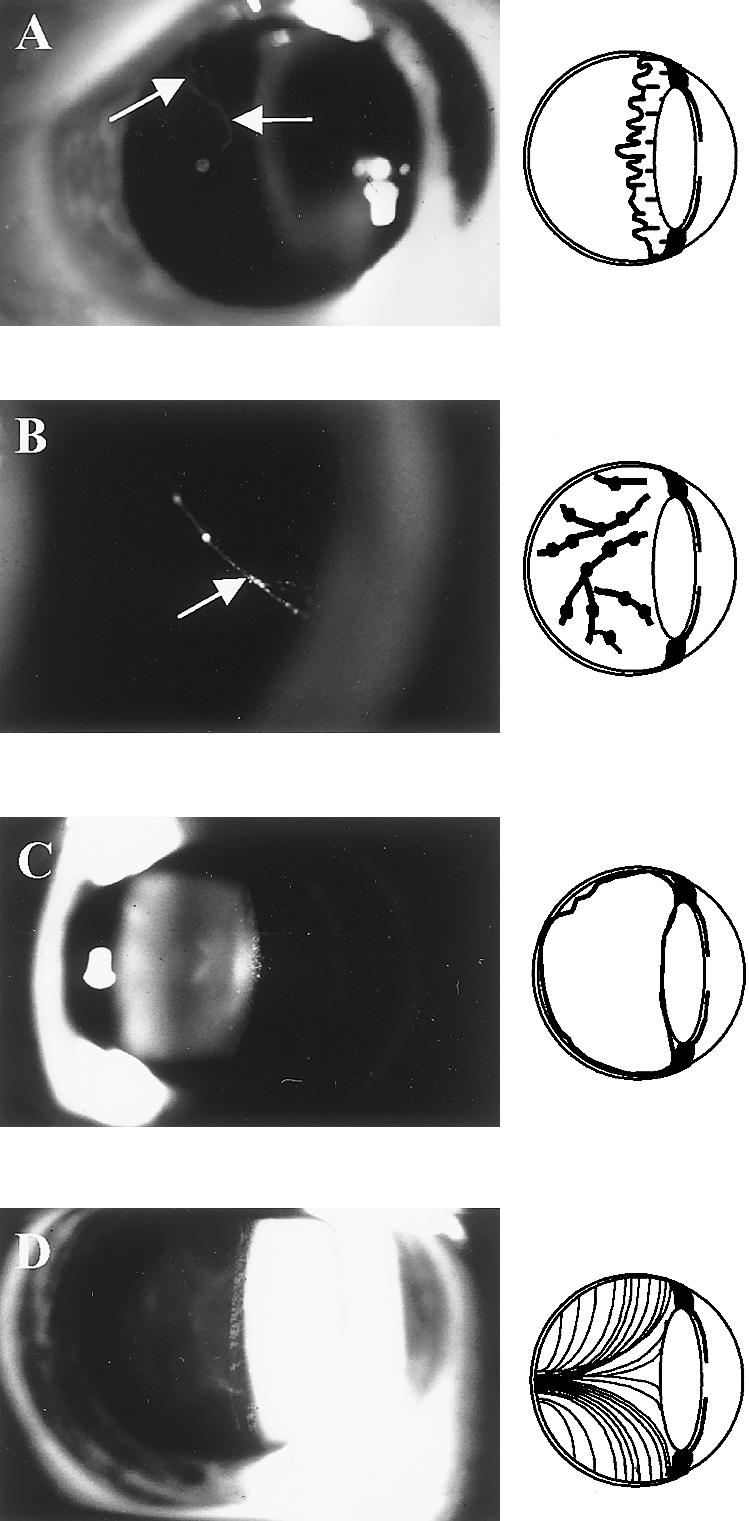
Vitreous phenotypes in Stickler syndrome. A, Membranous congenital vitreous anomaly seen in MS12 and MS16. Note vestigial gel occupying retrolental space and bordered by a distinct folded membrane (arrows). B, Beaded congenital vitreous anomaly: COL11A1 mutation (see Martin et al. 1999). Note irregularly thickened fiber bundles, giving a string-of-pearls appearance (arrows). C, Afibrillar congenital vitreous anomaly seen in MS25. Note complete absence of visible fiber bundles. D, Normal vitreous appearance. Note healthy compact homogenous fibrillar array.
In affected individuals, ophthalmic, aural, skeletal, and orofacial features were assessed and compared with age- and sex-matched controls, as described elsewhere (Richards et al. 2000). The results are summarized in table 1, which also includes individuals MS12 and MS16, for comparison.
