

Abstract
It is not known to what extent glucocorticoid hormones cause their anti-inflammatory actions and their undesirable side effects by the same or different molecular mechanisms. Glucocorticoids combine with a cytoplasmic receptor that alters gene expression in two ways. One way is dependent on the receptor's binding directly to DNA and acting (positively or negatively) as a transcription factor. The other is dependent on its binding to and interfering with other transcription factors. Both mechanisms could underlie suppression of inflammation. The liganded receptor binds and inhibits the inflammatory transcription factors activator protein-1 and NF-κB. It also directly induces anti-inflammatory genes such as that encoding the protein inhibitor of NF-κB. Recent work has shown that glucocorticoids inhibit signalling in the mitogen-activated protein kinase pathways that mediate the expression of inflammatory genes. This inhibition is dependent on de novo gene expression. It is important to establish the significance of these different mechanisms for the various physiological effects of glucocorticoids, because it may be possible to produce steroid-related drugs that selectively target the inflammatory process.
Keywords: glucocorticoid, inflammation, JNK, MAP kinase, NF-κB
Introduction
The anti-inflammatory action of glucocorticoid hormones was discovered by Hench and colleagues over 50 years ago [1]. Hench had noted that the symptoms of rheumatoid arthritis were often improved in pregnancy and when a patient had jaundice, both situations in which, he reasoned, there was an increase in steroids in the body. The active substances of the adrenal cortex had then recently been isolated by Reichstein and Kendall and shown to be steroids. It seemed possible that these might alleviate inflammatory symptoms. Hench and his co-workers found that small doses of cortisone dramatically improved the symptoms of patients with rheumatoid arthritis. Hench, Kendall, and Reichstein were jointly awarded the Nobel Prize in physiology and medicine in 1950. Powerful synthetic glucocorticoids were then developed, which, despite their unwelcome side effects, became and remain mainstays of anti-inflammatory and immunosuppressive therapy.
The side effects of glucocorticoids that severely limited their use were osteoporosis, diabetes, hypertension, cataracts, thinning of the skin, and the characteristic appearance of Cushing's syndrome. They also suppressed the hypothalamic–pituitary–adrenal axis and arrested growth. Their potency in suppressing inflammation stimulated much investigation into their mechanism of action. Glucocorticoids inhibit expression of many of the genes involved in inflammatory and immune responses. These include those encoding cytokines, chemokines, cell-surface receptors, adhesion molecules, tissue factor, degradative proteinases, and enzymes such as cyclooxygenase-2 and inducible nitric oxide synthase, which produce inflammatory mediators.
The glucocorticoid receptor (GR) [2] binds to specific DNA sequences – glucocorticoid response elements (GREs) – which may be positive or negative, either activating or repressing transcription, respectively (Fig. 1). In the absence of ligand, the receptor is cytoplasmic and is complexed with chaperones, including hsp90, hsp56 (immunophilin), and calreticulin. Upon ligand binding to the receptor, the chaperones are shed, exposing nuclear localisation signals. The receptor dimerises when it binds to a GRE. Genes that are positively regulated by GREs include those involved in gluconeogenesis, such as those for tyrosine aminotransferase, alanine aminotransferase, and phosphoenolpyruvate carboxykinase. Examples of genes negatively regulated by GREs are those for pro-opiomelanocortin (the ACTH precursor) and prolactin. The genes responsible for such side effects of glucocorticoids as osteoporosis, diabetes, and hypertension are largely unknown. Understanding the transcriptional basis of the different physiological effects is important because it may be possible to develop more selective agents, which target the inflammatory process.
Figure 1.
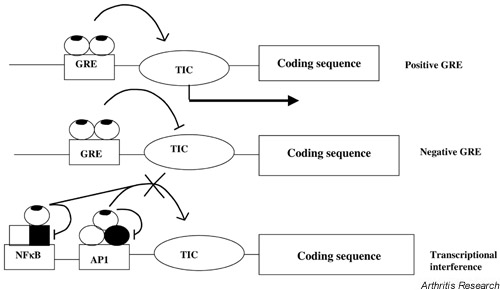
Mechanisms of transcriptional regulation by glucocorticoids (adapted from M Karin [3]). Liganded GRs (ovals with patches) regulate transcription by direct binding to DNA elements (GREs) or by binding to other transcription factors (tethering). GRE-regulated and NF-κB/AP-1-regulated promoters are shown schematically with single binding sites; typically, there may be one or several sites. Interference with the activity of AP-1 (open and dark ovals) or NF-κB (open and dark squares) after tethering could be a direct effect on the factors' function, or it could be an interaction with coactivators or components of the TIC. AP-1 = activator protein-1; GR = glucocorticoid receptor; GRE = glucocorticoid response element; NF-κB = nuclear factor-κB; TIC = transcriptional initiation complex.
