

Abstract
Erythrokeratodermia variabilis (EKV) is an autosomal dominant keratinization disorder characterized by migratory erythematous lesions and fixed keratotic plaques. All families with EKV show mapping to chromosome 1p34-p35, and mutations in the gene for connexin 31 (Cx31) have been reported in some but not all families. We studied eight affected and three healthy subjects in an Israeli family, of Kurdish origin, with EKV. After having mapped the disorder to chromosome 1p34-p35, we found no mutations in the genes for Cx31, Cx31.1, and Cx37. Further investigation revealed a heterozygous T→C transition leading to the missense mutation (F137L) in the human gene for Cx30.3 that colocalizes on chromosome 1p34-p35. This nucleotide change cosegregated with the disease and was not found in 200 alleles from normal individuals. This mutation concerns a highly conserved phenylalanine, in the third transmembrane region of the Cx30.3 molecule, known to be implicated in the wall formation of the gap-junction pore. Our results show that mutations in the gene for Cx30.3 can be causally involved in EKV and point to genetic heterogeneity of this disorder. Furthermore, we suggest that our family presents a new type of EKV because of the hitherto unreported association with erythema gyratum repens.
Connexins (denoted by the prefix “Cx”) are a family of polypeptides that form the subunits of the gap-junction channels. Members of the connexin family are characterized by four hydrophobic transmembrane domains (M1–M4) that are linked by one cytoplasmic and two extracellular (E1 and E2) loops. The N and C termini are located on the cytoplasmic membrane face. Extracellular-loop and transmembrane domains display the highest homology between the connexin family members, whereas the cytoplasmic loop and the C-terminal region are highly variable. Six connexin polypeptides assemble into a connexon, a hemichannel that interacts with its counterpart on adjacent cells to form a complete intercellular channel, thereby connecting the cytoplasm of neighboring cells (Yeager and Nicholson 1996). Gap junctions are composed of numerous aggregated connexons. Functionally, gap junctions allow rapid transfer, between adjacent cells, of ions and second messenger molecules of size <1 kD, thereby permitting a coordinated response of groups of cells to external stimuli. This cell-cell communication is crucial for growth control and differentiation, as well as for maintenance of tissue homeostasis (Goodenough et al. 1996). Signal transmission via gap junction is modulated by a number of molecular and physiological conditions. Channel permeability and voltage-gating properties depend on the composition of the connexons. So far, >14 connexin genes have been described, and each tissue expresses a specific subset of it. The importance of the physiological functions of connexins is illustrated by the identification of connexin mutations as the molecular cause of different human diseases (Krutovskikh and Yamasaki 2000). These results from human diseases are further corroborated by the findings obtained in knockout mice (White and Paul 1999).
Erythrokeratodermia variabilis (EKV [MIM 133200]) is a rare, autosomal dominant genodermatosis characterized by the coexistence of two morphological features: usually transient areas of erythema and keratotic lesions that are more or less fixed. The erythematous patches vary in size, shape, and location and seem to be provoked by exposure to wind, cold, heat, and emotional stress (Rappaport et al. 1986). There are also fixed brownish-red hyperkeratotic plaques—particularly over the knees, elbows, groin, and axillae—in a symmetric distribution. There is a high intra- and interfamilial variability of the phenotype. Generally, EKV presents at birth or begins during early life. All families with EKV so far have shown mapping to chromosome 1p34-35, between the markers D1S496 and D1S186, with a maximum LOD score (Zmax) of 12.88 for D1S472 (Richard et al. 1997). These markers define a 2.6-cM candidate interval that contains a cluster of three genes for Cx: GJB3, encoding Cx31, GJA4, encoding Cx37, and GJB5, encoding Cx31.1 (van der Schroeff et al. 1988; Richard et al. 1997). Subsequently, six distinct Cx31 mutations were found—but only in 8 of 20 EKV families investigated, suggesting that EKV is genetically heterogeneous (Richard et al. 1998, 2000; Wilgoss et al. 1999). The human candidate region is orthologous to mouse chromosome 4, which harbors, in addition to the three genes described above, the gene for Cx30.3 (Hennemann et al. 1992). This suggests that this gene, GJB4, may be a good candidate. In this report, we describe an Israeli family, of Kurdish origin, with EKV, which we have also mapped to chromosome 1p34-35. Although we found no mutations in genes for Cx31, Cx31.1, and Cx37, we detected one, F137L, in the gene for Cx30.3. These findings provide the first and clear evidence for the causal involvement of a second connexin gene in this disorder and may delineate a specific subset.