Linkage Analysis
Linkage analysis was performed in family MS25, for markers close to the COL2A1, COL11A1, COL11A2, and COL5A2 genes, as described elsewhere (Martin et al. 1999). Additional polymorphic markers were used to investigate the relationship between MS12 and MS16.
Amplification and Sequencing
Regions of the COL2A1 gene were amplified and sequenced, as described elsewhere (Richards et al. 2000). COL2A1 cDNA was amplified after two rounds of PCR, essentially as described elsewhere for COL11A1 (Martin et al. 1999). Reverse transcription of RNA from cultured skin fibroblasts was performed using Superscript II (Stratagene). After an initial 40 cycles of amplification, reaction products were purified and reamplified with the same sense primer and an antisense primer internal to that used in the primary amplification. Primers used for amplification are described in table 2. To amplify a region containing exon 42, primers X36F, X53R, and X44R were used. This reaction product was sequenced with X43R. Similarly, a region containing exon 31 was amplified with X20F, X43R, and X34R and was sequenced with X33R. A region containing exon 26 was amplified with X20F, X34R, and X33R and was sequenced with X24F. All primers were based on the sequence for the COL2A1 gene (Genome Database accession number L10347)
Table 2.
Primer Sequences Used for RT-PCR
| Primer | Position | Sequence |
| 20F | Exon 20 sense | CAGATGGAATTCCTGGAGCCAAAG |
| 24F | Exon 24 sense | CAAGATGGTCTGGCAGGTCCCAAG |
| 36F | Exon 36 sense | CCAGCTGGTGCTAACGGCGAGAAG |
| 33R | Exon 33 antisense | CTGGGCACCGGGAGAGCCACGTTC |
| 34R | Exon 34 antisense | AGCGATACCAGCTGCTCCCCTCTC |
| 43R | Exon 43 antisense | AGGGAATCCTCTCTCACCACGTTG |
| 44R | Exon 44 antisense | GGGTTCACCTGCAGGACCCGTCAG |
| 53R | Exon 53 antisense | TGAACCTGCTATTGCCCTCTGCCC |
Family Testing
Restriction digestion with the enzyme BbvI was used to test the family and controls for the R365C mutation. A region of COL2A1 containing exon 26 was amplified and incubated with the enzyme at 37°C for 16 h. The products were analyzed by electrophoresis in an 8% polyacrylamide gel, stained with ethidium bromide, and viewed under UV light. Hybridization of allele-specific oligonucleotides was also used, to examine possible mosaicism in MS12 and MS16. The mutant allele was cloned into the Bluescript vector (Stratagene). Amplified mutant product was mixed with an equal amount of normal PCR product, to produce a complete heterozygote. Blood and skin were used to amplify DNA from affected individuals, as were blood samples from family members and normal controls. Amplified products were slot blotted in triplicate, hybridized to allele-specific oligonucleotides (cactggccgccctgg R365 and cactggctgccctgg C365), and washed according to conditions used by Wood et al. (1985). A single dideoxynucleotide-sequencing reaction was used to test for the L467F mutation in MS25 and controls, as described elsewhere (Richards et al. 2000).
Results
Slit-lamp examination of three cases of Stickler syndrome identified two sporadic cases (pedigrees MS12 and MS16) with the congenital membranous anomaly characteristic of mutations in COL2A1. In the third case, a large family (MS25) had an unusual vitreous phenotype in which an afibrillar vitreous gel devoid of all normal lamella structures filled the posterior segment. This appearance also differed from the phenotype associated with mutations in COL11A1 (fig. 2). Systemic features in MS25 were relatively mild (fig. 1 and table 1). In family MS25, linkage analysis that used the COL2A1 3′ VNTR was uninformative. However, flanking markers D12S85 and D12S1703 were consistent with COL2A1's being the disease locus, with LOD scores of 2.4 and 2.8, respectively, at 0 recombination. Linkage to COL11A1, COL11A2, and COL5A2 was excluded by recombination between affected individuals and polymorphic markers (data not shown). Loci were excluded if affected individuals were recombinant for markers very close to the candidate genes or if affected individuals had inherited different haplotypes for flanking markers.
