

Abstract
Defining the relationship between multiple polymorphisms in a small genomic region and an underlying quantitative trait locus (QTL) represents a major challenge in human genetics. Pedigree analyses have shown that angiotensin I–converting enzyme (ACE) levels are influenced by a QTL located within or close to the ACE gene and most likely resides in the 3′ region of this locus. We genotyped seven polymorphisms spanning 13 kb in the 3′ end of ACE in 159 Afro-Caribbean subjects to evaluate the linkage disequilibrium between these sites and to narrow the genomic region associated with an elevated ACE level using a cladistic analysis. The linkage disequilibrium measurement D′ and a haplotype tree revealed three distinct haplotype segments, presumably because of recombination. The value of the linkage disequilibrium parameter pexcess was highest for site 22982, which is located in the middle segment. A series of nested, cladistic analyses confirmed that the other two regions are unlikely to be the ACE-linked QTL and that the variant resides in the middle region. Analyses of the same polymorphisms in 98 unrelated Europeans in the Monitoring Trends and Determinants in Cardiovascular Diseases (MONICA) study resulted in fewer haplotypes than were observed among the Afro-Caribbean subjects, suggesting that populations with greater genetic diversity may be especially informative for fine-scale mapping.
Introduction
Angiotensin I–converting enzyme (ACE, DCP1 [MIM 106180]) is one of the most intensely studied candidate genes in human cardiovascular pathophysiology (Williams 1988; Weber et al. 1995). Strong evidence exists for an association between the ACE Alu insertion/deletion (I/D) found in intron 16 and plasma ACE activity, with increased levels found among persons with the “deletion” allele (Rigat et al. 1990; Tiret et al. 1992). Several studies of more-complex cardiovascular phenotypes have suggested that the D allele confers increased susceptibility to cardiovascular disease (Evans et al. 1994; Soubrier et al. 1994; Schunkert 1997; Oren et al. 1999; Taittonen et al. 1999), although many other reports have been published showing inconsistent or even beneficial effects (Galinsky et al. 1997; Keavney et al. 2000). The Alu marker does not have a known functional effect but is considered an “anonymous” marker in linkage disequilibrium with a functional variant directly affecting plasma ACE level. It is proposed that other candidate polymorphisms within this gene contribute to the mechanism affecting plasma ACE activity and the putative health sequelae. Identification of such polymorphism(s) would allow accurate genotyping of a “functional” variant and may clarify the role of ACE as a susceptibility gene for cardiovascular disease.
The ACE Alu element is in near-complete disequilibrium with many other single-nucleotide polymorphisms (SNPs) in the surrounding region, defining two major haplotype backgrounds (Keavney et al. 1998; Rieder et al. 1999). Conflicting evidence exists as to whether or not the functional variants are upstream or downstream from the Alu element. In a French study, ACE levels were found to be influenced by polymorphisms 5′ to the Alu element (Villard et al. 1996). However, a measured haplotype analysis performed in British families (a) tested subdivisions within the major haplotypes that were created through recombination and (b) appeared to exclude the ACE promoter and ∼8 kb upstream from containing a functional DNA variant (Keavney et al. 1998). A recent analysis using a more extensive set of markers (Farrall et al. 1999) also reported that an additional 3.9 kb of 5′ segment of this gene was not associated with ACE levels. We therefore chose to focus this study on the remaining 3′ region. Further narrowing of this region would rely on haplotypes derived from multiple recombination events. Several studies have demonstrated a greater genetic diversity among populations of African descent (Castiglione et al. 1995; Tishkoff et al. 1996, 1998) that should increase the likelihood of observing recombinant haplotypes and assist in fine mapping. Accordingly, we genotyped individuals of African descent at multiple markers, resolved haplotypes, analyzed linkage disequilibrium among 3′ sites, and tested ACE haplotypes against plasma ACE levels using a cladistic analysis. In addition, we typed the same markers in unrelated German participants in the World Health Organization Monitoring Trends and Determinants in Cardiovascular Diseases (MONICA) study for the purpose of comparison across populations.
Material and Methods
Subjects
Subjects in this study included 159 Afro-Caribbeans (66 men, 93 women; mean age 49 years), randomly sampled from the metropolitan area of Kingston, Jamaica, as part of an international collaborative study of hypertension in blacks (Cooper et al. 1997). Approval was obtained from the institutional review board of the University of the West Indies, Mona, Jamaica. In addition, 98 samples from healthy, normotensive individuals of European descent were selected from the World Health Organization MONICA study in Augsburg, Germany for comparison with the Afro-Caribbean data (Keil et al. 1988). This study was approved by the institutional review board of the University of Regensburg.
Phenotyping
The Jamaican participants were examined using a standardized epidemiologic protocol, and a range of anthropometric and sociocultural variables were collected (Cooper et al. 1997). Determination of ACE concentration was performed in the Jamaican samples using a previously published sandwich ELISA (Danilov et al. 1996). In the MONICA samples, ACE activity was measured by a fluorescent assay as previously described (Schunkert et al. 1990).
