

Abstract
Matrix metalloproteinase (MMP)-1, MMP-8 and MMP-13 are interstitial collagenases that degrade type II collagen in cartilage; this is a committed step in the progression of rheumatoid arthritis and osteoarthritis. Of these enzymes, the expression of MMP-1 and MMP-13 is substantially increased in response to IL-1 and tumor necrosis factor-α, and elevated levels of these collagenases are observed in arthritic tissues. Therefore, cytokine-mediated MMP-1 and MMP-13 gene regulation is an important issue in arthritis research. In this review, we discuss current models of MMP-1 and MMP-13 transcriptional regulation, with a focus on signaling intermediates and transcription factors that may be future targets for the development of new arthritis drugs.
Keywords: arthritis, matrix metalloproteinases, mitogen-activated protein kinases, nuclear factor κB, transcription
Introduction
The matrix metalloproteinase (MMP) family members are the major enzymes that degrade the components of the extracellular matrix [1,2]. At the time of writing this article, 20 members of this family have been identified [3]. All are active at neutral pH, require Ca2+ for activity and contain a central zinc atom as part of their structure. Most MMPs are secreted into the extracellular space in a latent proform, and require proteolytic cleavage for enzymatic activity. A few MMPs, however, are activated intracellularly by a furin-like mechanism and therefore, these enzymes are fully active when they reach the extracellular space [2].
Most cells in the body express MMPs, even though some enzymes are often associated with a particular cell type. For example, the principle substrate of MMP-2 (also known as gelatinase A) and MMP-9 (also known as gelatinase B) is the type IV collagen in basement membrane and thus, these enzymes are usually expressed by endothelial cells, although other cells (e.g. stromal fibroblasts, macrophages, tumor cells) also express them [1,4].
MMP-3 (also known as stromelysin) activates MMP-1 (also known as collagenase-1) and cleaves a broad range of matrix proteins [5]; MMP-1, which is an interstitial collagenase, and MMP-3 are among the most ubiquitously expressed MMPs. In contrast, MMP-13 (also known as collagenase-3) has a more restricted pattern of expression within connective tissue, and is usually produced only by cartilage and bone during development, and by chondrocytes in osteoarthritis (OA) [6,7,8].
Expression of MMPs is low in normal cells, and these low levels allow for healthy connective tissue remodeling. In pathologic conditions, however, the level of MMP expression increases considerably, resulting in aberrant connective tissue destruction. Excess MMP production is associated with the pathology of many diseases, including periodontitis, atherosclerosis, tumor invasion/metastasis and arthritic disease [1,4,9]. In rheumatoid arthritis (RA) and OA, connective tissue destruction is mediated primarily by chondrocytes, by synovial fibroblasts and on occasion, by osteoclasts [7,10,11,12].
The interstitial collagens (types I, II and III), are the principle targets of destruction, and the secreted collagenases (MMP-1 and MMP-13) have the major role in this process. These MMPs are induced in response to the cytokines and growth factors usually found in arthritic joints. MMP-9 is also an inducible MMP, but its role in connective tissue destruction in arthritis appears to be secondary, since it contributes to the degradation of collagen only after the chains of the triple helix have been cleaved by the interstitial collagenases [2]. In contrast, MMP-2 and MMP-14 (membrane type 1-MMP), are constitutively expressed, with minimal regulation, and they may have a relatively minor role in the pathophysiology of arthritis. Thus, the collagenases (MMP-1, MMP-8 and MMP-13) have the unique ability to cleave the triple helix of collagen, thereby allowing the chains to unwind; the chains then become susceptible to further degradation by other MMPs. Recently, MMP-8 (traditionally termed neutrophil collagenase) has been found in arthritic lesions, even in the absence of neutrophils, indicating that chondrocytes, and perhaps synovial cells, can produce this enzyme [13,14]. MMP-13 may have a particular role in cartilage degradation because it is expressed by chondrocytes, and because it hydrolyzes type-II collagen more efficiently than the other collagenases [15]. However, MMP-1 is more abundant and it also degrades interstitial collagens effectively [1,2]. We will, therefore, focus this discussion on the mechanisms controlling transcription of MMP-1 and MMP-13 in arthritic disease, although the concepts may be applicable to other members of this gene family and to other pathologic conditions.