After DNA amplification, exon sequencing of COL2A1 identified identical mutations in the two sporadic cases. Each had a C→T base change in exon 26, which converted a codon for arginine to cysteine, at position 365 of the collagen helix (fig. 3). According to convention, the initial glycine of the collagen triple helix was assigned to position 1. The R365C C→T mutation created a new BbvI restriction site; this was used to confirm that the mutation was absent in the DNA from both unaffected parents in MS12 (fig. 4A) and from an additional 25 controls (50 chromosomes; data not shown). Slot-blot analysis that used allele-specific oligonucleotides and PCR products amplified from blood and cultured skin cells did not indicate mosaicism in either affected individual (data not shown). Analysis of polymorphic markers confirmed parentage in MS12 and confirmed that family MS16 was unrelated to this family (data not shown). Sequencing of exons 2–54, amplified from an affected individual in MS25, identified a C→T change in exon 31, which converted leucine to phenylalanine at amino acid position 467 (fig. 3). For MS25, a single dideoxynucleotide-sequencing reaction was used to demonstrate that all affected individuals had inherited the L467F mutation (fig. 4B). In addition to unaffected family members, 50 controls (100 chromosomes; data not shown) were shown to lack the C→T base change.
Figure 3 .

COL2A1 genomic sequencing. The heterozygous (arrows) mutation-containing sequences obtained from affected individuals in MS12, MS16, and MS25 are shown under the normal sequence (N). Intron sequences are in lowercase, and coding sequences are in uppercase.
Figure 4 .

DNA analysis of families. A, DNA containing exon 26, which was amplified from MS16 (1) and MS12 (2–5) and digested with BbvI. Extra bands in samples from affected individuals, visualized after electrophoresis, are marked with arrows. Undigested DNA (lane U) and standard size markers (bp) were included on the gel. B, Results of [33P]dideoxynucleotide sequencing reaction, which was used to detect the mutation in MS25. Samples correspond to the pedigree shown above the gel. Family members not included in the analysis are situated on either side of the gel. Nucleotide numbers correspond to the complete COL2A1 gene sequence (Genome Database accession number L10347).
These mutations were unlike the usual haploinsufficiency mutations that cause Stickler syndrome and should exert a dominant negative effect. Previous observations have shown that premature termination codons can lead to the degradation of the mRNA from the mutant allele (Baserga and Benz 1988; Maquet 1995). We compared the cDNA sequences of these three cases of missense mutations with cDNA amplified from an individual with a nonsense mutation in exon 42 (MS20; previously characterized by Richards et al. [2000]). The cDNA sequence from the individual with a COL2A1 nonsense mutation demonstrated the loss of heterozygosity that would be expected if mRNA transcribed from the mutant allele were specifically degraded (fig. 5). However, cDNA from MS12, MS16, and MS25 was heterozygous for the mutations seen in genomic DNA (data shown only for MS16 and MS25), which supports the notion of a dominant negative mutation causing the disorder in these families.
Figure 5 .

COL2A1 cDNA sequencing. Sequencing of cDNA amplified from an individual (MS20 C) with a nonsense mutation in exon 42 showed loss of heterozygosity when compared with the genomic sequence (MS20 G). Amplified cDNA from MS16 and MS25 showed heterozygosity for the mutations seen in genomic DNA (fig. 1).
Discussion
Stickler syndrome is a clinically and genetically heterogeneous disorder (Francomano et al. 1987; Scott 1989, 1998; Fryer et al. 1990; Vintiner et al. 1991; Brunner et al. 1994; Richards et al. 1996; Sirko-Osadsa et al. 1998; Annunen et al. 1999; Martin et al. 1999). A recent study of patients with Stickler syndrome, and of those with the closely related Marshall syndrome, showed that many clinical features are shared by the two, but that some are more common to Marshall syndrome (Annunen et al. 1999). We have noted that hearing loss is more common in patients who have mutations in COL11A1, whereas cleft palate is more common in patients who have COL2A1 mutations (Snead et al. 2000). Certain features, such as abnormal frontal sinuses, intracranial ossifications, and thickened calvarium, were specific to patients with Marshall syndrome who have COL11A1 mutations (Annunen et al. 1999). However, these were examined only in three cases of Marshal syndrome and two cases of Stickler syndrome. In addition, no data were given regarding vitreous phenotype, which is a key feature of the Stickler syndrome, so that a definitive difference between the two syndromes remains to be resolved.