Genotyping
Regions surrounding each polymorphic variant were amplified using a standard PCR buffer (25 ng genomic DNA, 10 mM Tris–HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2, and 0.001% gelatin) containing 0.16 mM of each of the four deoxynucleotide triphosphates, 0.5 mM of each primer, and 10 U/ml Taq polymerase (Perkin-Elmer-Cetus). Thermocycling conditions consisted of an initial denaturation at 95°C for 1 min, followed by 35 cycles of 95°C for 30 s, 58°C for 45 s, and 72°C for 2 min, with a final extension of 72°C for 5 min. After PCR, the ELISA-based oligonucleotide ligation assay (OLA) was used to genotype all sites (Tobe et al. 1996). Allele-specific oligonucleotides for each site were constructed with 5′ modifications of either digoxigenin or fluorescein, along with joining/capture probes modified with a 3′-Biotin-ON and chemically phosphorylated using 5′-Phosphate-ON (Clontech). Specific oligonucleotide primers used for each site were 10979-CCAAACCTGAGCATGTGCA(T/C), P-TACACACAGAGATGCTGTCC-BIO, 15214-GGGCAAGTCACCATGGAT(G/A), P-GGGGAAGAAGTTAATAATCTT-BIO, 20833-GACAGAACTGGGTTCAAAC(T/C), P-CCGTCTCCATTACTTTGTTT-BIO, 22982-CTGTCTCCTTGCTTCCC(A/G), P-CTCAGCTCGCTCAGAAGG-BIO, 23945-TGTACCAGCTCCATGA(CT)2/3, P-GCTCGGGTGAACAGCCTT-BIO, 24599-TGTCCTGTTCTGTGTGTG(C/T), and P-GTTGTACACAGTGCTTGGCT-BIO.
Spectrophotometric absorbances were taken at 490 and 655 nm to detect the fluorescein and digoxigenin reporters for each allele (Tobe et al. 1996), respectively, and were used to assign genotypes. Genotypes for the Alu element were determined using standard PCR primers flanking this region and were scored after visualization using ethidium bromide staining of PCR products.
Statistical Methods
The allele frequencies for each SNP were calculated by using the allele-counting method. Hardy-Weinberg equilibrium at each locus was assessed by the χ2 test with one degree of freedom (Weir 1996). We used Clark’s (1990) “subtracting” algorithm to determine each individual’s haplotypes, as follows. Complete homozygosity across a region or heterozygosity at a single site allowed unambiguous assignment of a haplotype over this region. After this determination, unresolved haplotypes were identified using the initial set of known haplotypes as the basis from which to infer unresolved individuals' putative haplotypes. This method is surprisingly effective at inferring the haplotype structure hidden in a set of unphased genotype data and does not require the assumption of Hardy-Weinberg equilibrium (Clark 1990). Haplotype frequencies were estimated by counting haplotypes. Linkage disequilibrium estimates for pairs of markers were obtained by calculating the frequently used measure D′ (Devlin and Risch 1995). We define the individuals above the 80th percentile of the ACE distribution as the case group and those below the 60th percentile as the control group. Linkage disequilibrium between a putative functional QTL and each polymorphic site was obtained by calculating pexcess=(pD-pN)/(1-pN), where pD is the frequency of an allele at a specific polymorphic site in the case group, and pN is its frequency in the controls (Bengtsson and Thomson 1981; Hastbacka et al. 1994; Risch et al. 1995). The standard deviation σexcess of pexcess caused by sampling variation is approximately given by:
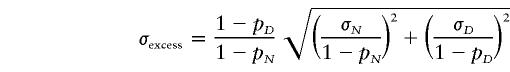 |
where σD and σN are the standard deviation due to sampling variation of pD and pN, respectively (Hastbacka et al. 1994). In general, pexcess can be used to determine the most likely location of the trait locus from among a collection of linked loci (Devlin and Risch 1995). To detect recombination, we use the test statistic proposed by Crandall and Templeton (1999).
Linear regression was used to model the relationship between ACE plasma level and sex, age, and haplotype effects, under the assumption of additivity, using the following model:
where Hi and Hj indicate an individual’s two haplotype effects. Since sex may have a nonlinear effect on how other factors contribute to a phenotype, we performed separate regression analyses within each sex group. Haplotype effects were estimated with ordinary least squares, which are equivalent to Fisher’s average effects (Templeton 1987). To test the null hypothesis of equal haplotype effects, we fitted the full model under the alternative hypothesis and then compared it to the reduced model, using the general F test (Neter et al. 1990).
Results
Participants were part of a randomly sampled community study in Kingston, Jamaica. Polymorphisms at seven sites (10979, Alu, 15214, 20833, 22982, 23945, and 24599) were genotyped across a 13-kb segment in the 3′ end of the ACE gene (fig. 1). The shaded region in the 5′ region of the figure represents the promoter and the 8-kb region excluded from containing a functional ACE variant (Keavney et al. 1998). No significant deviations from Hardy-Weinberg equilibrium were found among the sites tested (P>.18). The mean plasma ACE level was calculated for each genotype at all of the seven polymorphic sites (fig. 2 [left side]). Differences in ACE levels between genotypes at each variant site were tested for statistical significance by a one-way analysis of variance (ANOVA) followed by a Student-Newman-Keuls test between groups. No significant differences in ACE activities were found among genotypes at sites 10979, Alu, 15214, 23945, and 24599. Significant differences were found between the high ACE level homozygotes, heterozygotes, and the low homozygotes at sites 20833 and 22982 (P<.05 vs. homozygotes [white bar]).
