

Abstract
This paper reviews hypotheses about roles of angiogenesis in the pathogenesis of inflammatory disease in two organs, the synovial joint and the lung. Neovascularisation is a fundamental process for growth and tissue repair after injury. Nevertheless, it may contribute to a variety of chronic inflammatory diseases, including rheumatoid arthritis, osteoarthritis, asthma, and pulmonary fibrosis. Inflammation can promote angiogenesis, and new vessels may enhance tissue inflammation. Angiogenesis in inflammatory disease may also contribute to tissue growth, disordered tissue perfusion, abnormal ossification, and enhanced responses to normal or pathological stimuli. Angiogenesis inhibitors may reduce inflammation and may also help to restore appropriate tissue structure and function.
Keywords: angiogenesis, arthritis, interstitial lung disease, joint disease, neovascularisation
Introduction
Inflammation
Inflammation is a response of vascularised tissue to sub-lethal injury [1]. Inflammation can be classified according to duration as either acute or chronic. In acute inflammation, changes in small blood vessels result in fluid and granulocytic cells accumulating at the site of injury. This reaction may trigger a systemic response such as fever, leucocytosis, protein catabolism, and altered hepatic synthesis of plasma proteins such as C-reactive protein. Chronic inflammation is characterised by tissue infiltration by macrophages and lymphocytes.
Inflammation may be considered a homeostatic response designed to destroy or inactivate invading pathogens, remove waste and debris, and permit restoration of normal function, either through resolution or repair. Tissue structure is normal after resolution, whereas repair leads to a functional, but morphologically altered, organ. In acute inflammation, tissue damage is followed by resolution, whereas in chronic inflammation, damage and repair continue concurrently. The initial inflammatory response is usually acute, and may or may not evolve into chronic inflammation. However, chronic inflammation is not always preceded by an acute phase. Although usually beneficial to the organism, inflammation itself may lead to tissue damage, resulting in escalation of chronic inflammation.
Angiogenesis
Angiogenesis is the growth of new capillary blood vessels from pre-existing vasculature [2]. It is a fundamental process required for embryogenesis, growth, tissue repair after injury, and the female reproductive cycle. It may also contribute to the pathology of conditions such as cancer, psoriasis, diabetic retinopathy, and chronic inflammatory diseases in joints or lungs. Angiogenesis is stimulated when hypoxic, diseased, or injured tissues produce and release angiogenic promoters such as vascular endothelial growth factor (VEGF) or fibroblast growth factor (FGF)-1. These angiogenic factors stimulate the migration and proliferation of endothelial cells in existing vessels and, subsequently, the formation of capillary tubes and the recruitment of other cell types to generate and stabilise new blood vessels.
Inflammation may promote angiogenesis in a number of ways [3,4,5]. Inflammatory tissue is often hypoxic, and hypoxia can induce angiogenesis through upregulation of factors such as VEGF. Extravasated plasma proteins such as fibrinogen products may stimulate neovascularisation. Inflammatory cells such as macrophages, lymphocytes, mast cells, and fibroblasts, and the angiogenic factors they produce, can stimulate vessel growth. Many proinflammatory cytokines, such as tumour necrosis factor(TNF)-α, may have angiogenic activity in addition to proinflammatory activity. Increased blood flow itself may stimulate angiogenesis through shear stresses on the endothelium. Inflammation also may upregulate the expression of angiogenic growth factors such as VEGF and FGF-1 by resident cells such as fibroblasts [6,7].
Inflammation in the joint
The synovial joint has a specialised structure allowing both stability and movement. The normal synovial lining is highly vascular, providing support to the avascular articular cartilage (Fig. 1a) [8]. The synovial vasculature is fed by a variety of arteries, which anastomose and branch in arcades towards the synovial lining region. The deeper layers of the articular cartilage also may obtain metabolic support from blood vessels at the osteochondral junction. Vascular proliferation is not usually a feature of the normal joint [7].
Figure 1.