The vitreous phenotype can be an important indication of which genetic locus bears the mutation (Snead et al. 1994; Snead and Yates 1999; Richards et al. 2000). The vitreous also provides a unique opportunity to observe the effect of collagen mutations in vivo. Although the vitreous is 98% water, its collagen, proteoglycan, and cellular components impart a characteristic structural appearance on slit-lamp biomicroscopy. The healthy gel in a young patient has a dense compact homogenous lamella structure (fig. 2D). Ultrastructural examination of the rabbit eye has shown that these lamellae reflect the organization of collagen fibrils within the vitreous gel (Los et al. 1999). We have shown elsewhere that families with a vestigial congenital membranous anomaly of the vitreous consistently have mutations in the COL2A1 gene (Snead et al. 1994). Recent mutation detection in such families and sporadic cases in which linkage analysis was not possible has now confirmed this finding (Richards et al. 2000; authors' unpublished data). Families with mutations in COL11A1 have a different vitreous phenotype, of sparse, irregularly thickened lamellae (fig. 2B; Richards et al. 1996; Martin et al. 1999; Snead and Yates 1999). The family described here (MS25) was unique in that genetic analysis linked the disorder to COL2A1, but the vitreous appearance was different from that seen in other families with COL2A1 mutations. The pedigree also lacked the “string of pearls,” or beaded, appearance caused by mutations in COL11A1. Here we have shown that this family has a L467F substitution in the X position of the type II collagen Gly-X-Y triple helix. Taken together, the linkage data, absence of other amino acid substitutions, lack of nonsense-mediated decay, and the mutation's absence in normal chromosomes indicate that the L467F COL2A1 change described here is the causative mutation in this family. We have also shown that an additional X-position mutation (R365C) causes Stickler syndrome, but with the vitreous phenotype usually associated with premature termination codons in COL2A1.
Similar X- and Y-position Arg-Cys substitutions (fig. 6) have been described in types I, II, and XI collagen (Ala-Kokko et al. 1990; Chan et al. 1993; Williams et al. 1993; Ballo et al. 1998; McGuirt et al. 1999; Nuytinck et al. 2000). It now seems clear that this type of amino acid substitution in the fibrillar collagens results in connective-tissue disorders. Evidence for other X- or Y-position mutations causing disease is scarce. A Y-position R618Q substitution in α1(I) collagen resulted in abnormal migration of the protein in gel electrophoresis (Philips et al. 1990). This change was seen in a woman with features of Marfan syndrome, although the features were less pronounced in her father, who also had the change. Marfan syndrome has since been linked to mutations in fibrillin (Dietz et al. 1991), and it is unclear whether the amino acid substitution caused all of the clinical phenotype. A short report also described a R618H change in α2(I) collagen (Superti-Furga et al. 1994) but concluded that at most it caused “connective tissue weakness.” A similar report described a α1(III) L2F change in an individual with late-onset aneurysms (Anderson et al. 1994). Because of the sporadic nature of this latter case, it was unclear whether the change in COL3A1 was causative, but its similarity to the L467F mutation described here is interesting.
Figure 6 .

Position of Arg→Cys substitutions in fibrillar collagens. The position of cysteine substitutions in the α1(I), α1(II), and α2(XI) collagens are illustrated. X-position changes are indicated below the line representing each molecule, whereas Y-position changes are indicated above the lines. Numbers represent the amino acid position within the helix. The resulting phenotypes are osteoarthritis (OA), spondyloepipheseal dysplasia (SED), SED congenita (SEDC), Stickler syndrome (SS), mild chondrodysplasia (MC), Stickler-like syndrome (SLS), classical Ehlers-Danlos syndrome (CEDS), and nonsyndromic deafness (DFN).
The R365C mutations described here have arisen independently and appear to be a mutational hotspot, similar to other Arg→Cys mutations that occur in COL2A1 (Ala-Kokko et al. 1990; Eyre et al. 1991; Holderbaum et al. 1993; Williams et al. 1993, 1995; Reginato et al. 1994). These cysteine substitutions have been identified in cases of either precocious osteoarthritis/mild SED/SED congenita (Y position) or a Stickler-like syndrome (X position). The family with Stickler syndrome also had brachydactyly, which may be due to the C-terminal location of the mutation rather than to its X position in the Gly-X-Y repeat, since neither of the patients with R365C mutations reported here has this trait.
The differing appearances that the R365C and L467F mutations cause in the vitreous suggests that they are having a different effect on the amount of type II collagen available for gel formation. Both of the R365C mutations resulted in the same vitreous phenotype that was, because of premature termination codons, seen in Stickler syndrome. Our working hypothesis for the vitreous phenotypes seen with Stickler syndrome's COL2A1 and COL11A1 mutations is that, at a specific stage of fetal eye development, a critical mass of collagen is required for proper formation of the secondary vitreous. Haploinsufficiency of type II collagen results in this threshold not being met, and only a vestigial gel forms in the retrolental space. This anomaly is congenital and appears to be clinically static, suggesting that subsequent accumulation of type II collagen cannot compensate for this stage-specific shortfall in the major constituent of the vitreous. Type XI collagen is a quantitatively minor component, and mutations in COL11A1 do not stop the bulk formation of the vitreous gel. However, because of the role of type XI collagen in the control of fibril diameter, COL11A1 mutations result in abnormal fibrillogenesis, and we suggest that this is reflected in the variability of lamella bundle organization seen on slit-lamp examination of the vitreous. In these cases, irregularly thickened fiber bundles are seen, indicating a reduction in the level of type XI collagen, compared with type II collagen, allowing fibrils of irregular diameter to form. The α3 chain of type XI collagen is transcribed from the COL2A1 gene (Wu and Eyre 1995). However, because expression of type II collagen is much higher than that of type XI, it is unclear whether COL2A1 haploinsufficiency mutations will affect type XI collagen and, subsequently, the lamellar morphology. Such an effect would be difficult to distinguish by slit-lamp examination, since, in these cases, the vitreous gel is so abnormal.