An Israeli family of Kurdish origin that had already been described in 1978 (Hacham-Zadeh and Even-Paz 1978) was the object of our study. The pedigree included 32 individuals in four generations, of whom 8 affected and 3 healthy subjects were available for study (fig. 1). The dermatosis started at birth or shortly thereafter, and the patients were usually in good general health. Migratory erythematous lesions tended to gradually turn into more or less fixed keratotic plaques. Some of these erythematous lesions appeared as erythema gyratum repens, characterized by rapidly migrating figurate erythema 1–2 cm wide in an annular, garland, or spiral arrangement (Braun-Falco et al. 1984), which so far has not been reported in EKV (fig.2B and C). Generally, the palms and soles were intact. Lesions tended to become worse in summer and to improve in winter. In one of the patients, lesions also worsened during her pregnancies (fig. 2). Audiograms in two affected patients (III-6 and IV-1) were normal.
Figure 1 .

Pedigree and haplotype analysis of the family with EKV. Haplotypes are given (top to bottom) for markers D1S496, D1S472, and D1S186. Haplotype 1-5-4 was found to segregate with the disease. x = meiotic recombination.
Figure 2.

Clinical pictures. A, III-6, a 37-year-old man. The nape of the neck has brownish, lichenification-like hyperkeratotic plaque with mostly clear-cut borders. B, IV-1, a 19-year-old man. The periaxillary region has well-demarcated polycyclic plaque composed of diffuse scaling and erythema gyratum repens–like migratory lesions. C, IV-2, a 9.5-year-old girl. The anterior trunk has erythema gyratum repens–like migratory lesions.
To see whether EKV in this family also shows linkage to 1p34-p35, as described elsewhere (Richard et al. 1997), linkage analysis with the three Généthon microsatellite markers D1S186, D1S496, and D1S472 was performed (Dib et al. 1996). The Zmax (2.343, at recombination fraction 0) of the two-point analyses, when MLINK from the LINKAGE 5.2 package (Lathrop et al. 1984) is used, was obtained with marker D1S472, and all affected individuals shared a common allele (fig. 1). The coding region of the genes for Cx37 (GJA4) and Cx31 (GJB3) were amplified as described elsewhere (Richard et al. 1997, 1998). The gene for Cx31.1 (GJB5) was amplified using primers Cx31.1-F (5′-AGAGCAAGTCTGTGATAAATGTAGG-3′) and Cx31.1-R (5′-CCCCTACCTCATGGCTTAGC-3′). Sequence analysis of the genes for Cx31 and Cx31.1 did not reveal any pathogenic mutation in two patients (II-1 and III-8) and one unaffected individual (II-5) in the family with EKV. In one patient (III-2), we identified, at position 388 of the gene for Cx37, a G→A transition that abolishes a BsiEI restriction site. However, this previously reported polymorphism (Val130Ile) (Krutovskikh et al. 1996) did not cosegregate with the disease in the kindred with EKV, thus excluding the possibility that the disease is caused by this nucleotide change.