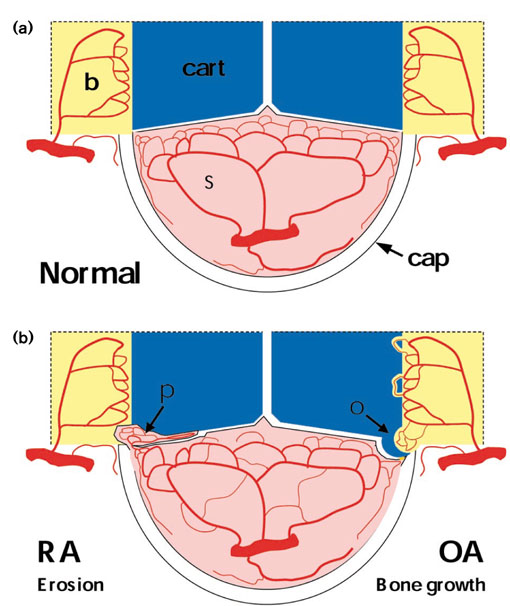
Vascular plasticity in rheumatoid arthritis and osteoarthritis. (a) Vascular distribution in the normal joint. Blood vessels in the synovium (s) are highly organised, with larger vessels sparsely distributed in the sublining region, branching to form dense microvascular networks adjacent to the synovial surface and capsule (cap). An additional vascular network in the subchondral bone (b) does not normally cross into the articular cartilage (cart), which remains avascular. (b) In rheumatoid arthritis (RA), hyperplastic synovial pannus (p) attaches to and invades articular cartilage and adjacent bone. The synovial vascular network is reorganised, leading to reduced vascular densities adjacent to the joint space and increased vascular densities in the deeper synovium. Similar changes in vascular organisation occur in the synovium in osteoarthritis (OA). However, rather than destruction of bone and cartilage by pannus, vascular invasion of cartilage in the developing osteophyte (o) and at the osteochondral junction may lead to advancing endochondral ossification. Innervation of new vessels by fine, unmyelinated sensory nerves may contribute to increased pain sensation.
Trauma and disease are both causes of joint inflammation. Joint swelling due to inflammation may be caused by intra-articular effusion, synovial thickening, periarticular soft-tissue inflammation (bursitis or tendonitis), and eventually bony enlargement. Injury may lead to chronic conditions in some instances, such as osteoarthritis induced by trauma, although the initiating factor is unknown in most forms of arthritis. The diseases in which synovial inflammation is common include rheumatoid arthritis and osteoarthritis, seronegative spondylarthropathies, and septic arthritis.
During joint inflammation, increased angiogenesis leads to reorganisation of articular blood vessels [9,10]. There are reduced vascular densities adjacent to the synovial lining region, increased vascular densities in the deeper synovium, growth and invasion of vascularised pannus into articular cartilage and subchondral bone, and vascularisation and endochondral ossification at the osteochondral junction (Fig. 1b).
Inflammatory conditions in the lung
The lung is a highly specialised and vital organ, which has evolved to maximise gas exchange. It possesses a dual circulation. The bronchial, systemic circulation provides for the metabolic requirements of the lung tissues. The pulmonary circulation transmits deoxygenated blood through the interface with the alveolar airspace. Each circulation is highly organised and tightly regulated in the normal lung, with few anastomoses between the two.
Each compartment of the lung may be affected by chronic inflammation, with functional consequences that reflect the role of that structure. Mural inflammation in muscular airways contributes to limitation of airflow, which may be variable (asthma) or fixed. Intraluminal inflammation is a feature of bronchiectasis and bronchiolitis obliterans organising pneumonia (BOOP). Inflammation of the lung parenchyma may lead to thickening of the alveolar septa or filling of alveolar spaces with granulation tissue and subsequent fibrosis. This pulmonary fibrosis may be a result of a variety of lung injuries, including neonatal hyperoxia, adult respiratory distress syndrome, infection, or extrinsic allergic alveolitides. Pulmonary fibrosis is often idiopathic, as in interstitial organising pneumonia or idiopathic pulmonary fibrosis. Pleural inflammation may result from infection or be idiopathic. Inflammation of the large airways may be a particular challenge in lung transplantation. Finally, unlike the joint, the lung is a common site for primary and secondary neoplasia, the inflammatory response to which may modify clinical features.
Inflammatory lung disease is not uncommon in rheumatic conditions [11,12]. Patients with rheumatoid arthritis have an increased susceptibility to chronic suppurative lung disease and BOOP. Pulmonary fibrosis and pleural inflammation may be specific features of a variety of rheumatological conditions, including rheumatoid arthritis, systemic lupus erythematosus, and systemic sclerosis.
The extent to which these disease associations reflect common pathological mechanisms or linked genetic or environmental susceptibility remains unknown. At present it seems unlikely that primary abnormalities of vascular growth will explain the incidence and association of pulmonary and articular inflammation. However, both synovium and lung have evolved as highly vascular tissues. Vascular turnover is extremely low in normal synovium and lung. Synovial and lung microvasculatures are finely organised, with highly developed neurovascular regulatory systems. Destabilising their microvasculatures through angiogenesis may have consequences that are shared between the two organs.
Methods
This review is directed at hypotheses rather than observations. In any area of research, the original ideas are the initiating events, and from these, experiments are designed and observations collected. This review addresses hypotheses about the role of angiogenesis in the pathogenesis of inflammatory disease. Hypotheses from two separate areas have been compared, the joint and the lung. The mechanisms by which inflammation may cause angiogenesis are not addressed in detail, as these have been extensively reviewed elsewhere [2,3,13,14]. Similarly, readers are referred to the referenced articles and reviews for more detailed discussions of the quality of evidence supporting the identified hypotheses.