Previous studies (Eyre et al. 1991) have shown that the type II collagen R519C substitution reduces the amount of protein secreted, therefore reducing the amount of collagen available for matrix assembly. Similarly, the COL1A1, X-position, R134C mutation (Nuytinck et al. 2000) has been shown to form disulfide-bonded collagen dimers, which were not secreted by cultured cells. A skin biopsy from this patient also showed dilatation of the rough endoplasmic reticulum, indicating retention of mutant collagen within the cell. A mutation of glycine to cysteine in type II collagen also formed dimers and was poorly secreted (Mundlos et al. 1996). Because the COL2A1 R365C mutations described here both resulted in the phenotype associated with haploinsufficiency, this suggests that this mutation also causes a reduction in secretion of type II collagen, which disrupts secondary vitreous embryogenesis. As yet, we have only limited tissue available to test this hypothesis or to perform ultrastructural studies.
In MS25, the vitreous appears to fill the entire posterior segment, but with an appearance different from that seen in families with COL11A1 mutations. The lack of the type 1 vitreous anomaly suggests that, for the L467F mutation, the amount of collagen available for gel formation is greater than both that for the R365C change and that in cases of haploinsufficiency. The relatively mild systemic features seen in this family also suggest that the L467F change has less of an effect than does haploinsufficiency. The mutation in MS25 will be incorporated into both types II and XI collagen, although, because of the stochiometry of chain composition, it will be expected to affect each to a different extent. In the homotrimeric type II collagen, α1(II)3, 7/8 of molecules will contain at least one mutant α chain, whereas only half of the molecules of type XI heterotrimer collagen, α1(XI)α2(X)α1(II), will contain a mutant α chain. Other forms of type XI collagen exist in the eye, including α1(XI)2α1(V) (Mayne et al. 1993); these molecules will be unaffected by mutations in COL2A1. In MS25, the vitreous is diaphanous and devoid of the normal fibrillar lamellar structure. A widely observed effect of mutations that disrupt collagen helices is reduced secretion of the mutant protein (Prockop and Kivirikko 1995). However, at present it is unclear whether the vitreous phenotype in MS25 is due to altered secretion of mutant type II collagen or whether the mutation changes the process of fibrillogenesis. If secretion of the mutant L467F collagen is reduced, the change will affect type II collagen more than it affects type XI, because of the chain stochiometry of the different molecules. Thus, the vitreous phenotype may be due to an increase in the ratio of type XI to type II collagen, resulting in thinner structures not easily seen by slit-lamp examination. Alternatively, the mutation may either disrupt association between collagen molecules or affect interaction with other components of the vitreous extracellular matrix. Ultrastructural studies of tissue from these patients will be necessary to determine changes to collagen-fiber diameter and conformation.
In summary, we have shown that dominant negative mutations in COL2A1 can result in Stickler syndrome. An R365C substitution resulted in the same vitreous phenotype as was seen in cases of haploinsufficiency, and with similar systemic features. A novel and unique mutation in COL2A1, L467F, caused a different afibrillar vitreous. This contrasted with the thickened beaded bundles seen elsewhere in patients with COL11A1 mutations. Patients with this mutation had only mild systemic features, but, in both groups, ocular features were severe, including extreme myopia and bilateral retinal detachment.