Figure 1.

Polymorphic markers genotyped in ACE. The genomic region of ACE covers ∼24 kb on chromosome 17 (Rieder et al. 1999). Six single-nucleotide polymorphisms, in addition to the Alu insertion/deletion, were typed in 159 Jamaicans. The shaded area in the 5′ region represents the promoter and 8-kb region excluded from containing a functional ACE variant (Keavney et al. 1998).
Figure 2.

Site-specific ACE levels in Jamaicans (left bars) and Europeans (right bars). All site genotypes are plotted against ACE activity normalized to the lowest level (100%) at each site. Two sites showed a significant change in ACE levels on the basis of genotypes (20833 and 22982) for Jamaicans. All sites showed a significant change for Europeans. An asterisk (*) denotes that P<.05 versus the lowest homozygote (white bar).
A total of 28 haplotypes were inferred from 318 chromosomes in Afro-Caribbean subjects and were assigned to each individual, using Clark’s inferential algorithm (Clark 1990), on the basis of the genotyping results. Among these haplotypes, eight occurred with a frequency ⩾4.4%, accounting for 82.7% of the total (table 2); the remaining group was lumped as residual (R). Linkage disequilibrium was computed in the Afro-Caribbean subjects for each pair of sites using the measure D′ (table 1 [above the diagonal]). The markers at position 10979, Alu, and 15214 were in almost complete linkage disequilibrium (D′ > .94), whereas the pairs of markers at 20833 and 22982 and at 23945 and 24599 shared similar, lower degrees of linkage disequilibrium (D′ = .855 and .836, respectively). We then defined sites 10979, Alu, and 15214 as segment 1, 20833 and 22982 as segment 2, and 23945 and 24599 as segment 3.
Table 2.
Haplotype Frequencies[Note]
|
Site |
Frequency(%) |
||||||||
| Haplotype | 10979 | Alu | 15214 | 20833 | 22982 | 23945 | 24599 | Jamaicans | Europeans |
| 1 | C | I | A | C | G | 3 | C | 21.7 | 41.3 |
| 2 | T | D | G | T | A | 3 | C | 13.5 | 0 |
| 3 | C | I | A | C | G | 2 | T | 10.4 | 0 |
| 4 | T | D | G | T | A | 2 | T | 10.4 | 35.2 |
| 5 | T | D | G | C | G | 2 | T | 9.7 | 0 |
| 6 | T | D | G | T | A | 3 | T | 7.9 | 0 |
| 7 | C | I | A | C | G | 3 | T | 4.7 | 0 |
| 8 | T | D | G | C | G | 3 | T | 4.4 | 0 |
| R | 17.3 | 23.5 | |||||||
Note.— R indicates all the remaining haplotypes.
Table 1.
Linkage Disequilibrium (D′) of Each Pair of Markers in the Jamaican (above Diagonal) and European Samples (below Diagonal)
| 10979 | Alu | 15214 | 20833 | 22982 | 23945 | 24559 | |
| 10979 | … | .974 | .948 | .798 | .689 | .178 | .162 |
| Alu | 1.0 | … | 1.0 | .819 | .711 | .190 | .193 |
| 15214 | 1.0 | 1.0 | … | .852 | .742 | .233 | .206 |
| 20833 | .978 | .978 | .978 | … | .855 | .100 | .084 |
| 22982 | .913 | .913 | .914 | .897 | … | .100 | .121 |
| 23945 | .956 | .956 | .956 | .958 | .979 | … | .836 |
| 24599 | .968 | .968 | .969 | .970 | .94 | .941 | … |
To provide a comparison to the European populations used in earlier studies, we investigated the linkage-disequilibrium characteristics in this region by genotyping 98 individuals randomly sampled in the MONICA study. The haplotypes were also inferred using Clark’s method. Haplotypes 1 (CIACG3C) and 4 (TDGTA2T) were the most frequent in this sample. They accounted for 41% and 35% of chromosomes, respectively (table 2), and correspond closely to the findings for haplotype 1 and 2 reported by Keavney et al. (1998). The third-most-frequent haplotype was TDGTA2C, which accounted for 16% of the European sample but only 0.9% of the Jamaican sample. The seven sites were in almost complete linkage disequilibrium in the Germans (table 1 [below the diagonal]), indicating that populations of European origin are unlikely to have the additional ancestral breakpoints necessary to refine the location of the ACE-linked mutation. Differences in ACE activities between genotypes at each variant site were also tested in the German samples (fig. 2 [right]). Significant differences were found between the high–ACE activity homozygotes, heterozygotes, and the low homozygotes at all seven sites (P<.05 vs. homozygotes; see fig. 2 [white bar, right panel]). As in the Afro-Caribbean subjects, the largest difference in ACE activities between genotypes was observed at site 22982.