Articles were identified using the MEDLINE database 1966 to July 2000, and references cited within these articles. Search terms for lung or joint diseases were combined with terms for angiogenesis. One hundred and forty abstracts relating to lungs and 126 relating to joints were identified, leading to detailed review of 49 and 41 articles respectively.
Reviewed hypotheses
Potentiation of inflammation by angiogenesis
It is commonly hypothesised with regard to both lungs and joints that angiogenesis potentiates inflammation. Angiogenic factors are often proinflammatory; expression of adhesion molecules such as integrin αvβ3 and e-selectin is upregulated in the neovasculature, and new vessels appear particularly sensitive to inflammogens [15,16,17,18,19]. The angiogenic factor FGF-1 enhances antigen-induced synovitis in rabbits but is not proinflammatory when administered alone [20]. Hypotheses that angiogenesis potentiates inflammation are supported by the observation that synovitis in laboratory animals is inhibited by agents that inhibit angiogenesis, such as TNP470, integrin αv antagonists, 2-methoxyestradiol, paclitaxel, and P38 mitogen-activated protein kinase inhibitors [20,21,22,23,24,25]. However, angiogenesis can occur in the absence of inflammation, for example during embryonic growth and in the female reproductive cycle. Furthermore, all agents so far reported may have anti-inflammatory activity independent of their anti-angiogenic activity. Inflammation and angiogenesis can occur independently, although it seems likely that their coexistence may lead to more severe, damaging, and persistent inflammation [26].
Tissue growth
A role for angiogenesis in enhancing tumour growth is now widely accepted, and a variety of anti-angiogenic agents are in clinical development. Folkman championed the concept that vascular growth permits tumour growth, and that tumours cannot grow beyond a few millimetres in diameter in the absence of angiogenesis [27]. Mechanisms for angiogenesis-dependent tumour growth were originally conceived in terms of the neovasculature meeting the metabolic requirements of the expanding tissue. Extensions to the hypothesis have proposed other mechanisms, including generation of growth factors by vascular cells and inhibition of apoptosis in the growing tissue [28,29]. Preventing the function of growth factors such as VEGF can abolish pathological angiogenesis in tumours.
Angiogenesis may contribute to thickening of airways in asthma and of lung parenchyma in pulmonary fibrosis, and to growth of sarcoid granulomas [30,31,32,33]. Growth of granulation tissue into airspaces also may be angiogenesis-dependent in bronchi after lung transplantation, in bronchioles in BOOP, and in alveoli after acute lung injury or in other forms of pulmonary fibrosis [34]. In each of these situations, the expanded tissue may impair function.
Subepithelial thickening of the airway wall results in reduced luminal diameter that is exacerbated by bronchoconstriction [35]. Obstruction of airways or replacement of alveolar airspaces by granulation tissue impairs circulation of gases and gas exchange. Pathophysiological enhancement of the tissue's normal function may lead to increased bronchial responsiveness to exercise and allergens [36,37].
Angiogenesis may also contribute to growth of the synovial pannus in rheumatoid arthritis [20,24,38,39,40,41,42]. Mass effects of increased synovial volume have rarely been considered in detail. Although synovial swelling is clinically apparent, obstructive effects on movement of synovial fluid or the joint itself may not be of great importance. Interposition of expanded, innervated synovium between articulating surfaces may contribute to pain on movement.
Rheumatologists have focused more on pathophysiological than mechanical consequences of tissue growth. Enhancement of the tissue's normal function may contribute to excessive formation of synovial fluid. The expanded synovium, furthermore, is recognised as an abnormal tissue that has acquired novel activities such as antibody and cytokine production, adhesion, and invasion of articular cartilage and bone [43].
Inhibitors of angiogenesis can reduce tumour bulk, by shifting the balance from tumour cell proliferation to apoptosis [29]. Increased cell turnover is also a feature of inflammatory diseases in both joints and lung, raising the possibility that inhibiting angiogenesis may reduce tissue mass in those organs also [7,40,44]. Clinical benefit from debulking synovium has already been indicated through surgical and radiation synovectomy. Inhibition of angiogenesis may permit repeated polyarticular medical synovectomy.
Vascular reorganisation
Normal lung and synovium both have highly organised and regulated microvascular beds. Increased vascular turnover leads to reorganisation of the microvasculature and to changes in vasoregulatory systems, characterised by reduced innervation and increased sensitivity to inflammatory mediators [4].
In pulmonary fibrosis, vascular redistribution may result in reduced vascular densities in alveolar walls adjacent to the airspace, with increased numbers of vessel profiles located further from the airspaces than the diffusion distance for oxygen. Capacity for vascular proliferation may be greater in bronchial vessels than in pulmonary vessels, leading to a greater contribution of systemic blood flow to lung perfusion [45,46].