Acknowledgments
The authors gratefully acknowledge the financial support of the Guide Dogs for the Blind Association, the Medical Research Council, the Stanley Thomas Johnson Foundation, and Michael and Elizabeth Greig. We also wish to thank the following: Maureen Laidlaw (tissue culture); Katherine Haslam and Medical Illustration, Addenbrooke’s Hospital, Cambridge (photography); Gillian Whitmore (Hereditary Vitreous Disorders Clinic); Chris Maddren, Department of Genetics (sequencing); and our medical colleagues who kindly referred patients to the Vitreoretinal Service at Addenbrooke's Hospital.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Genome Database, The, http://gdbwww.gdb.org (for COL2A1 [accession number L10347])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for Marshall syndrome [MIM 154780] and Stickler syndrome [MIM 108300, MIM 184840, and MIM 120280])
References
- Ahmad NN, Ala-Kokko L, Knowlton RG, Jimenez SA, Weaver EJ, Maguire JI, Tasman W, Prockop DJ (1991) Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy). Proc Natl Acad Sci USA 88:6624–6627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ala-Kokko L, Baldwin CT, Moskowitz RW, Prockop DJ (1990) Single base mutation in the type II procollagen gene (COL2A1) as a cause of primary osteoarthritis associated with a mild chondrodysplasia. Proc Natl Acad Sci USA 87:6565–6568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DW, Tromp G, Kuivaniemi H, Ricketts M, Pope FM, Stolle CA, Boyd CD (1994) A type III procollagen gene mutation in a patient with late onset aneurysms. Matrix Biol 14:3927866652
- Annunen S, Korkko J, Czarny M, Warman ML, Brunner HG, Kaariainen H, Mullikan JB, Tranebjarg L, Brooks DG, Cox GF, Cruysberg JR, Curtis MA, Davenport SL, Friedrich CA, Kaitila I, Krawczynski MR, Latos-Bielenska A, Mukai S, Olsen BR, Shinno N, Somer M, Vikkula M, Zlotogora J, Prockop DJ, Ala-Kokko L (1999) Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet 65: 974–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballo R, Beighton PH, Ramesar RS (1998) Stickler-like syndrome due to a dominant negative mutation in the COL2A1 gene. Am J Med Genet 80:6–11 [DOI] [PubMed] [Google Scholar]
- Baserga SJ, Benz EJ (1988) Nonsense mutations in the human β-globin gene affect mRNA metabolism. Proc Natl Acad Sci USA 85:2056–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beighton P (ed) (1993) The Ehlers Danlos syndrome. In: McKusick's heritable disorders of connective tissue, 5th ed. Mosby, St. Louis, pp 199–206 [Google Scholar]
- Brown DM, Nichols BE, Weingeist TA, Sheffield VC, Kimura AE, Stone EM (1992) Procollagen II gene mutation in Stickler syndrome. Arch Ophthalmol 110:1589–1593 [DOI] [PubMed] [Google Scholar]
- Brown DM, Vandenburgh K, Kimura AE, Weingeist TA, Sheffield VC, Stone EM (1995) Novel frameshift mutations in the procollagen 2 gene (COL2A1) associated with Stickler syndrome (hereditary arthro-ophthalmopathy). Hum Mol Genet 4:141–142 [DOI] [PubMed] [Google Scholar]
- Brunner HG, van Beersum SEC, Warman ML, Olsen BR, Ropers H-H, Mariman ECM (1994) A Stickler syndrome gene is linked to chromosome 6 near the COL11A2 gene. Hum Mol Genet 3:1561–1564 [DOI] [PubMed] [Google Scholar]
- Chan D, Thomas KF, Cole WG (1993) Characterization of an arginine 789 to cysteine substitution in α1(II) collagen chains of a patient with spondyloepiphyseal dysplasia. J Biol Chem 268:15238–15245 [PubMed] [Google Scholar]
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, Stetten G, Meyers DA, Francomano CA (1991) Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352:337–339 [DOI] [PubMed] [Google Scholar]
- Eyre DR, Weis MA, Moskowitz RW (1991) Cartilage expression of a type II collagen mutation in an inherited form of osteoarthritis associated with mild chondrodysplasia. J Clin Invest 87:357–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francomano CA, Rowan BG, Liberfarb RM, Hirose T, Maumenee IH, Stoll HU, Pyeritz RE (1987) The Stickler and Wagner syndromes: evidence for genetic heterogeneity. Am J Hum Genet Suppl 43:A83 [Google Scholar]