A putative haplotype tree was constructed from the eight common haplotypes in Afro-Caribbean subjects, using the principle of maximum parsimony (Wagner’s parsimony method, PHYLIP software v. 3.57c). Figure 3 describes the evolutionary relationships among these haplotypes and their grouping into similar clades (A, B, and C), according to previously defined methods in which all haplotypes can be joined together by moving back one mutational step (Templeton et al. 1987, 1988). Each solid line between two haplotypes corresponds to a single mutational event that occurs at the polymorphic site indicated under or beside the line. From this figure, it can be seen that site 23945 “mutates” three times in this tree, the greatest number for any site.
Figure 3.

The eight most frequent haplotypes were used to infer a maximum-parsimony tree. Haplotypes were then grouped with neighboring haplotypes into clades A, B, and C. The maximum-parsimony mutational connections among the haplotypes are indicated by solid lines, with the 0s representing all intermediate haplotypes that are missing from the sample. The number under the haplotypes corresponds to the haplotype, and the number under or beside the solid lines is the mutation site. The phenotypic effects attributed to these groups then were tested under different model assumptions against plasma ACE activity by use of a measured-haplotype analysis.
Haplotypes 1 (CIACG3C), 2 (TDGTA3C), 3 (CIACG2T), and 4 (TDGTA2T) were the most frequent. Haplotypes 1 and 4 were complementary at all seven sites; this was true for haplotypes 2 and 3 as well. Haplotypes 1 and 4 can be generated from haplotypes 2 and 3 through a recombination event occurring between sites 22982 and 23945 or through mutational events. The site 23945 is a simple dinucleotide repeat characterized by the length of the polymorphism. At such sites, it is well known that mutations to and from particular lengths may occur more than once. This results in homoplasy. However, the strength of linkage disequilibrium between 23945 and 24599 suggests that recombination between 22982 and 23945 may be a more plausible explanation than multiple mutational hits. All the other haplotypes (5, 6, 7, and 8) can be obtained from haplotypes 1, 2, 3, or 4 by hypothesizing a simple recombination or mutation event. This pattern suggests that the two principal ancestral lineages of the Jamaican population included haplotypes 1/4 and 2/3.
Although D′ values (table 1) do not provide strong evidence for recombination between site 15214 and 20833, it seems plausible that such a breakpoint does exist, since the estimated haplotype tree (fig. 3) has only two long branches (the one connecting A to C and the one connecting A to B). Moreover, all mutations are 3′ on the branch connecting A to B and are 5′ on the branch connecting A to C. The null hypothesis of no recombination is not rejected (P=.1) by the test proposed by Crandall and Templeton (1999), but this might be a consequence of the small number of sites under consideration. Overall, these data are entirely consistent with the hypothesis that a recombination event occurred between sites 15214 and 20833 and that clade A is the recombinant clade produced from clade B and C.
A series of regression analyses were performed to isolate specific regions with SNPs on various haplotype backgrounds (table 3). Additive models describing the contribution of each haplotype to plasma ACE level were fitted after age was adjusted for. (We dropped sex because that term was not significant [P=.44]). The most general model was [1, 2, 3, 4, 5, 6, 7, 8, R], which considers each haplotype to have a different phenotypic effect. Using the haplotype classification from figure 3, a goodness-of-fit test between the model [A, B, C, R] and the most general model was not significant (P=.38), indicating that the phenotypic values are similar within each of the three clades (A, B, and C), therefore suggesting that the segment beyond 23945 is unlikely to harbor the ACE-linked QTL. A possible reason for this phenotypic similarity could be the shared allelic states at sites between 10979 and 22982 within each of the clades, which would imply that the ACE-linked QTL is located in that interval. We then tested the model [A = B, C, R] against the most general model, to determine whether the effect of clade A resembled that of clade B. No significant difference was found between these models (P=.4), indicating phenotypic similarity because of shared allelic states at sites 20833 and 22982. We further tested the model [A = C, B, R] against the most general model and found significant differences (P=.016), perhaps because of the different allelic states at sites 20833 and 22982. Since there are only two tests needed at this level (A = B and A = C), the result is still significant even after multiple comparisons are adjusted for. These tests suggest that differences in the allelic states of the SNPs (20833 and 22982) account for differences in the observed phenotype between the clade groupings, indicating that the ACE-linked QTL may be located in the region containing sites 20833 and 22982.
Table 3.
Model Comparisons
|
P Value Obtained against Model [1, 2, 3, 4, 5, 6, 7, 8, R] |
|||
| Model | Overall | Male | Female |
| A, B, C, R (2=6=4, 5=8, 1=7=3, R) | .3809 | .1028 | .9592 |
| A=B, C R | .4344 | .1508 | .9770 |
| A=C, B, R | .016 | .016 | .6244a |
P value is .0636 if test is against model [A, B, C, R].