Signal transduction of transcription factor activation
The etiologies of RA and OA are quite different, in that RA is caused by immune dysfunction and chronic inflammation, while OA is the consequence of years of mechanical stress on the articular cartilage. A common feature, however, of these two diseases is the proteolytic degradation and ultimate destruction of articular cartilage, which results in loss of joint function. In RA, inflammatory cytokines such as IL-1 and tumor necrosis factor-α (TNF-α) are produced by activated macrophages in the synovium. These cytokines stimulate connective tissue cells such as synovial fibroblasts and articular chondrocytes to produce MMP-1 and MMP-13 [15,16,17,18]. In OA, the mechanical insult causes cytokine expression by articular chondrocytes, with subsequent autocrine MMP expression [14]. Upon ligand binding, IL-1 and TNF-α receptors each recruit a unique set of receptor-associated proteins that transduce the stimulus into the cell. Beyond the particularities of their receptors, however, IL-1 and TNF-α elicit a series of shared phosphorylation events within the cells that facilitate transcriptional induction of MMPs.
One group of proteins that mediate some of these phosphorylation events is the mitogen-activated protein kinase (MAPK) group [19]. The MAPK family of serine/threonine kinases consists of the c-Jun N-terminal kinases (JNKs), the extracellular signal-regulated kinases (ERKs) and the p38 kinases. The JNKs and p38 kinases are activated in response to inflammatory cytokines, osmotic stress and apoptotic signals [20], while the ERKs respond to cytokines, growth factors and phorbol esters [19,21] (Fig. 1). These stimuli first activate a group of protein kinases (MAPK kinase kinases [MAPKKKs]) that phosphorylate other kinases (MAPK kinases [MAPKKs]), which in turn are responsible for phosphorylation and activation of MAPK. Upon activation by MAPKKs, MAPKs translocate to the nucleus to phosphorylate and activate various transcription factors. Of particular relevance to MMP transcription, JNKs and ERKs phosphorylate and activate the activating protein-1 (AP-1) family member c-Jun [22,23], which dimerizes with c-Fos to drive transcription of multiple MMP genes. In addition to c-jun, the ERK pathway regulates the activity of erythroblastosis twenty-six (Ets) transcription factors [24,25], which cooperate with AP-1 proteins in multiple MMP promoters. To date, there are no known targets of p38 that directly regulate MMP promoters. However, p38 phosphorylates activating transcription factor-2, which then drives both the c-jun promoter and the ternary complex factor Elk-1, which activates the c-fos promoter [20]. Thus, by promoting expression of AP-1 genes, p38 may indirectly contribute to MMP transcription.
Figure 1.
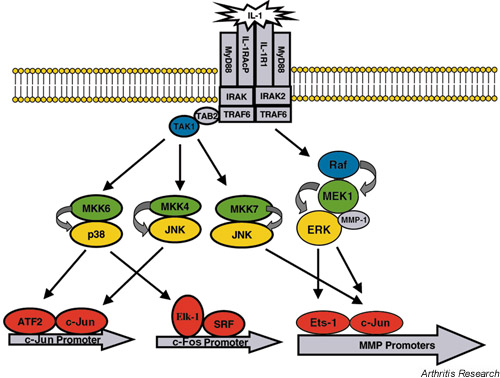
Activation of mitogen-activated protein kinase (MAPK; shown in yellow) pathways by IL-1. Stimulation by IL-1 activates the MAPK kinase kinases (MAPKKKs; shown in blue), transforming-growth-factor-β-activated kinase-1 (TAK 1) and Raf, which then phosphorylate and activate several MAPK kinases (MAPKKs; shown in green): MKK6, MKK4, MKK7, MEK. These MAPKKs in turn phosphorylate and activate the MAPKs, p38, c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK), which translocate to the nucleus. There, these MAPKs phosphorylate and activate the transcription factors (activating transcription factor 2 [ATF2], c-Jun, Elk-1 and erythroblastosis twenty-six [Ets]-1; shown in red) that contribute to matrix metalloproteinase (MMP) transcription. IL-1R, IL-1 receptor; IL-1RAcP, IL-1 receptor-associated protein; IRAK, IL-1 receptor activated kinase; SRF, serum response factor; TAB, Tak-binding protein; TRAF, TNF receptor-associated factor.