Mouse Cx30.3 transcripts are strongly expressed in keratinocytes, and the gene is localized within the connexin cluster on mouse chromosome 4 (Hennemann et al. 1992; Schwarz et al. 1992). This region is orthologous to human 1p34-p35, suggesting that the gene for Cx30.3 might be a candidate in some cases of EKV. Searching the nonredundant database by BLASTN, at the UK Human Genome Mapping Project Resource Centre Web site, with the mouse gene for Cx30.3 (GenBank accession number M91443), we identified the human PAC clone RP1-34M23 (GenBank accession number AL121988), which contains the four genes for Cx—Cx31, Cx31.1, Cx37, and Cx30.3. Expression analysis by reverse transcription (RT)–PCR showed that the gene for Cx30.3 is indeed expressed both in human skin in vivo and in cultured epidermal keratinocytes but not in cultured skin fibroblasts and leukocytes (fig. 3). Therefore, a 952-bp fragment containing the entire coding region of the gene for Cx30.3 was amplified using primers Cx30.3-F (5′-CAATCGCACCAGCATTAAGGG-3′; nt −126 to −106) and Cx30.3-R (5′-TGATCTTATCTGCTGATCTCGCAG-3′; nt 803–826) in one affected individual (III-2) in the family with EKV. Sequence analysis detected a heterozygous T→C nucleotide transversion at position 409, resulting in a phenylalanine-to-leucine change at position 137 (F137L) (fig. 4A and B). This heterozygous missense mutation, which creates a new SmlI restriction site, was found only in affected individuals in this family and was not detected in either unaffected family members (fig. 4C) or 200 alleles from normal white individuals. Sequence analysis of the gene for Cx30.3 did not reveal any other nucleotide variations in 10 control alleles.
Figure 3 .

Expression analysis of the Cx30.3 transcript. Lanes 1–4, RT-PCR with Cx30.3 primers. Lanes 5–8, PCR with Cx30.3 primers, omitting RT. Lanes 9–12, RT-PCR with actin primers. Total RNA for the reactions was derived from human skin biopsy (lanes 1, 5, and 9), cultured keratinocytes (lanes 2, 6, and 10), cultured skin fibroblasts (lanes 3, 7, and 11), and peripheral leukocytes (lanes 4, 8, and 12).
Figure 4.
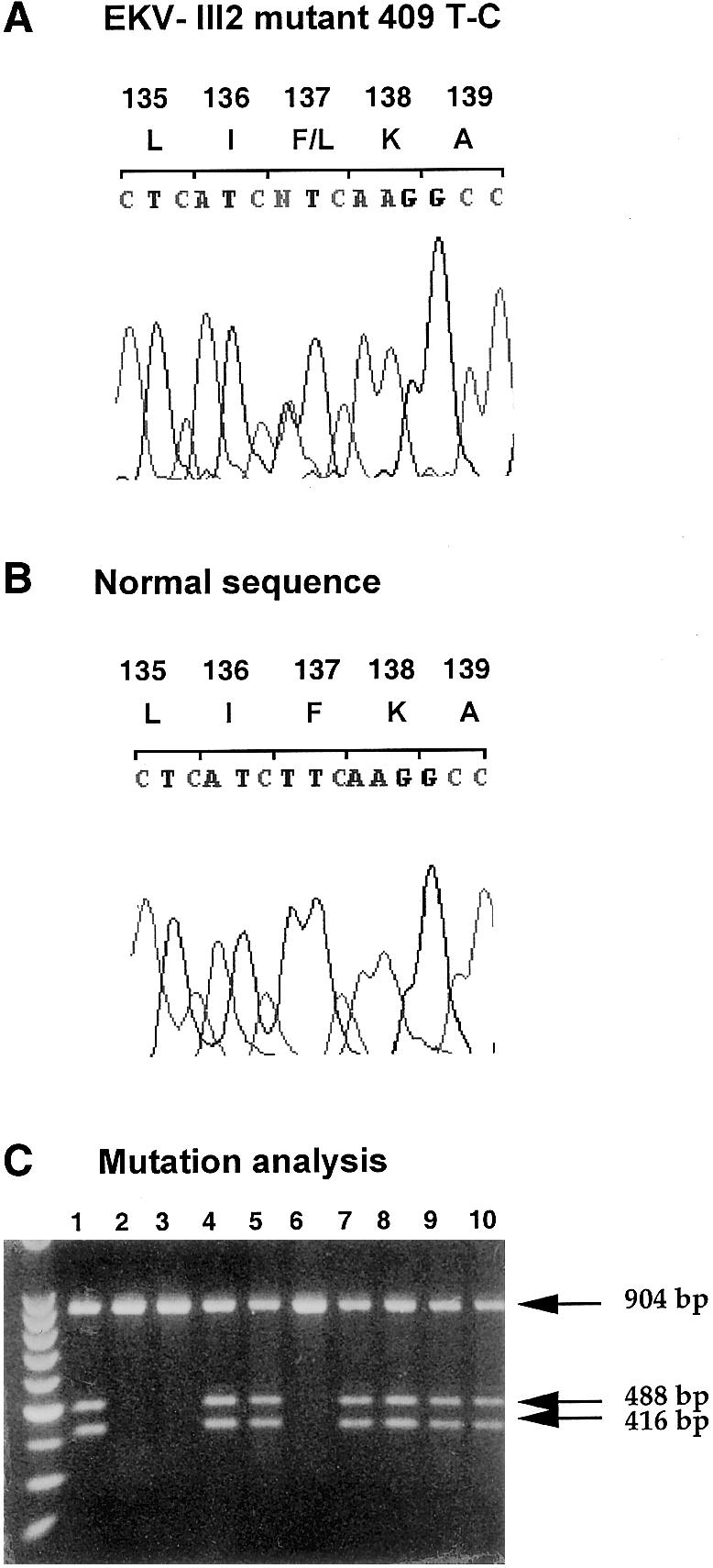