Although sex was not significantly associated with ACE in this data set, it may have a nonlinear effect on how haplotypes contribute to ACE. We therefore conducted the regression analysis described above within each sex group, and the results are also reported in table 3. The results in men were similar to those for the whole sample. However, no significant result was found in women when models [A, B, C, R], [A = B, C, R] and [A = C, B, R] were tested against the most general model (P=.96, .98, and .62, respectively). A possible reason could be the decrease in the sample size caused by stratification and the large number of degrees of freedom. To reduce the degrees of freedom, we tested model [A = C, B, R] against model [A, B, C, R], obtaining a result of marginal significance (P=.06) for women.
The effect of haplotype combinations was further evaluated. Table 4 lists the individuals' genotypes as a combination of the three clades and their corresponding ACE levels in Afro-Caribbean subjects. (Only those genotypes with more than five individuals are reported.) We observed that genotype C/C had the highest mean ACE level and that clade C was associated with an increased ACE level, which is consistent with the nested cladistic analysis. Furthermore, the nested cladistic analysis implies that A/C and B/C should be the same phenotypically and, likewise, that B/B and A/B should be the same. These inferences are confirmed in table 4, where A/C and B/C have an almost identical phenotype, as do B/B and A/B. Table 4 also shows that the A = B/C genotype class is almost exactly intermediate between C/C and A = B/A = B, which is consistent with our assumption of an additive model. Genotype B/R had the second-highest mean ACE level, which is apparently different from the phenotype of A/R. However, since the R category was not included in the nested analysis, this may be due to heterogeneity within the R category. For example, among the individuals with genotype B/R, 11 had ACE activity >100; among these, 7 had allele A at site 22982, a genotype strongly associated with ACE level. In the category A/R, only three of nine individuals had allele A at site 22982. Hence, this result would be reasonable if the ACE-linked mutation were located around 22982. This direct analysis of haplotype combinations supports the assertion from the nested haplotype analysis that the differences in clades B and C in the 3′ region described above contribute directly to changes in plasma ACE levels.
Table 4.
Genotypic Means of ACE Levels for Jamaicans[Note]
| “Genotype” | Mean ± SD | N |
| C/C | 127.9 ± 55.1 | 17 |
| B/R | 105.7 ± 43.1 | 21 |
| A/C | 101.9 ± 36.0 | 11 |
| B/C | 101.6 ± 43.3 | 38 |
| C/R | 82.9 ± 29.8 | 17 |
| B/B | 75.0 ± 29.4 | 21 |
| A/B | 74.6 ± 41.8 | 15 |
| A/R | 65.9 ± 25.0 | 9 |
Note.— Only groupings with >5 individuals are reported.
The ACE levels corresponding to the individual's genotype as a combination of the three clades in German subjects were also analyzed. Since haplotype TDGTA2C has one site difference from haplotype TDGTA2T at site 24599, this haplotype was incorporated into clade C. The result was consistent with that in table 4 (data not shown).
We then compared individuals from the upper fifth of the ACE distribution (ACE > 120) to those with low values (ACE < 100) as cases and controls. Since sex and age were not significantly associated with ACE level (P=.44 and 0.14, respectively), we did not adjust for them. Linkage disequilibrium was measured by pexcess between ACE and each site. Figure 4 (solid line) shows that pexcess increased from 10979 to 22982, then subsequently decreased. The pexcess value at site 22982 appears to achieve the maximum value, presumably because of the strength of linkage disequilibrium between 22982 and the ACE-linked QTL. Similarly, pexcess was also calculated for German subjects (fig. 4 [dotted line]). Sites 22982 and 23945 also have the largest pexcess value.
Figure 4.

Maximum pexcess with error bars denoting one standard deviation for each site. Solid and dotted lines represent the pexcess values for the Jamaican and European samples, respectively. The offset for the two curves represents the higher average level of linkage disequilibrium among the Europeans.
Finally, linear regression analysis was performed between the ACE level and each site, after age and sex were adjusted for, in Afro-Caribbean subjects. Again, only alleles T and A at markers 20833 and 22982 were associated with high ACE levels (P=.006 and .0002, respectively), further confirming the strong linkage disequilibrium between 22982 and the ACE-linked QTL.
Discussion
Several studies suggest that a functional polymorphism strongly influencing plasma ACE levels is contained within or near the ACE locus on chromosome 17. A previous study from the United Kingdom that relied on 10 SNP markers confirmed a single-variant model and indicated that the putative functional region was not in the promoter and 5′ region (Keavney et al. 1998; Farrall et al. 1999). However, this study did not permit precise localization of the region. By assuming a single-variant model, we have conducted a similar study in a more genetically diverse population and have found two potential ancestral breakpoints in a 9-kb region of the 3′ portion of ACE that likely harbors a functional variant. We used five SNPs and the commonly typed Alu insertion/deletion, chosen from the set of 78 variants in the 24 kb of genomic ACE sequence (Rieder et al. 1999) and a newly discovered SNP (24599) in the 3′ region of this gene.