Another major cytokine-induced signaling pathway involves translocation of nuclear factor-κB (NF-κB) family members from the cytoplasm to the nucleus (Fig. 2). Upon binding of IL-1 to its cognate receptor, transforming-growth-factor-β-activated kinase becomes active, leading to the activation of the NF-κB-inducing kinase (NIK) [26]. In turn, NIK is responsible for the phosphorylation and activation of the inhibitor of κB (IκB) kinases (IKKs), which then phosphorylate IκB [27]. In resting cells, IκB binds to, and sequesters, dimers of the NF-κB1/p50 and c-rel-related factor A (RelA)/p65 NF-κB subunits in the cytoplasm. Upon phosphorylation, however, IκB becomes ubiquitinated and is targeted for proteosome-mediated degradation. Loss of IκB leaves the p50/p65 dimers free to translocate to the nucleus and transactivate several genes including those for some MMPs. Indeed, when NF-κB is maintained in the cytoplasm by constitutive levels of IκB, reduced expression of MMP-1, MMP-3 and MMP-13 is observed in cytokine-stimulated cells [7,28,29,30].
Figure 2.
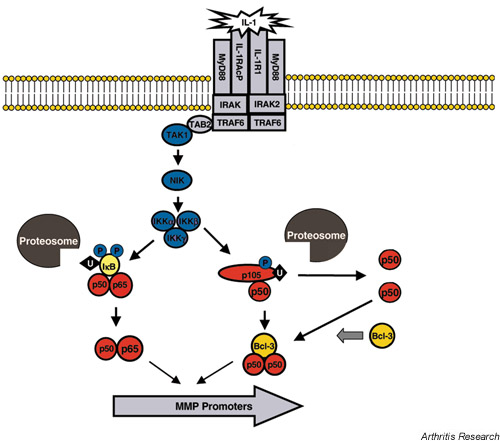
Activation of the NF-κB pathway by IL-1. IL-1 binds to its receptor (IL-1R1) and receptor-associated protein (IL-1RAcP), causing conformational changes in multiple receptor-bound proteins (MyD88, IRAK, TRAF6, TAB2; see Figure 1 for definition). This results in recruitment and activation of transforming-growth-factor-κ-activated kinase-1 (TAK 1), which phosphorylates and activates NF-κB-inducing kinase (NIK). In turn, NIK activates the inhibitor of κB kinase (IKK) complex, which is responsible for phosphorylation of inhibitor of κB (IκB) and NF-κB1 (a 105 kDa protein, p105). Upon phosphorylation, IκB and p105 become polyubiquitinated (U), which targets these proteins for degradation by the proteosome. Degradation of these cellular inhibitors allows translocation of NF-κB subunits to translocate to the nucleus and transactivate matrix metalloproteinase (MMP) promoters. The IκB-like protein, Bcl-3, promotes dimerization of the 50 kDa NF-κB subunit (p50) and regulates the transcriptional activity of p50.
In their latent forms, some NF-κB family members function as IκB proteins. NF-κB1 is a 105 kDa protein that has its carboxyl terminus cleaved to yield the 50 kDa NF-κB subunit (p50). NF-κB2 is a 100 kDa protein that is cleaved similarly to yield the 52 kDa NF-κB subunit (p52) [27]. In their latent states, both NF-κB1/p105 and NF-κB2/p100 can sequester p50 and p52 in the cytoplasm. Recent evidence suggests that the NIK and IKK activation leads to phosphorylation, ubiquitination and degradation of NF-κB1 and NF-κB2 [31,32,33]. This process results in the release of p50 and p52, so that they can translocate to the nucleus. Heissmeyer et al. reported, however, that IKK-dependent degradation of NF-κB1 is independent of NF-κB1 processing [32], so that changes in the total amount of p50 and p52 may be controlled by a different mechanism. The functional consequence of this alternative pathway is not completely understood, since liberation of p50 from p105 leads to the association of p50 homodimers in the nucleus [34], and p50 homodimers can repress NF-κB-dependent transcription by p50/p65 heterodimers [35].