The numbers of bronchopulmonary anastomoses are increased in a wide variety of inflammatory lung diseases, including after acute lung injury and in pulmonary fibrosis, bronchiectasis, and lung abscess [46,47,48]. Obliteration of airspaces, vascular redistribution, and bronchopulmonary anastomoses combine to impair gas transfer. Together they produce physiological shunting of blood from bronchial to pulmonary circulations, retrograde blood flow within the pulmonary vessels, increased pulmonary artery pressure and oxygen tension, and an increase in bronchial blood flow from the normal 1% to as much as 35% of cardiac output [33,48]. New blood vessels respond with increased plasma extravasation to proinflammatory agents such as substance P, and the resulting tissue oedema may further contribute to increased tissue mass and damage [37,49,50].
Synovial angiogenesis also leads to vascular reorganisation. Chronic synovitis is associated with vascular rarefaction adjacent to the synovial surface and increased vascular densities further away from the joint space [9,10]. The microvasculature of chronic synovitis resembles that of other neovascular beds with reduced innervation and increased plasma extravasation [7,51]. The role of shunting in the inflamed synovium, however, has received little attention. Structures resembling arteriovenous anastomoses have been described in normal synovium, and during synovitis, reduction of temperature gradients from the synovial cavity (normally warmer) to the skin suggests that diversion of the flow of oxygenated blood away from the synovial surface may be a feature of inflammatory synovitis [8].
Changes in the distribution of blood flow may contribute to synovial hypoxia and to the maintenance of synovial inflammation and impaired cartilage metabolism [7,9,52]. Indeed, neovascularisation may generate an immature, disregulated microvascular bed with a reduced capacity to match perfusion to local metabolic demand [40]. Inhibition of angiogenesis in the early stages of inflammation may retain organisation and maturity of the microvasculature to enable resolution with matching of vascular function to tissue requirements. Identification of factors that facilitate regression of aberrant vessels or promote the development of vasoregulatory systems may be helpful where vascular reorganisation has already occurred.
Tissue phenotype
As well as permitting tissue growth, neovascularisation can alter tissue phenotype. Angiogenesis in tumours has been associated with a phenotypic switch associated with rapid growth and metastasis. This angiogenic switch may precede oncogenic transformation, with increased vascular densities associated with 'preneoplastic', dysplastic bronchial epithelium [28]. Osteoblastic transformation and ossification follow vascular invasion of cartilage [53,54]. Inhibition of angiogenesis can prevent endochondral ossification and may have potential therapeutic application in inhibiting new bone formation in osteoarthritis and spondylarthropathies. Further research may identify specific, vascular-derived factors that facilitate phenotypic changes from cartilage to bone, or from dysplastic to neoplastic epithelium.
Tissue repair
Much attention has focused on potentially damaging consequences of angiogenesis, although another school of thought emphasises the essential role of angiogenesis in tissue repair. Bronchial dehiscence remains an important complication of lung transplantation and appears related to ischaemia in the transplanted tissue [34,48,55]. Enhancement of neovascularisation has been considered to minimise the risk of dehiscence, using, for example, vascularised omental grafts or angiogenic growth factors [34,48,55].
A reparative role of angiogenesis has also been proposed in nontraumatic inflammation. Greater luminal angiogenesis in BOOP than in idiopathic pulmonary fibrosis has been proposed as an explanation for the better prognosis of BOOP and its greater responsiveness to therapy [45]. We similarly proposed that angiogenesis in the very early phase of synovitis might help to maintain joint nutrition and facilitate resolution or repair [40]. In an animal model of synovitis, however, angiogenesis at day 3 predicted persistence rather than the resolution of inflammation, leading us to prefer an alternative hypothesis that disruption of the normal synovial vasculature impairs recovery from acute inflammatory synovitis [40].
The outcome of chronic inflammation can be viewed as a balance between inflammation/injury and repair. The effects of angiogenesis inhibition on this balance will require careful study in each clinical situation.
Haemorrhage
New vessels have incomplete walls and may be particularly susceptible to disruption and extravasation of blood. This has been proposed as a cause of haemoptysis and pulmonary haemorrhage in inflammatory lung disease [5,48,56]. Haemosiderin deposition and extravasated red cells are commonly observed in inflammatory synovitis, although the contribution of angiogenesis to synovial microhaemorrhage is unknown, and its contribution to synovial inflammation remains unclear. The inflammatory potential and algesic effect of haemarthrosis is evident, however, in patients with haemophilia.