In order to maximize our ability to detect a recombinant breakpoint, these markers were chosen to be spaced over the region beyond site 10696, where the presence of a large degree of linkage disequilibrium has been identified in individuals of European descent. Linkage disequilibrium was tested between these seven markers, indicating that, among Afro-Caribbeans, this region can be broken into three linked chromosomal regions: one including 10979, Alu, and 15214, one including 20833 and 22982, and the other including 23945 and 24599. In the Afro-Caribbean subjects, a recombinant breakpoint between 22982 and 23945 appears to define four major haplotypes: 1 (CIACG3C), 2 (TDGTA3C), 3 (CIACG2T), and 4 (TDGTA2T). These major haplotypes were grouped into three clades, A–C (fig. 3). The haplotypes in clade A are likely generated from clades B and C through a later recombination event occurring between site 15214 and 20833. However, in the German subjects, we only observed three major haplotypes. The striking variation in the amount of linkage disequilibrium between the Europeans and the Afro-Caribbeans, if consistent at other regions, also has important implications for future mapping studies using these techniques.
Haplotypes derived for this region were analyzed using a cladistic analysis and a regression method that assumes additive effects, which is the model usually confirmed (McKenzie et al. 1995; Villard et al. 1996; Keavney et al. 1998). Additive effects can also be observed from the genotype means in table 4, in which the mean ACE level in the presence of one haplotype from clade C is intermediate between that of C/C and individuals having no C clade haplotypes. However, it is possible that the haplotypes will interact to affect the ACE level. Such interactions are potentially of great interest, but a full investigation will require a larger sample size than is available here.
Hypothesis tests showed that the individuals within clades A, B, and C are similar and that clades A and B have similar phenotypes that are different from clade C. This finding suggests that the differences in the interval between sites 23945 and 24599 could not account for the variation in ACE activity, nor could the differences in the interval between sites 10979 and 15214. However, the region including the sites 20833 and 22982 was shared between these clade groupings (i.e., clades A and B share CG, and clade C has TA), suggesting that this region could contain a functional variant. The plasma ACE means of the groupings in table 4 are consistent with the results from the nested haplotype analysis for both Afro-Caribbean and German samples, further confirming that the functional variant could be located between sites 15214 and 23945.
pexcess is usually considered to be the preferred measure of disequilibrium for fine-scale linkage disequilibrium mapping for case-control data, because it is directly related to the recombination fraction between the trait and the marker loci and it is less dependent on the frequency of the trait haplotype that is sampled (Devlin and Risch 1995). A larger pexcess value indicates a stronger association between the trait and a marker, suggesting, in turn, a smaller distance between the functional variant and the marker, although this is not always the case (Eaves et al. 2000; Taillon-Miller et al. 2000). The increase in pexcess from site 10979 to 22982 and the subsequent decrease, from 22982 to 24599, provided additional evidence that a functional variant may lie around site 22982.
It should be pointed out that the linkage disequilibrium D′ and the haplotype analysis are dependent on the accuracy of the frequencies inferred by Clark’s subtracting algorithm, which does not provide the degree of uncertainty. In our data, although only 39 individuals had unambiguous haplotypes, the estimated D′ values were consistent with that estimated from the maximum-likelihood method (Excoffier and Slatkin 1995; authors' unpublished data).
It should also be pointed out that our conclusion depends on the strength of the evidence for the two ancestral recombinant breakpoints, which is not definitive. Further study, with more SNPs and a larger sample size, will be required.
Taken together, these results for linkage disequilibrium, cladistic analyses, and the single-site correlations with ACE activity, suggest that a region in the 3′ portion of this gene contains one (or more) variants in linkage disequilibrium with a functional variant affecting plasma ACE activities. Functional studies related to the SNPs in the 9-kb region need to be undertaken to identify the mechanism for elevated plasma ACE level. Several SNPs in this region are present in or near functionally relevant sites. For example, sites 22251 and 22982 (genotyped in this study) both occur within 6 bp of exon-splice junctions and may affect recently described alternative spliced forms of ACE that are directly solubilized (Sugimura et al. 1998).
In conclusion, these results provide strong evidence that the I/D polymorphism is unlikely to be the ACE-linked QTL and that at least one of the ACE-linked variants is likely to be close to site 22982. In addition, these studies demonstrate the advantages of using diverse ethnic groups and measured-haplotype analysis to help dissect the genetic bases of phenotypic traits.
Acknowledgments
Professor Robert C. Elston is thanked for helpful discussions. The authors also thank two anonymous referees for their constructive comments. This work was supported by the National Heart, Lung, and Blood Institute (grants HL45508 and HL53353), National Institutes of Health–National Human Genome Research Institute Fellowship 1 F32 HG00187-01 (to M.J.R.), the Reynold's Scholar program from the American Heart Association and the Deutsche Forschungsgemeinschaft (Schu 672/3-1, 9-1, and 10-1 [to H.S.] and Br 1626/2-1 [to U.B.]).