Transcriptional regulation by dimers of NF-κB containing p50 and/or p52 appears to require an IκB-related protein, Bcl-3. Following degradation of p105, Bcl-3 promotes p50 homodimer formation by creating a stable p50/p50/ Bcl-3 trimeric complex [34]. Bcl-3 can then act as a coactivator molecule for p50 and directly contribute to transcriptional activation by p50 homodimers. Alternatively, Bcl-3 can inhibit the binding of p50 homodimers to certain promoter elements, and this frees these sites for transactivation by p50/p65 heterodimers [36].
The MAPK and NF-κB pathways are coordinately activated by IL-1 and TNF-α, and are central pathways in RA and OA pathogenesis. While these kinase cascades lead to the transcription of an array of inflammatory genes, their direct regulation of MMP transcription is just beginning to be elucidated. In the remainder of this review, we address how these pathway-specific signals lead to the recruitment of a cohort of transcription factors that cooperate to initiate MMP-1 and MMP-13 transcription.
Regulation of transcription
The promoters of MMP-1 and MMP-13 (and most other MMPs) contain a TATA box, the core transcriptional unit, at approximately -30 bp, and an AP-1 site at approximately -70 bp [37]. The AP-1 site (5'-TGAG/CTCA-3') binds dimers of the Fos and Jun families. Several additional AP-1 sites are present throughout the MMP promoters, and may contribute to gene expression. One site (5'-TTAATCA-3') is found at -186 bp in the rabbit and human MMP-1 promoters [38]. In contrast to the proximal AP-1 site at -70 bp, this upstream site has only a modest role in basal transcription, but it increases transcription in response to phorbol esters [38]. A third AP-1 site has been identified in the human MMP-1 promoter that cooperates with an adjacent Ets site. Thus, there may be distinct roles for various AP-1 elements, and these functions may depend, at least in part, on the particular AP-1 family members that bind to each site [38,39].
Although initial studies demonstrated the pivotal role of the AP-1 site in MMP transcription in many cells, later studies have clearly shown that it must cooperate with a variety of cis-acting sequences found in the upstream regions of the MMP promoters. For example, induction of MMP-1 by IL-1 in rabbit fibroblasts requires interaction between the AP-1 site at -77 bp and a NF-κB-like element located upstream at -3030 bp [29,30]. Interestingly, while both IL-1 and TNF-α activate NF-κB in primary rabbit synovial fibroblasts, only IL-1 is capable of inducing MMP-1 transcription [29]. This is due, in part, to the inability of TNF to activate the ERK pathways in these cells, both of which contribute to IL-1 induction. Recently, Firestein and colleagues have demonstrated that the JNK pathway, but not the p38 pathway is required for cytokine induction of MMP-1 in human rheumatoid synovial fibroblasts [40], and for MMP-13 induction in murine inflammatory arthritis [11]. In contrast to synovial fibroblasts, chondrocytes rely on the p38, JNK and NF-κB pathways to activate MMP-13 [7]. A working model [7,29] that explains these observations is that MAPKs, either directly or indirectly, activate AP-1 family members, which cooperate with NF-κB to activate MMP-1 and MMP-13 transcription. Additional transcription factors, however, such as Ets family members, may also be nuclear targets of MAPKs in this complex transcriptional program.
The AP-1 site at -1602 bp is of interest because it cooperates with an Ets site created by a single nucleotide polymorphism at -1607 bp. This single nucleotide polymorphism is represented by the insertion of an extra guanine base (G), which creates a core binding site for the Ets family of transcription factors (5'-GGA-3'). At least one copy of this '2G allele' is present in about 75% of the human population, where it has the potential to enhance MMP-1 transcription in both normal fibroblasts and some tumor cells [41,42,43]. Thus, this Ets site may contribute substantially to MMP-1 gene expression, at least under certain conditions. Ets sites have critical roles in MMP transcription and they often interact with AP-1 sites. In addition, multiple Ets sites are present in all the MMP promoters, except MMP-2. The number of these sites and their location within a given promoter vary among the MMP family members, and these variations influence the regulation of these genes [37].
The interaction of AP-1 with other transcription factors may also regulate tissue-specific expression of MMPs. The transcription factor Runt domain factor-2 (Runx-2) appears to be expressed almost exclusively in developing cartilage and bone [44,45,46,47], and these are precisely the cells that normally express MMP-13. Among the MMPs, a Runx-2 binding site is unique to the MMP-13 promoter, and it cooperates with the AP-1 site to mediate MMP-13 transcription [48,49,50]. Important future studies will define the role of MAPK in Runx-2 activation, and how NF-κB interacts with the AP-1/Runx-2 complex.