Angiogenesis, the innocent bystander
Although rarely expounded in print, an alternative hypothesis should be considered: that angiogenesis plays no part in the pathogenesis or clinical consequences of chronic inflammation. Angiogenesis has been described as a 'side effect' of granuloma formation, or a 'bystander' to growth factors in asthma [48,57]. However, evidence that angiogenesis is important in chronic inflammation is no weaker than was that for the pivotal role for TNF-α at the beginning of development of anti-TNF therapies. More evidence should be sought before the more exciting hypotheses can be rejected.
Conclusions
Angiogenesis and inflammation are codependent processes [58]. Some forms of inflammation, especially chronic inflammation, can stimulate vessel growth. New vessels may contribute to a tissue's altered inflammatory response. Angiogenesis and inflammation, however, remain distinct processes that can occur independently of each other. Inhibition of chronic inflammation may be expected to inhibit angiogenesis where the stimulus for vascular growth is derived from inflammatory cells such as macrophages [20]. Some anti-inflammatory agents may also have anti-angiogenic activity that is independent of their effects on inflammation [58,59,60,61,62]. Evidence is now accumulating that agents that have been designed to specifically inhibit angiogenesis may also inhibit chronic inflammation [20,24,25].
Perhaps the most important application of angiogenesis inhibitors in inflammation will be in preventing the clinico-pathological consequences of vascular growth. Angiogenesis inhibitors may be able to inhibit tissue growth and remodelling, impair the formation of vascular anastomoses, shunting, and mismatch between blood flow and metabolic demand, and normalise vascular reactivity. Other proposed therapeutic applications in arthritis have included reducing joint damage, preventing the development or persistence of synovitis, and reducing inflammation and pannus growth.
In this review we have focused on hypotheses, rather than the evidence that supports or refutes those hypotheses. It was not possible to identify the origins or authorship of most hypotheses in published articles. Despite this limitation, the current review draws attention to the intellectual background that has led the researchers cited to investigate diverse potential consequences of angiogenesis in inflammation.
Reviewing the possible roles of angiogenesis in inflammatory lung disease suggests additional possible mechanisms by which angiogenesis may contribute to human joint diseases. Does fragility of new vessels within the synovium lead to recurrent microhaemorrages that sustain inflammation? Does the growing vasculature generate factors that transform the surrounding tissue? Do these factors increase proliferation or endochondral ossification, and do they increase invasiveness in the rheumatoid pannus, or increase synovial fibrosis? Does angiogenesis generate vascular anastomoses within the synovium, with shunting of blood away from the synovial surface, impairment of cartilage metabolism, and potentiation of joint damage?
General reduction of vessel growth by nonselective angiogenesis inhibitors may have limited therapeutic application in chronic diseases such as rheumatoid arthritis because of predictable adverse effects of inhibiting physiological and reparative angiogenesis [4]. The nature and regulation of angiogenesis may differ in different physiological and pathological states. Refining hypotheses about the specific aspects of angiogenesis that it would be desirable to inhibit in inflammation should lead to the development of more specific and less toxic pharmacological agents that can complement other therapeutic strategies in human arthritis and lung disease.
Abbreviations
BOOP = bronchiolitis obliterans organising pneumonia; FGF = fibroblast growth factor; TNF = tumour necrosis factor; VEGF = vascular endothelial growth factor.
References
- Ballou SP, Kushner I. Laboratory evaluation of inflammation. In Textbook of Rheumatology Edited by Kelley WN, Harris ED Jr, Ruddy S, Sledge CB Philadelphia: W B Saunders Co, 2000. pp. 697–703.