Molecular analysis of the gene for Cx30.3 (GJB4) in the family with EKV. A and B, Chromatograms of an affected individual with the Cx30.3 mutation, T409C, and a normal individual. C, Inheritance of the T409C mutation by restriction-enzyme digestion with SmlI. Normal alleles produce a 904-bp band that is cleaved into 488- and 416-bp fragments in mutant alleles. Lanes 1–10, Individuals II-1, II-5, III-1, III-2, III-3, III-5, III-6, III-8, III-9, and IV-1, respectively (see pedigree in fig. 1).
This mutation occurs within the third transmembrane domain, M3, of Cx30.3 and alters a phenylalanine residue that is highly conserved within the Cx family of proteins (fig. 5). The same mutation was previously reported in the homologous residue of the gene for Cx32 (residue F141L) in X-linked Charcot-Marie-Tooth disease (Rouger et al. 1997) and of the gene for Cx31 (residue F137L) in a sporadic case of severe EKV (Richard et al. 2000), providing evidence for the crucial function of this amino acid residue. Indeed, the M3 domain is known to be involved in the formation of the wall of the gap-junction pore and is at least partly implicated in the voltage gating of the channels. Closure of the channel occurs by the tilt of the M3 domain, which blocks the channel by moving small polar groups out of the lumen and permitting bulky phenylalanines into it (Bennett et al. 1991). It has been proposed that the kinetics of channel closure in response to voltage changes is inversely correlated with the number of phenylalanines in the M3 domain. Cx32 and Cx26, which harbor three phenylalanines, display slower kinetics than do Cx37 and Cx40, which have only one phenylalanine each. Cx30.3, with two phenylalanines, should have, according to this model, an intermediate channel-closure time (Unwin 1989; Hennemann et al. 1992). Hence, replacement of phenylalanine 137 by leucine may lead to faster closure of the channel, inhibiting the propagation of signals important for normal epidermal growth and differentiation. Two other mutations in the vicinity of Phe137, I141V and a deletion of Ile141, were identified in the M3 domain of Cx31 in recessive nonsyndromic hearing loss (Liu et al. 2000; Richard et al. 2000). Thus, mutations within the M3 domain may produce different phenotypes, such as hearing or skin disorders. This is not surprising, since Cx30.3 mRNA expression is restricted to skin and kidney in rodents, whereas the gene for Cx31 (GJB3) is also expressed in the inner ear and in the CNS (Hoh et al. 1991; Hennemann et al. 1992; Tucker and Barajas 1994).