Glucocorticoid receptors interact with inflammatory transcription factors
No genes responding positively to glucocorticoid hormones have been identified that explain the profound suppression of the inflammatory response. Furthermore, genes of the inflammatory response lack repressing GREs, so other explanations for the suppression of inflammation have been sought [3,4]. Currently, it is thought that a major mechanism is transcriptional interference (see Fig. 1). Inflammatory-response genes are typically regulated by the transcription factors NF-κB and activator protein-1 (AP-1). The liganded GR interacts with AP-1 complexes (generally c-Jun/c-Fos heterodimers) and prevents their transcriptional activity [3,4]. Similar tethering occurs with NF-κB [5,6]. This interference between transcription factors is reciprocal: not only can GR prevent the function of AP-1 and NF-κB, but also AP-1 and NF-κB can prevent transcriptional activation by GR. The physiological significance of these mechanisms is still speculative and the interactions between the endogenous proteins have been difficult to demonstrate in vivo. Another possible explanation for the functional competition between AP-1, NF-κB, and GR is that they compete for the transcriptional coactivators CREB-binding protein (CBP) and p300 [7,8]. However, this notion is controversial.
An additional mechanism of transcriptional interference has been recently proposed [9]. Corticosteroid interferes with IL-1-induced gene activation by inhibiting the histone acetylation that loosens chromatin structure, and enables access of transcription factors to their DNA binding sites [9]. The liganded GR bound to the transcriptional complexes inhibits their acetyltransferase activity. This could be an adjunct to the mechanism of tethering of transcription factors.
Glucocorticoids may also interfere with NF-κB activation by a mechanism dependent upon de novo gene expression. NF-κB is sequestered in the cytoplasm with an inhibitor, IκBα, whose degradation is induced by inflammatory stimuli. Production of this inhibitor is increased by dexamethasone [10,11]. This effect is slow and its physiological significance remains to be established.
Dexamethasone interferes with pathways of mitogen-activated protein kinase
Recently, another possible mechanism for glucocorticoid action has become apparent: dexamethasone inhibits signalling in mitogen-activated protein kinase (MAP kinase) pathways, which are activated by inflammatory stimuli [12]. This suggests that glucocorticoids may block inflammatory signalling at a level above transcription factor activation. The effect, unlike transcriptional interference, requires gene induction, possibly of a MAP kinase phosphatase, and could inhibit both transcriptional and post-transcriptional mechanisms controlled by the MAP kinases (Fig. 2).
Figure 2.
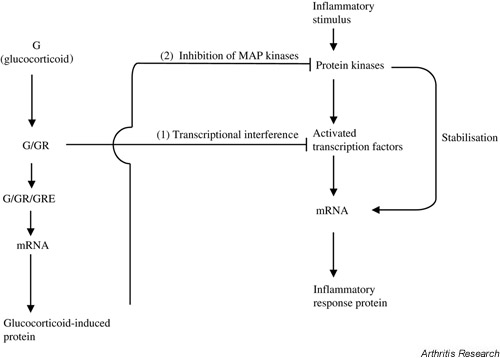
Glucocorticoids may act (1) by transcriptional interference and (2) by inhibiting MAP kinases. The left side represents glucocorticoid (G) combining with its receptor (GR) and inducing gene expression by binding to DNA glucocorticoid response elements (GREs). The right side shows an inflammatory stimulus (e.g. lipopolysaccharide, IL-1, tumour necrosis factor) activating protein kinase cascades (see Fig. 3) and inducing inflammatory response genes. (1) Transcriptional interference is due to the liganded GR directly binding the transcription factors activator protein-1 and nuclear factor-κB and inhibiting their action. (2) Glucocorticoid-induced genes, possibly MAP kinase phosphatases, inhibit MAP kinase signalling pathways by keeping them in the dephosphorylated state. This would inhibit both transcriptional and post-transcriptional mechanisms underlying inflammatory gene expression. MAP = mitogen-activated protein.
There are three types of MAP kinase [13] and they participate in distinct phosphorylation cascades (Fig. 3). The prototype MAP kinase is the extracellularly regulated kinase (ERK), or p42, which is activated by many stimuli. The other two, the c-Jun N-terminal kinase (JNK) and p38, are very strongly activated by inflammatory stimuli such as IL-1, tumour necrosis factor, and microbial products (e.g. lipopolysaccharide), and by cellular stress (e.g. UV). The MAP kinases are activated by their specific MAP kinase kinases (MKKs) by dual phosphorylation of a motif comprising a threonyl and a tyrosyl residue separated by a single amino acid. The MAP kinases phosphorylate various substrates, including other protein kinases. At the bottom of the cascades lie proteins controlling gene expression and other processes.