- Fryer AE, Upadhyaya M, Littler M, Bacon P, Watkins D, Tsipouras P, Harper PS (1990) Exclusion of COL2A1 as a candidate gene in a family with Wagner-Stickler syndrome. J Med Genet 27:91–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JG, Froster-Iskenius UG, Allanson JE (1989) Handbook of normal physical measurements. Oxford Medical Publications, Oxford University Press, New York [Google Scholar]
- Holderbaum D, Malemud C, Moskowitz R, Haqqi T (1993) Human cartilage from late stage familial osteoarthritis transcribes type II collagen mRNA encoding a cysteine in position 519. Biochem Biophys Res Commun 192:1169–1174 [DOI] [PubMed] [Google Scholar]
- Kuivaniemi H, Tromp G, Prockop DJ (1997) Mutations in fibrillar collagens (type I, II, III and XI, fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage and blood vessels. Hum Mut 9:300–315 [DOI] [PubMed] [Google Scholar]
- Li Y, Lacerada DA, Warman ML, Beier DR, Yoshioka H, Ninomiya Y, Oxford JT Morris NP, Andrikopoulos K, Ramirez F, Wardell BB, Lifferth GD, Teuscher C, Woodward SR, Taylor BA, Seegmiller RE, Olsen BR (1995) A fibrillar collagen gene, COL11A1, is essential for skeletal morphogenesis. Cell 80:423–430 [DOI] [PubMed] [Google Scholar]
- Los LI, van Luyn MJA, Nieuwenhuis P (1999) Organization of the rabbit vitreous body: lamellae, Cloquet’s channel and a novel structure, the “alae canalis Cloqueti.” Exp Eye Res 69:343–350 [DOI] [PubMed]
- Maquat LE (1995) When cells stop making sense: effects of nonsense codons on RNA metabolism in vertebrate cells. RNA 1:453–465 [PMC free article] [PubMed] [Google Scholar]
- Marchant JK, Hahn RA, Linsenmayer TF, Birk DE (1996) Reduction of type V collagen using a dominant-negative strategy alters the regulation of fibrillogenesis and results in the loss of corneal-specific fibril morphology. J Cell Biol 135:1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Richards AJ, Yates JRW, Scott JD, Pope FM, Snead MP (1999) Stickler syndrome: further mutations in COL11A1 and evidence for additional locus heterogeneity. Eur J Hum Genet 7:807–814 [DOI] [PubMed] [Google Scholar]
- Mayne R, Brewton RG, Mayne PM, Baker JR (1993) Isolation and characterization of the chains of type V/type XI collagen present in bovine vitreous. J Biol Chem 268:9381–9386 [PubMed] [Google Scholar]
- McGuirt WT, Prasad SD, Griffith AJ, Kunst HPM, Green GE, Shpargel KB, Runge C, Huybrechts C, Mueller RF, Lynch E, King MC, Brunner HG, Cremers CWRJ, Takanosu M, Li SW, Arita M, Mayne R, Prockop DJ, Van Camp G, Smith RJH (1999) Mutations in COL11A2 cause non-syndromic hearing loss (DFNA13). Nat Genet 23:413–419 [DOI] [PubMed] [Google Scholar]
- Michalickova K, Susic M, Willing MC, Wenstrup RJ, Cole WG (1998) Mutations of the α2(V) chain of type V collagen impair matrix assembly and produce Ehlers-Danlos syndrome type I. Hum Mol Genet 7:249–255 [DOI] [PubMed] [Google Scholar]
- Mundlos S, Chan D, McGill J, Bateman JF (1996) An α1(II) Gly913Cys substitution prevents the matrix incorporation of type II collagen which is replaced with type I and type III collagens in cartilage from a patient with hyperchondrogenesis. Am J Med Genet 63:129–136 [DOI] [PubMed] [Google Scholar]
- Nicholls AC, Oliver JE, McCarron S, Harrison JB, Greenspan DS, Pope FM (1996) An exon skipping mutation of a type V collagen gene (COL5A1) in Ehlers-Danlos syndrome. J Med Genet 33:940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuytinck L, Freund M, Lagae L, Pierard GE, Hermanns-Le T, De Paepe A (2000) Classical Ehlers-Danlos syndrome caused by a mutation in type I collagen. Am J Hum Genet 66:1398–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips CL, Shrago-Howe AW, Pinnell SR, Wenstrup RJ (1990) A substitution at a non-glycine position in the triple helical domain of proα2(I) collagen presents in an individual with a variant of the Marfan syndrome. J Clin Invest 86:1723–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prockop DJ, Kivirikko KI (1995) Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem 64:403–434 [DOI] [PubMed] [Google Scholar]