Figure 5 .

Sequence alignment of the M3 and partial E2 domains of human Cx30.3 with other β-class gap-junction proteins. The arrowhead indicates the position of the Cx30.3 mutation, F137L, in the family with EKV. GenBank accession numbers are CAB90270 (for hCx30.3), AAA37428 (for mCx30.3), CAA53762 (for rCx30.3), CAA06165 (for hCx31), AAD18005 (for hCx31.1), NP_003995 (for hCx26), B29005 (for hCx32), and NP_006774 (for hCx30). The sequence of pCx30.3 was obtained from Itahana et al. (1996). h = human, m = mouse, r = rat, and p = porcine.
The similarity in clinical pictures of patients with EKV who have mutations in the genes for Cx31 and Cx30.3 suggests that these two connexins are functional partners in the formation of keratinocyte gap junctions. Nevertheless, the cutaneous phenotype of EKV with erythema gyratum repens without palmoplantar involvement, which was observed in some patients from the family that we studied, may define a specific clinical subset and indicate subtle molecular and biological differences. Our results show that the connexin protein Cx30.3 is important for both the regulation of epidermal differentiation and the maintenance of epidermal homeostasis. Further investigations will be necessary to elucidate the specific role that each of the epidermally expressed connexins plays in the physiology and pathophysiology of epidermal tissues and in the clinical phenotype of connexin mutants.
Acknowledgments
This project was supported by Swiss National Science Foundation grant 31-55849.98 (to D.H. and M.H.) and a grant by the Faculty of Medcine, University of Lausanne (to D.H. and D.S.). We thank F. Peneveyre for technical help, P. Meda for reviewing the manuscript before submission, and J.A. Häfliger for helpful discussions.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for mouse gene for Cx30.3 and human clone RP1-34M23 [accession numbera M91443 and AL121988])
- Généthon, http://www.genethon.fr (for genetic markers)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for EKV [MIM 133200]). [PubMed]
- UK Human Genome Mapping Project Resource Center, http://www.hgmp.mrc.ac.uk (for the BLAST search)
References
- Bennett MV, Barrio LC, Bargiello TA, Spray DC, Hertzberg E, Saez JC (1991) Gap junctions: new tools, new answers, new questions. Neuron 6:305–320 [DOI] [PubMed] [Google Scholar]
- Braun-Falco O, Plewig G, Wolff HH, Winkelmann R (1991) Erythematous and erythematosquamous skin diseases. In: Brann-Falco O, Plewig G, Wolff HH, Winkelmann R (eds) Dermatology. Springer, Berlin, pp 403–466 [Google Scholar]
- Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, Lathrop M, Gyapay G, Morissette J, Weissenbach J (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380:152–154 [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Goliger JA, Paul DL (1996) Connexins, connexons, and intercellular communication. Annu Rev Biochem 65:475–502 [DOI] [PubMed] [Google Scholar]
- Hacham-Zadeh S, Even-Paz Z (1978) Erythrokeratodermia variabilis in a Jewish Kurdish family. Clin Genet 13:404–408 [DOI] [PubMed] [Google Scholar]
- Hennemann H, Dahl E, White JB, Schwarz HJ, Lalley PA, Chang S, Nicholson BJ, Willecke K (1992) Two gap junction genes, connexin 31.1 and 30.3, are closely linked on mouse chromosome 4 and preferentially expressed in skin. J Biol Chem 267:17225–17233 [PubMed] [Google Scholar]
- Hoh JH, John SA, Revel JP (1991) Molecular cloning and characterization of a new member of the gap junction gene family, connexin-31. J Biol Chem 266:6524–6531 [PubMed] [Google Scholar]