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Polverini PJ. Role of the macrophage in angiogenesis-dependent diseases. In Regulation of angiogenesis Edited by Goldberg ID, Rosen EM Berlin: Birkhauser, 1997. pp. 11–28. [DOI] [PubMed]
- Walsh DA. Angiogenesis and arthritis. Rheumatology (Oxford) 1999;38:103–112. doi: 10.1093/rheumatology/38.2.103. [DOI] [PubMed] [Google Scholar]
- McColley SA, Stellmach V, Boas SR, Jain M, Crawford SE. Serum vascular endothelial growth factor is elevated in cystic fibrosis and decreases with treatment of acute pulmonary exacerbation. Am J Respir Crit Care Med. 2000;161:1877–1880. doi: 10.1164/ajrccm.161.6.9905022. [DOI] [PubMed] [Google Scholar]
- Qu Z, Huang XN, Ahmadi P, Andresevic J, Planck SR, Hart CE, Rosenbaum JT. Expression of basic fibroblast growth factor in synovial tissue from patients with rheumatoid arthritis and degenerative joint disease. Lab Invest. 1995;73:339–346. [PubMed] [Google Scholar]
- Walsh DA, Wade M, Mapp PI, Blake DR. Focally regulated endothelial proliferation and cell death in human synovium. Am J Pathol. 1998;152:691–702. [PMC free article] [PubMed] [Google Scholar]
- Haywood L, Walsh DA. Vasculature of the normal and arthritic synovial joint. Histol Histopathol. 2001;16:277–284. doi: 10.14670/HH-16.277. [DOI] [PubMed] [Google Scholar]
- Stevens CR, Blake DR, Merry P, Revell PA, Levick JR. A comparative study by morphometry of the microvasculature in normal and rheumatoid synovium. Arthritis Rheum. 1991;34:1508–1513. doi: 10.1002/art.1780341206. [DOI] [PubMed] [Google Scholar]
- Ceponis A, Konttinen YT, MacKevicius Z, Solovieva SA, Hukkanen M, Tamulaitiene M, Matulis A, Santavirta S. Aberrant vascularity and von Willebrand factor distribution in inflamed synovial membrane. J Rheumatol. 1996;23:1880–1886. [PubMed] [Google Scholar]
- Tanoue LT. Pulmonary manifestations of rheumatoid arthritis. Clin Chest Med. 1998;19:667–685. doi: 10.1016/s0272-5231(05)70109-x. [DOI] [PubMed] [Google Scholar]
- Wallaert B, Dugas M, Dansin E, Perez T, Marquette CH, Ramon P, Tonnel AB, Voisin C. Subclinical alveolitis in immunological systemic disorders. Transition between health and disease? Eur Respir J. 1990;3:1206–1216. [PubMed] [Google Scholar]
- Koch AE. Review: angiogenesis: implications for rheumatoid arthritis. Arthritis Rheum. 1998;41:951–962. doi: 10.1002/1529-0131(199806)41:6<951::AID-ART2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Stooker W, Wildevuur CR, van Hinsbergh VW, Eijsman L. Let's understand nature better: de- and regeneration of autologous and artificial small caliber vascular grafts. Artif Organs. 1998;22:63–67. doi: 10.1046/j.1525-1594.1998.06067.x. [DOI] [PubMed] [Google Scholar]
- Fava RA, Olsen NJ, Spencer-Green G, Yeo KT, Yeo TK, Berse B, Jackman RW, Senger DR, Dvorak HF, Brown LF. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med. 1994;180:341–346. doi: 10.1084/jem.180.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgstrom P, Hughes GK, Hansell P, Wolitsky BA, Sriramarao P. Leukocyte adhesion in angiogenic blood vessels. Role of E-selectin, P-selectin, and beta2 integrin in lymphotoxin-mediated leukocyte recruitment in tumor microvessels. J Clin Invest. 1997;99:2246–2253. doi: 10.1172/JCI119399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DM, Schoeb TR, Lindsey JR. Mycoplasma pulmonis infections cause long-lasting potentiation of neurogenic inflammation in the respiratory tract of the rat. J Clin Invest. 1991;87:787–799. doi: 10.1172/JCI115082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA. Definition of two angiogenic pathways by distinct alpha v integrins. Science. 1995;270:1500–1502. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storgard CM, Stupack DG, Jonczyk A, Goodman SL, Fox RI, Cheresh DA. Decreased angiogenesis and arthritic disease in rabbits treated with an alphavbeta3 antagonist. J Clin Invest. 1999;103:47–54. doi: 10.1172/JCI3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsson E, Tarkowski A. Suppression of type II collagen-induced arthritis by the endogenous estrogen metabolite 2-methoxyestradiol. Arthritis Rheum. 1997;40:154–163. doi: 10.1002/art.1780400120. [DOI] [PubMed] [Google Scholar]
- Jackson JR, Bolognese B, Hillegass L, Kassis S, Adams J, Griswold DE, Winkler JD. Pharmacological effects of SB 22 a selective inhibitor of P38 mitogen-activated protein kinase, in angiogenesis and chronic inflammatory disease models. J Pharmacol Exp Ther. 0025;284:687–692. [PubMed] [Google Scholar]