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for ACE [MIM 106180]) [PubMed]
References
- Bengtsson BO, Thomson G (1981) Measuring the strength of associations between HLA antigens and diseases. Tissue Antigens 18:356–363 [DOI] [PubMed] [Google Scholar]
- Castiglione CM, Deinard AS, Speed WC, Sirugo G, Rosenbaum HC, Zhang Y, Grandy DK, Grigorenko EL, Bonne-Tamir B, Pakstis AJ, Kidd JR, Kidd KK (1995) Evolution of haplotypes at the DRD2 locus. Am J Hum Genet 57:1445–1456 [PMC free article] [PubMed] [Google Scholar]
- Clark AG (1990) Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol 7:111–122 [DOI] [PubMed] [Google Scholar]
- Cooper R, Rotimi C, Ataman S, McGee D, Osotimehin B, Kadiri S, Muna W, Kingue S, Fraser H, Forrester T, Bennett F, Wilks R (1997) Hypertension prevalence in seven populations of African origin. Am J Public Health 87:160–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall KA, Templeton AR (1999) Statistical approaches to detecting recombination. In: Crandall KA (ed) The evolution of HIV. The Johns Hopkins University Press, Baltimore, pp 153–176 [Google Scholar]
- Danilov S, Savoie F, Lenoir B, Jeunemaitre X, Azizi M, Tarnow L, Alhenc-Gelas F (1996) Development of enzyme-linked immunoassays for human angiotensin I converting enzyme suitable for large-scale studies. J Hypertens 14:719–727 [DOI] [PubMed] [Google Scholar]
- Devlin B, Risch N (1995) A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29:311–322 [DOI] [PubMed] [Google Scholar]
- Evans AE, Poirier O, Kee F, Lecerf L, McCrum E, Falconer T, Crane J, O’Rourke DF, Cambien F (1994) Polymorphisms of the angiotensin-converting-enzyme gene in subjects who die from coronary heart disease. Q J Med 87:211–214 [PubMed] [Google Scholar]
- Eaves IA, Merriman TR, Barber RA, Nutland S, Tuomilehto-Wolf E, Tuomilehto J, Cucca F, Todd JA (2000) The genetically isolated populations of Finland and Sardinia may not be a panacea for linkage disequilibrium mapping of common disease genes. Nat Genet 25:320–323 [DOI] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M (1995): Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 12:921–927 [DOI] [PubMed] [Google Scholar]
- Farrall M, Keavney B, McKenzie C, Delepine M, Matsuda F, Lathrop GM (1999) Fine-mapping of an ancestral recombination breakpoint in DCP1. Nat Genet 23:270–271 [DOI] [PubMed] [Google Scholar]
- Galinsky D, Tysoe C, Brayne CE, Easton DF, Huppert FA, Dening TR, Paykel ES, Robinsztein DC (1997) Analysis of the apo E/apo C-I, angiotensin converting enzyme and methylenetetrahydrofolate reductase genes as candidates affecting human longevity. Atherosclerosis 129:177–183 [DOI] [PubMed] [Google Scholar]
- Hastbacka J, de la Chapelle A, Mahtani M, Clines G, Reeve-Daly M, Daly Mark, Hamilton BA, Kusumi K, Trivedi B, Weaver A, Colloma A, Lovett M, Buckler A, Kaitila I, Lander ES (1994) The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell 78:1073–1087 [DOI] [PubMed] [Google Scholar]
- Keavney B, McKenzie C, Connell JMC, Julier C, Ratcliffe PJ, Sobel E, Lathrop M, Farrall M (1998) Measured haplotype analysis of the angiotensin-1 converting enzyme (ACE) gene. Hum Mol Genet 7:1745–1751 [DOI] [PubMed] [Google Scholar]
- Keavney B, McKenzie C, Parish S, Palmer A, Clark S, Youngman L, Delepine M, Lathrop M, Peto R, Collins R (2000) For the ISIS Collaborators. Large-scale test of hypothesised associations between the angiotensin-converting-enzyme insertion/deletion polymorphism and myocardial infarction in about 5000 cases and 6000 controls. Lancet 355:434–444 [DOI] [PubMed] [Google Scholar]
- Keil U, Stieber J, Doring A, Chambless L, Hartel U, Filipatrik B (1988) The cardiovascular risk factor profile in the study area Augsburg: results from the first MONICA survey 1984/85. Acta Med Scand Suppl 728:119–128 [DOI] [PubMed] [Google Scholar]
- McKenzie CA, Julier C, Forrester T, McFarlane-Anderson N, Keavney B, Lathrop GM, Ratcliffe PJ, Farrall M (1995) Segregation and linkage analysis of serum angiotensin I converting enzyme levels: evidence for two quantitative-trait loci. Am J Hum Genet 57:1426–1435 [PMC free article] [PubMed] [Google Scholar]
- Neter J, Wasserman W, Kutner MH (1990) Applied linear statistical models: regression, analysis of variance, and experimental designs. 3d ed. Irwin, Homewood, IL [Google Scholar]
- Oren I, Brook JG, Gershoni-Baruch R, Kepten I, Tamir A, Linn S, Wolfovitz E (1999) The D allele of the angiotensin-converting enzyme gene contributes towards blood LDL-cholesterol levels and the presence of hypertension. Atherosclerosis 145:267–271 [DOI] [PubMed] [Google Scholar]