Repression of MMP transcription
Since joint destruction in arthritic diseases is not reversible, the inhibition of the proteases responsible for cartilage degradation is an important part of therapy. While potent inhibitors of MMP enzymatic activity have been developed, their use has been limited due to safety issues [51]. Inhibition of MMP gene expression at the transcriptional level may be a viable alternative option. With the exception of the glucocorticoid hormones, however, none of the several compounds now available to reduce MMP transcription are in clinical use, although some have been used successfully in animal models of arthritis.
The glucocorticoids bind to their receptors (GRs), which then usually interact with glucocorticoid response elements present in the promoters of many genes [52]. Because the MMP promoters do not contain glucocorticoid response elements, inhibition of transcription occurs through an indirect mechanism. This 'transrepression' involves binding of the activated receptor to Fos and Jun proteins present at the proximal AP-1 site, with a subsequent change in their conformation and a reduction in transcription [53]. Activated GRs can also potently repress NF-κB-dependent transcription by two separate mechanisms. First, GRs physically interact with RelA/p65, resulting in inhibition of NF-κB-dependent transcription [54,55]. This interaction is specific for p65, and is distinct from the domain involved in AP-1 transrepression [56]. Second, glucocorticoids enhance IκBα synthesis, resulting in sequestration of NF-κB in the cytoplasm.
The vitamin A analogues, retinoids, also block MMP transcription through the AP-1 site [57,58,59,60]. Ligand activated receptors (e.g. the retinoic acid receptors α, β and γ, and the retinoid × receptors α, β and γ) reduce MMP transcription by binding to Fos and Jun proteins at the AP-1 site, sequestering these proteins away from the promoter and/or reducing the level of Fos and Jun mRNAs [61]. Although retinoids have reduced joint destruction in animal models of arthritis [62], they have not been used in patients. In addition, both glucocorticoids and retinoids affect a broad number of genes, and this lack of specificity may contribute to the side effects associated with these compounds.
There is one novel compound that may block the expression of specific MMPs in arthritis. This is a synthetic triter-penoid, 2-cyano-3,12-dioxoolean-1,9,dien-28-oic acid (CDDO) [63,64]. At nanomolar concentrations, CDDO selectively inhibits the induction of MMP-1 and MMP-13 by inflammatory cytokines in IL-1-stimulated chondrosar-coma cells, without affecting basal expression [63]. In addition, the expression of other MMPs is not affected, and thus the low constitutive levels of MMPs required for normal physiology may remain untouched. CDDO inhibits MMP-1 and MMP-13 gene expression, at least in part, by reducing IL-1-induced transcription [63]. While this mechanism is not fully understood, this drug is known to be a ligand for peroxisome proliferator-activated receptor-γ (PPAR-γ) [65]; other PPAR-γ agonists such as 15-deoxyprostaglandin J2 can also inhibit MMP-13 synthesis [66]. Since PPAR-γ can physically interact with c-Jun, it is tempting to speculate that CDDO treatment induces an AP-1/PPAR-γ association that is transcriptionally repressive. Additional work is required to determine if CDDO represses in an AP-1-dependent manner, or if it works through a novel mechanism to repress MMPs.
Finally, increased knowledge of the specific signal/transduction pathways driving MMP-1 and MMP-13 expression in arthritic chondrocytes and synovial cells has led to the search for agents that can inhibit these pathways. Blocking MAPK pathways inhibits gene expression of MMPs in tissue culture experiments, and prevents progression of arthritis in animal models. For example, the p38 MAPK inhibitor, SB203580, blocked MMP-13 gene expression in cultured chondrocytes [7] and inhibited IL-1 mediated collagen degradation in cartilage explants [67]. In the collagen-induced arthritis model of rheumatoid arthritis, SB203580 significantly inhibited TNF-α and IL-6 production, reduced paw inflammation, and inhibited the formation of joint lesions [68]. In addition, orally active p38 inhibitors were also effective in animal models of inflammatory arthritis [69,70] presumably by blocking MMP synthesis. Inhibition of JNK by the novel inhibitor SP600125 inhibited bone destruction in adjuvant-induced arthritis, suggesting a role for this MAPK in disease pathogenesis [11].