- Itahana K, Morikazu Y, Takeya T (1996) Differential expression of four connexin genes, Cx-26, Cx-30.3, Cx-32, and Cx-43, in the porcine ovarian follicle. Endocrinology 137:5036–5044 [DOI] [PubMed] [Google Scholar]
- Krutovskikh V, Mironov N, Yamasaki H (1996) Human connexin 37 is polymorphic but not mutated in tumours. Carcinogenesis 17:1761–1763 [DOI] [PubMed] [Google Scholar]
- Krutovskikh V, Yamasaki H (2000) Connexin gene mutations in human genetic diseases. Mutat Res 462:197–207 [DOI] [PubMed] [Google Scholar]
- Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XZ, Xia XJ, Xu LR, Pandya A, Liang CY, Blanton SH, Brown SD, Steel KP, Nance WE (2000) Mutations in connexin31 underlie recessive as well as dominant non-syndromic hearing loss. Hum Mol Genet 9:63–67 [DOI] [PubMed] [Google Scholar]
- Rappaport IP, Goldes JA, Goltz RW (1986) Erythrokeratodermia variabilis treated with isotretinoin: a clinical, histologic, and ultrastructural study. Arch Dermatol 122:441–445 [PubMed] [Google Scholar]
- Richard G, Brown N, Smith LE, Terrinoni A, Melino G, Mackie RM, Bale SJ, Uitto J (2000) The spectrum of mutations in erythrokeratodermias—novel and de novo mutations in GJB3. Hum Genet 106:321–329 [DOI] [PubMed] [Google Scholar]
- Richard G, Lin JP, Smith L, Whyte YM, Itin P, Wollina U, Epstein E Jr, Hohl D, Giroux JM, Charnas L, Bale SJ, DiGiovanna JJ (1997) Linkage studies in erythrokeratodermias: fine mapping, genetic heterogeneity and analysis of candidate genes. J Invest Dermatol 109:666–671 [DOI] [PubMed] [Google Scholar]
- Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Epstein EH Jr, DiGiovanna JJ, Compton JG, Bale SJ (1998) Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet 20:366–369 [DOI] [PubMed] [Google Scholar]
- Rouger H, LeGuern E, Birouk N, Gouider R, Tardieu S, Plassart E, Gugenheim M, Vallat JM, Louboutin JP, Bouche P, Agid Y, Brice A (1997) Charcot-Marie-Tooth disease with intermediate motor nerve conduction velocities: characterization of 14 Cx32 mutations in 35 families. Hum Mutat 10:443–452 [DOI] [PubMed] [Google Scholar]
- Schwarz HJ, Chang YS, Hennemann H, Dahl E, Lalley PA, Willecke K (1992) Chromosomal assignments of mouse connexin genes, coding for gap junctional proteins, by somatic cell hybridization. Somat Cell Mol Genet 18:351–359 [DOI] [PubMed] [Google Scholar]
- Tucker MA, Barajas L (1994) Rat connexins 30.3 and 31 are expressed in the kidney. Exp Cell Res 213:224–230 [DOI] [PubMed] [Google Scholar]
- Unwin N (1989) The structure of ion channels in membranes of excitable cells. Neuron 3:665–676 [DOI] [PubMed] [Google Scholar]
- van der Schroeff JG, van Leeuwen-Cornelisse I, van Haeringen A, Went LN (1988) Further evidence for localization of the gene of erythrokeratodermia variabilis. Hum Genet 80:97–98 [DOI] [PubMed] [Google Scholar]
- White TW, Paul DL (1999) Genetic diseases and gene knockouts reveal diverse connexin functions. Annu Rev Physiol 61:283–310 [DOI] [PubMed] [Google Scholar]
- Wilgoss A, Leigh IM, Barnes MR, Dopping-Hepenstal P, Eady RA, Walter JM, Kennedy CT, Kelsell DP (1999) Identification of a novel mutation R42P in the gap junction protein β-3 associated with autosomal dominant erythrokeratoderma variabilis. J Invest Dermatol 113:1119–1122 [DOI] [PubMed] [Google Scholar]
- Yeager M, Nicholson BJ (1996) Structure of gap junction intercellular channels. Curr Opin Struct Biol 6:183–192 [DOI] [PubMed] [Google Scholar]