Figure 3.
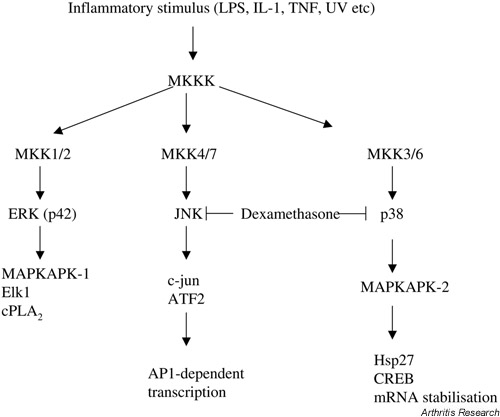
The MAP kinase cascades. This highly simplified scheme shows the three major MAP kinase pathways of mammalian cells. The MAP kinases are ERK, JNK, and p38. A few downstream substrates are shown. ATF = activating transcription factor; cPLA2 = cytosolic phospholipase A2; CREB = cAMP-response-element-binding protein; ERK = extracellularly regulated kinase; JNK = c-Jun N-terminal kinase; LPS = lipopolysaccharide; MAP = mitogen-activated protein; MAPKAPK = MAP kinase-activated protein kinase; MKK = MAP kinase kinase; MKKK = MAP kinase kinase kinase; TNF = tumour necrosis factor.
Serving to modulate the activity of MAP kinases and to switch off the pathways after the response to a stimulus, are MAP kinase phosphatases [14]. These dual-specificity phosphatases remove phosphates from both the threonyl and tyrosyl residues in the MAP kinase activation motifs. More than 12 of these phosphatases are known to exist, differing in expression, subcellular localisation, and specificity for the different MAP kinases. Since removal of phosphate from either threonyl or tyrosyl residues inactivates the enzymes, they may also be regulated by phosphoserine/phosphothreonine protein phosphatases and protein phosphotyrosine phosphatases.
The three MAP kinase pathways, in addition to the protein kinase system that activates NF-κB by phosphorylating the inhibitor IκB, are the major signalling systems through which genes of the inflammatory response are activated. Such genes are activated by several transcription factors and typically, as mentioned earlier, NF-κB and AP-1 are crucial. The JNK pathway is a major route for activation of c-Jun, and therefore of AP-1 complexes. The mRNAs of many cytokines and key regulators of the inflammatory response are unstable by virtue of clustered AUUUA motifs in their 3' untranslated regions [15]. The p38 MAP kinase pathway stabilises such mRNAs [16-21]. It is presumed that downstream kinases regulate proteins that bind to the instability motifs.
The role of the p38 MAP kinase pathway has been elucidated by studying its effect on the function of the 3' untranslated regions of the cyclooxygenase-2 [20] and IL-8 mRNAs [21]. Because dexamethasone destabilises some mRNAs, including the mRNA of cyclooxygenase-2 [22], its mechanism was investigated. It antagonised the stabilising action of the p38 MAP kinase on the cyclooxygenase-2 3' UTR. This raised the possibility that dexamethasone was inhibiting p38 MAP kinase. There had been reports that glucocorticoids inhibited JNK activity in cells [23,24] and suppressed activity of the ERK cascade in mast cells [25]. Treating cells with dexamethasone for 1–2 hours before stimulation inhibited the activation of both p38 MAP kinase and JNK by UV light, by IL-1, or by bacterial lipopolysaccharide [12]. We know that this inhibition was at the level of the MAP kinase, because dexamethasone had no effect on the p38 activator, MKK6, and the inhibition could be circumvented by transfecting an active mutant of the substrate of p38, the MAP kinase-activated protein kinase-2 [12]. The effect of dexamethasone on the p38 MAP kinase was dependent upon GR and de novo mRNA synthesis.
An explanation for these findings is that steroids induce a protein phosphatase that keeps the p38 MAP kinase (and JNK) in the dephosphorylated state. Alternatively, the steroid could be inducing a molecule that inhibits the MKKs. It will be interesting to know the basis of the MAP kinase inhibition, its dependence on GRE-regulated genes, and its importance in inflammation relative to the mechanism of transcriptional interference.