- Rieder MJ, Taylor SL, Clark AG, Nickerson DA (1999) Sequence variation in the human angiotensin converting enzyme. Nat Genet 22:59–62 [DOI] [PubMed] [Google Scholar]
- Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F (1990) An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 86:1343–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Leon D, Ozelius L, Kramer P, Almasy L, Singer B, Fahn S, Breakefield X, Bressman S (1995) Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat Genet 2:152–159 [DOI] [PubMed] [Google Scholar]
- Schunkert H (1997) Polymorphism of the angiotensin-converting enzyme gene and cardiovascular disease. J Mol Med 75:867–875 [DOI] [PubMed] [Google Scholar]
- Schunkert H, Dzau VJ, Tang SS, Hirsch AT, Apstein CS, Lorell BH (1990) Increased rat cardiac angiotensin converting enzyme activity and mRNA expression in pressure overload left ventricular hypertrophy. Effects on coronary resistance, contractility, and relaxation. J Clin Invest 86:1913–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubrier F, Nadaud S, Williams TA (1994) Angiotensin I converting enzyme gene: regulation, polymorphism and implications in cardiovascular diseases. Eur Heart J 15:24–29 [DOI] [PubMed] [Google Scholar]
- Sugimura K, Tian XL, Hoffmann S, Ganten D, Bader M (1998) Alternative splicing of the mRNA coding for the human endothelial angiotensin-converting enzyme: a new mechanism for solubilization. Biochem Biophys Res Commun 247:466–472 [DOI] [PubMed] [Google Scholar]
- Taillon-Miller P, Bauer-Sardina I, Saccone NL, Putzel J, Laitinen T, Cao A, Kere J, Pilia G, Eice JP, Kwok PY (2000) Juxtaposed regions of extensive and minimal linkage disequilibrium in human Xq25 and Xq28. Nat Genet 25:324–328 [DOI] [PubMed] [Google Scholar]
- Taittonen L, Uhari M, Kontula K, Kainulainen K, Miettinen H, Turtinen J, Nuutinen M (1999) Angiotensin converting enzyme gene insertion/deletion polymorphism, angiotensinogen gene polymorphisms, family history of hypertension, and childhood blood pressure. Am J Hypertens 12:858–866 [DOI] [PubMed] [Google Scholar]
- Templeton AR (1987) The general relationship between average effect and average excess. Genet Res 49:69–70 [DOI] [PubMed] [Google Scholar]
- Templeton AR, Boerwinkle E, Sing CF (1987) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. I. Basic theory and an analysis of alcohol dehydrogenase activity in drosophila. Genetics 117:343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton AR, Sing CF, Kessling A, Humphries S (1988) A cladistic analysis of phenotype associations with haplotypes inferred from restriction endonuclease mapping. II. The analysis of natural populations. Genetics 120:1145–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiret L, Rigat B, Visvikis S, Breda C, Corvol P, Cambien F, Soubrier F (1992) Evidence, from combined segregation and linkage analysis, that a variant of the angiotensin I–converting enzyme (ACE) gene controls plasma ACE levels. Am J Hum Genet 51:197–205 [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung K, Bonné-Tamir B, Santachiara-Benerecetti AS, Moral P, Krings M, Pa¨a¨bo S, Watson E, Risch N, Jenkins J, Kidd KK (1996) Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science 271:1380–1387 [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Goldman A, Calafell F, Speed WC, Deinard AS, Bonne-Tamir B, Kidd JR, Pakstis AJ, Jenkins T, Kidd KK (1998) A global haplotype analysis of the myotonic dystrophy locus: implications for the evolution of modern humans and for the origin of myotonic dystrophy mutations. Am J Hum Genet 62:1389–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe VO, Taylor SL, Nickerson DA (1996) Single-well genotyping of diallelic sequence variations by a two-color ELISA-based oligonucleotide ligation assay. Nucleic Acids Res 24:3728–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villard E, Tiret L, Visvikis S, Rakotovao R, Cambien F, Soubrier F (1996) Identification of new polymorphisms of the angiotensin I-converting enzyme (ACE) gene, and study of their relationship to plasma ACE levels by two-QTL segregation-linkage analysis. Am J Hum Genet 58:1268–1278 [PMC free article] [PubMed] [Google Scholar]
- Weber B, Nussberger J, Brunner H (1995) Angiotensin-converting enzyme inhibitors in hypertension. In: Laragh JH, Brenner BM (eds) Hypertension: pathophysiology, dianosis, and management. Raven Press, New York, pp 2861–2875 [Google Scholar]
- Weir BS (1996) Genetic data analysis II. Sinauer Associates, Sunderland, MA [Google Scholar]
- Williams GH (1988) Converting-enzyme inhibitors in the treatment of hypertension. N Engl J Med 319:1517–1525 [DOI] [PubMed] [Google Scholar]