- Arsenault AL, Lhotak S, Hunter WL, Banquerigo ML, Brahn E. Taxol involution of collagen-induced arthritis: ultrastructural correlation with the inhibition of synovitis and neovasculariza-tion. Clin Immunol Immunopathol. 1998;86:280–289. doi: 10.1006/clin.1997.4479. [DOI] [PubMed] [Google Scholar]
- Peacock DJ, Banquerigo ML, Brahn E. A novel angiogenesis inhibitor suppresses rat adjuvant arthritis. Cell Immunol. 1995;160:178–184. doi: 10.1016/0008-8749(95)80025-e. [DOI] [PubMed] [Google Scholar]
- Peacock DJ, Banquerigo ML, Brahn E. Angiogenesis inhibition suppresses collagen arthritis. J Exp Med. 1992;175:1135–1138. doi: 10.1084/jem.175.4.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risau W. Angiogenesis is coming of age. Circ Res. 1998;82:926–928. doi: 10.1161/01.res.82.8.926. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis. Adv Cancer Res. 1985;43:175–203. doi: 10.1016/s0065-230x(08)60946-x. [DOI] [PubMed] [Google Scholar]
- Keith RL, Miller YE, Gemmill RM, Drabkin HA, Dempsey EC, Kennedy TC, Prindiville S, Franklin WA. Angiogenic squamous dysplasia in bronchi of individuals at high risk for lung cancer. Clin Cancer Res. 2000;6:1616–1625. [PubMed] [Google Scholar]
- Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- Keane MP, Wilke CA, Burdick MD, Morris SB, Glass MC, Strieter RM. CXC chemokines regulate angiogenic activity in acute lung injury. Chest. 1999;116:93S–94S. doi: 10.1378/chest.116.suppl_1.93s. [DOI] [PubMed] [Google Scholar]
- Henke C, Fiegel V, Peterson M, Wick M, Knighton D, McCarthy J, Bitterman P. Identification and partial characterization of angiogenesis bioactivity in the lower respiratory tract after acute lung injury. J Clin Invest. 1991;88:1386–1395. doi: 10.1172/JCI115445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdullah F, Ovadia P, Feuerstein G, Neville LF, Morrison R, Mathiak G, Whiteford M, Rabinovici R. The novel chemokine mob-1: involvement in adult respiratory distress syndrome. Surgery. 1997;122:303–312. doi: 10.1016/s0039-6060(97)90022-2. [DOI] [PubMed] [Google Scholar]
- Marinelli WA, Henke CA, Harmon KR, Hertz MI, Bitterman PB. Mechanisms of alveolar fibrosis after acute lung injury. Clin Chest Med. 1990;11:657–672. [PubMed] [Google Scholar]
- Schraufnagel DE, Arzouman DA, Sekosan M, Ho YK. The effect of transforming growth factor-alpha on airway angiogenesis. J Thorac Cardiovasc Surg. 1992;104:1582–1588. [PubMed] [Google Scholar]
- Moreno RH, Hogg JC, Pare PD. Mechanics of airway narrowing. Am Rev Respir Dis. 1986;133:1171–1180. doi: 10.1164/arrd.1986.133.6.1171. [DOI] [PubMed] [Google Scholar]
- Hogg JC. Vascularity in asthmatic airways: relation to inhaled steroid dose. Thorax. 1999;54:283. doi: 10.1136/thx.54.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wilson JW. Increased vascularity of the bronchial mucosa in mild asthma. Am J Respir Crit Care Med. 1997;156:229–233. doi: 10.1164/ajrccm.156.1.9607066. [DOI] [PubMed] [Google Scholar]
- Koch AE, Cho M, Burrows J, Leibovich SJ, Polverini PJ. Inhibition of production of macrophage-derived angiogenic activity by the anti-rheumatic agents gold sodium thiomalate and auranofin. Biochem Biophys Res Commun. 1988;154:205–212. doi: 10.1016/0006-291x(88)90671-7. [DOI] [PubMed] [Google Scholar]
- Firestein GS. Starving the synovium: angiogenesis and inflammation in rheumatoid arthritis. J Clin Invest. 1999;103:3–4. doi: 10.1172/JCI5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DA, Rodway HA, Claxson A. Vascular turnover during carrageenan synovitis in the rat. Lab Invest. 1998;78:1513–1521. [PubMed] [Google Scholar]
- Paleolog EM. Angiogenesis: a critical process in the pathogenesis of RA - a role for VEGF? Br J Rheumatol. 1996;35:917–919. doi: 10.1093/rheumatology/35.10.917. [DOI] [PubMed] [Google Scholar]
- FitzGerald O, Soden M, Yanni G, Robinson R, Bresnihan B. Morphometric analysis of blood vessels in synovial membranes obtained from clinically affected and unaffected knee joints of patients with rheumatoid arthritis. Ann Rheum Dis. 1991;50:792–796. doi: 10.1136/ard.50.11.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraan MC, Versendaal H, Jonker M, Bresnihan B, Post WJ, t Hart BA, Breedveld FC, Tak PP. Asymptomatic synovitis precedes clinically manifest arthritis. Arthritis Rheum. 1998;41:1481–1488. doi: 10.1002/1529-0131(199808)41:8<1481::AID-ART19>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Salmon M, Walsh DA, Huang TJ, Barnes PJ, Leonard TB, Hay DW, Chung KF. Involvement of cysteinyl leukotrienes in airway smooth muscle cell DNA synthesis after repeated allergen exposure in sensitized Brown Norway rats. Br J Pharmacol. 1999;127:1151–1158. doi: 10.1038/sj.bjp.0702669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappi-Blanco E, Kaarteenaho-Wiik R, Soini Y, Risteli J, Paakko P. Intraluminal fibromyxoid lesions in bronchiolitis obliterans organizing pneumonia are highly capillarized. Hum Pathol. 1999;30:1192–1196. doi: 10.1016/s0046-8177(99)90036-9. [DOI] [PubMed] [Google Scholar]