Note added in proof
Glucocorticoid increasing expression of MKP-1 has recently been reported by Kassel et al. [26].
Abbreviations
ACTH = adrenocorticotropic hormone; AP-1 = activator protein-1; CREB = cAMP-response-element-binding protein; ERK = extracellularly regulated kinase; GR = glucocorticoid receptor; GRE = glucocorticoid response element; IL = interleukin; JNK = c-Jun N-terminal kinase; MAP = mitogen-activated protein; MKK = MAP kinase kinase; NF = nuclear factor.
Acknowledgments
Acknowledgements
The author is grateful to Mrs Suzie Johns for preparation of the manuscript, to the many authors whose work has not been directly cited in this short review, and to the Arthritis Research Campaign and the Medical Research Campaign for financial support.
References
- Hench PS, Slocumb CH, Barnes AR, Smith HL, Polley HF, Kendall EC. The effect of a hormone of the adrenal cortex, 17-hydroxy-11-dehydrocorticosterone (compound E), on the acute phase of rheumatic fevers. Proceedings of the Staff Meetings of the Mayo Clinic. 1949;24:277–297. [PubMed] [Google Scholar]
- Beato M, Truss M, Chavez S. Control of transcription by steroid hormones. Ann N Y Acad Sci. 1996;784:93–123. doi: 10.1111/j.1749-6632.1996.tb16231.x. [DOI] [PubMed] [Google Scholar]
- Karin M. New twists in gene regulation by glucocorticoid receptor: is DNA binding dispensable? Cell. 1998;93:487–490. doi: 10.1016/s0092-8674(00)81177-0. [DOI] [PubMed] [Google Scholar]
- Newton R. Molecular mechanisms of glucocorticoid action: what is important? Thorax. 2000;55B:603–613. doi: 10.1136/thorax.55.7.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci U S A. 1994;91:752–756. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS., Jr Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol Cell Biol. 1995;15:943–953. doi: 10.1128/mcb.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Sheppard KA, Phelps KM, Williams AJ, Thanos D, Glass CK, Rosenfeld MG, Gerritsen ME, Collins T. Nuclear integration of glucocorticoid receptor and nuclear factor-kappaB signaling by CREB-binding protein and steroid receptor coactivator-1. J Biol Chem. 1998;273:29291–29294. doi: 10.1074/jbc.273.45.29291. [DOI] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS., Jr Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- Lasa M, Brook M, Saklatvala J, Clark AR. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol Cell Biol. 2001;21:771–80. doi: 10.1128/MCB.21.3.771-780.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–92. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- Shaw G, Kamen R. A conserved AU sequence from the 3' untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell. 1986;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- Ridley SH, Dean JL, Sarsfield SJ, Brook M, Clark AR, Saklatvala J. A p38 MAP kinase inhibitor regulates stability of interleukin-1-induced cyclooxygenase-2 mRNA. FEBS Lett. 1998;439:75–80. doi: 10.1016/s0014-5793(98)01342-8. [DOI] [PubMed] [Google Scholar]
- Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274:264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Mori A, Miyata H, Akahane M, Ajisawa Y, Okudaira H. Regulation of interleukin-1beta-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by p38 mitogen-activated protein kinase. J Biol Chem. 1998;273:24832–24838. doi: 10.1074/jbc.273.38.24832. [DOI] [PubMed] [Google Scholar]
- Brook M, Sully G, Clark AR, Saklatvala J. Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. FEBS Lett. 2000;483:57–61. doi: 10.1016/s0014-5793(00)02084-6. [DOI] [PubMed] [Google Scholar]
- Lasa M, Mahtani KR, Finch A, Brewer G, Saklatvala J, Clark AR. Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol Cell Biol. 2000;20:4265–4274. doi: 10.1128/mcb.20.12.4265-4274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. Embo J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristimaki A, Narko K, Hla T. Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem J. 1996;318:325–331. doi: 10.1042/bj3180325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swantek JL, Cobb MH, Geppert TD. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol Cell Biol. 1997;17:6274–6282. doi: 10.1128/mcb.17.11.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caelles C, Gonzalez-Sancho JM, Munoz A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. 1997;11:3351–3364. doi: 10.1101/gad.11.24.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider LG, Hirasawa N, Santini F, Beaven MA. Activation of the mitogen-activated protein kinase cascade is suppressed by low concentrations of dexamethasone in mast cells. J Immunol. 1996;157:2374–2380. [PubMed] [Google Scholar]
- Kassel O, et al. Glucocorticoids inhibit MAPkinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001;20:7108–7116. doi: 10.1093/emboj/20.24.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]