- Reginato A, Passano G, Neumann G, Falasca G, Diaz-Valdez M, Jimenez S, Williams C (1994) Familial spondyloepiphyseal dysplasia tarda, brachydactyly, and precocious arthritis associated with an arginine 75-cysteine mutation in the procollagen type II gene in a kindred of Chiloe Islanders: clinical, radiographic, and pathologic findings. Arthritis Rheum 37:1078–1086 [DOI] [PubMed] [Google Scholar]
- Richards AJ, Martin S, Yates JRW, Scott JD, Baguley DM, Pope FM, Snead MP (2000) COL2A1 exon 2 mutations: relevance to the Stickler and Wagner syndromes. Br J Ophthalmol 84:364–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AJ, Yates JRW, Williams R, Payne SJ, Pope FM, Scott JD, Snead MP (1996) A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in α1(XI) collagen. Hum Mol Genet 5:1339–1343 [DOI] [PubMed] [Google Scholar]
- Scott JD (1989) Prevention and perspective in retinal detachment: Duke-Elder lecture. Eye 3:491–515 [DOI] [PubMed] [Google Scholar]
- ——— (1998) Surgery for retinal and vitreous disease, 1st ed. Butterworth Heinemann, Hong Kong, pp 183–184 [Google Scholar]
- Seery CM, Pruett RC, Liberfarb RM, Cohen BZ (1990) Distinctive cataract in the Stickler syndrome. Am J Ophthalmol 110:143–148 [DOI] [PubMed] [Google Scholar]
- Sirko-Osadsa D, Murray MA, Scott JA, Lavery MA, Warman ML, Robin NH (1998) Stickler syndrome without eye involvement is caused by mutations in COL11A2, the gene encoding the α2(XI) chain of type XI collagen. J Pediatr 132:368–371 [DOI] [PubMed] [Google Scholar]
- Snead MP, Payne SJ, Barton DE, Yates JRW, Al-Imara L, Pope FM, Scott JD (1994) Stickler syndrome: correlation between vitreo-retinal phenotypes and linkage to COL2A1. Eye 8:609–614 [DOI] [PubMed] [Google Scholar]
- Snead MP, Yates JRW (1999) Clinical and molecular genetics of Stickler syndrome. J Med Genet 36:353–359 [PMC free article] [PubMed] [Google Scholar]
- Snead M, Yates JRW, Scott JD, Baguley DM, Pope FM, Martin S, Richards A (2000) Clinical phenotypes of families with Stickler syndrome and mutations in either COL2A1 or COL11A1. Proc Greenwood Genet Center 19:169 [Google Scholar]
- Superti-Furga A, Raghunath M, Steinmann B (1994) Heterozygosity for a point mutation in COL1A1 causing a R618H substitution in α1(I) chains does not result in Marfan syndrome but may produce mild weakness of connective tissues. Matrix Biol 14:3857866652
- Vikkula M, Mariman ECM, Lui VCH, Zhidkova NI, Tiller GE, Goldring MB, van Beersum SEC, de Waal Malefijt MC, Vandenhoogen FHJ, Ropers HH, Mayne R, Cheah KSE, Olsen BR, Warman ML, Brunner HG (1995) Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 80:431–437 [DOI] [PubMed] [Google Scholar]
- Vintiner GM, Temple IK, Middleton-Price HR, Baraitser M, Malcolm S (1991) Genetic and clinical heterogeneity of Stickler syndrome. Am J Med Genet 41:44–48 [DOI] [PubMed] [Google Scholar]
- Williams CJ, Considine E, Knowlton RG, Reginato A, Neuman G, Harrison D, Buxton P, Jimenez S, Prockop DJ (1993) Spondyloepiphyseal dysplasia and precocious arthritis in a family with an arg 75-cys mutation in the procollagen type II gene (COL2A1). Hum Genet 92:499–505 [DOI] [PubMed] [Google Scholar]
- Williams CJ, Ganguly A, Considine E, McCarron S, Prockop DJ, Walsh-Vockley C, Michels VV (1996) An A→G transition at the 3′ acceptor splice site of IVS17 characterizes the COL2A1 gene mutation in the original Stickler syndrome kindred. Am J Med Genet 63: 461–467 [DOI] [PubMed] [Google Scholar]
- Williams CJ, Rock M, Considine E, McCarron S, Gow P, Ladda R, McLain D, Michels VM, Murphy W, Prockop DJ, Ganguly A (1995) Three new point mutations in type II procollagen (COL2A1) and identification of a fourth family with the COL2A1 arg519-cys base substitution using conformation sensitive gel electrophoresis. Hum Mol Genet 4:309–312 [DOI] [PubMed] [Google Scholar]
- Wood WI, Gitschier J, Lasky LA, Lawn RM (1985) Base composition-independent hybidization in tetramethylammonium chloride: a method for oligonucleotide screening of highly complex gene libraries. Proc Natl Acad Sci USA 82:1585–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Eyre DR (1995) Structural analysis of cross-linking domains in cartilage type XI collagen. J Biol Chem 270:18865–18870 [DOI] [PubMed] [Google Scholar]