- Keane MP, Belperio JA, Arenberg DA, Burdick MD, Xu ZJ, Xue YY, Strieter RM. IFN-gamma-inducible protein-10 attenuates bleomycin-induced pulmonary fibrosis via inhibition of angiogenesis. J Immunol. 1999;163:5686–5692. [PubMed] [Google Scholar]
- Keane MP, Arenberg DA, Lynch JP, 3rd, Whyte RI, Iannettoni MD, Burdick MD, Wilke CA, Morris SB, Glass MC, DiGiovine B, Kunkel SL, Strieter RM. The CXC chemokines, IL-8 and IP-10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J Immunol. 1997;159:1437–1443. [PubMed] [Google Scholar]
- Charan NB, Baile EM, Pare PD. Bronchial vascular congestion and angiogenesis. Eur Respir J. 1997;10:1173–1180. doi: 10.1183/09031936.97.10051173. [DOI] [PubMed] [Google Scholar]
- Baluk P, Bowden JJ, Lefevre PM, McDonald DM. Upregulation of substance P receptors in angiogenesis associated with chronic airway inflammation in rats. Am J Physiol. 1997;273:L565–L571. doi: 10.1152/ajplung.1997.273.3.L565. [DOI] [PubMed] [Google Scholar]
- Orsida BE, Li X, Hickey B, Thien F, Wilson JW, Walters EH. Vascularity in asthmatic airways: relation to inhaled steroid dose. Thorax. 1999;54:289–295. doi: 10.1136/thx.54.4.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DA, Hu DE, Mapp PI, Polak JM, Blake DR, Fan TP. Innervation and neurokinin receptors during angiogenesis in the rat sponge granuloma. Histochem J. 1996;28:759–769. doi: 10.1007/BF02272149. [DOI] [PubMed] [Google Scholar]
- Koch AE, Szekanecz Z, Friedman J, Haines GK, Langman CB, Bouck NP. Effects of thrombospondin-1 on disease course and angiogenesis in rat adjuvant-induced arthritis. Clin Immunol Immunopathol. 1998;86:199–208. doi: 10.1006/clin.1997.4480. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- Mori S, Yoshikawa H, Hashimoto J, Ueda T, Funai H, Kato M, Takaoka K. Antiangiogenic agent (TNP-470) inhibition of ectopic bone formation induced by bone morphogenetic protein-2. Bone. 1998;22:99–105. doi: 10.1016/S8756-3282(97)00248-2. [DOI] [PubMed] [Google Scholar]
- Mayer E, Cardoso PF, Puskas JD, De Campos K, Oka T, Dardick I, Patterson GA. The effect of basic fibroblast growth factor and omentopexy on revascularization and epithelial regeneration of heterotopic rat tracheal isografts. J Thorac Cardiovasc Surg. 1992;104:180–188. [PubMed] [Google Scholar]
- Wiseman NE, Reed MH. Bronchopulmonary arterial malformation occurring in aspergillus lung infection complicating chronic granulomatous disease. J Pediatr Surg. 1981;16:457–460. doi: 10.1016/s0022-3468(81)80006-1. [DOI] [PubMed] [Google Scholar]
- Tolnay E, Kuhnen C, Voss B, Wiethege T, Muller KM. Expression and localization of vascular endothelial growth factor and its receptor flt in pulmonary sarcoidosis. Virchows Arch. 1998;432:61–65. doi: 10.1007/s004280050135. [DOI] [PubMed] [Google Scholar]
- Jackson JR, Seed MP, Kircher CH, Willoughby DA, Winkler JD. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997;11:457–465. [PubMed] [Google Scholar]
- Matsubara T, Ziff M. Inhibition of human endothelial cell proliferation by gold compounds. J Clin Invest. 1987;79:1440–1446. doi: 10.1172/JCI112972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata S, Matsubara T, Saura R, Tateishi H, Hirohata K. Inhibition of in vitro vascular endothelial cell proliferation and in vivo neovascularization by low-dose methotrexate. Arthritis Rheum. 1989;32:1065–1073. doi: 10.1002/anr.1780320903. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Saura R, Hirohata K, Ziff M. Inhibition of human endothelial cell proliferation in vitro and neovascularization in vivo by D-penicillamine. J Clin Invest. 1989;83:158–167. doi: 10.1172/JCI113853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhok R, Wijelath E, Smith J, Watson J, Sturrock RD, Capell HA. Is the beneficial effect of sulfasalazine due to inhibition of synovial neovascularization? J Rheumatol. 1991;18:199–202. [PubMed] [Google Scholar]