Since NF-κB activation is required for the expression of MMP-1 and MMP-13, as well as inflammatory stimuli such as IL-1, IL-6 and TNF-α [7,29,71,72], this pathway is another potential therapeutic target. This is supported by the work of Bondeson et al. [73], in which over-expression of IκBα reduced expression of inflammatory cytokines and MMPs, but did not reduce anti-inflammatory cytokines or tissue inhibitor of metalloproteinases. Furthermore, mice deficient in the p50 subunit are refractory to collagen-induced arthritis [74], indicating that this factor has a prominent role in arthritic disease. Indeed, p50 was the only subunit found to bind to an IL-1-responsive element of the MMP-1 promoter [30]. Thus, direct blockade of the NF-κB pathway, at least in joint cells, may be a viable therapy to reduce MMP transcription in arthritis.
Conclusion
MMP-1 and MMP-13 play dominant roles in the progression of RA and OA, making them prime targets for arthritis therapies. Since the genes for both of these enzymes are transcriptionally activated by inflammatory cytokines, an understanding of the molecular pathways involved is crucial. As our knowledge of these molecular mechanisms increases, our ability to use this knowledge to develop novel and effective therapies will also increase. In the twenty-first century, the era of molecular medicine will surely include strategies targeted at the control MMP gene transcription.
Abbreviations
AP-1 = activating protein-1; bp = base pairs; CDDO = 2-cyano-3,12-dioxoolean-1,9,dien-28-oic acid; ERK = extracellular signal-regulated kinase; Ets = erythroblastosis twenty-six; GR = glucocorticoid receptor; IκB = inhibitor of κB; IKK = inhibitor of κB kinase; IL = interleukin; JNK = c-Jun N-terminal kinase; MAPK = mitogen-activated protein kinase; MAPKK = MAPK kinase; MAPKKK = MAPKK kinase; MMP = matrix metalloproteinase; NF-κB = nuclear factor κB; NIK = NF-κB-inducing kinase; OA = osteoarthritis; PPAR-γ = peroxisome proliferator-activated receptor-γ; RA = rheumatoid arthritis; Runx-2 = Runt domain factor-2; TNF-α = tumor necrosis factor-α.
Acknowledgments
Acknowledgements
The authors would like to acknowledge the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR-46977 and AR-02024 to MPV; AR-26599 to CEB), the National Cancer Institute (CA-77267 to CEB) the RGK Foundation, Austin Texas (to CEB), the Susan B Komen Foundation (to CEB) and the Department of Defense (BC991121 to CEB) for funding of this research.
References
- Vincenti MP. The matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP) genes. Transcriptional and posttranscriptional regulation, signal transduction and cell-type-specific expression. Methods Mol Biol. 2001;151:121–148. doi: 10.1385/1-59259-046-2:121. [DOI] [PubMed] [Google Scholar]
- Nagase H, Woessner JF., Jr Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- Stetler-Stevenson WG, Yu AE. Proteases in invasion: matrix metalloproteinases. Semin Cancer Biol. 2001;11:143–152. doi: 10.1006/scbi.2000.0365. [DOI] [PubMed] [Google Scholar]
- Borden P, Heller RA. Transcriptional control of matrix metalloproteinases and the tissue inhibitors of matrix metalloproteinases. Crit Rev Eukaryotic Gene Expr. 1997;7:159–178. doi: 10.1615/critreveukargeneexpr.v7.i1-2.90. [DOI] [PubMed] [Google Scholar]
- Vincenti MP, White LA, Schroen DJ, Benbow U, Brinckerhoff CE. Regulating expression of the gene for matrix metalloproteinase-1 (collagenase): mechanisms that control enzyme activity, transcription, and mRNA stability. Crit Rev Eukaryotic Gene Expr. 1996;6:391–411. doi: 10.1615/critreveukargeneexpr.v6.i4.40. [DOI] [PubMed] [Google Scholar]
- Borden P, Solymar D, Sucharczuk A, Lindman B, Cannon P, Heller RA. Cytokine control of interstitial collagenase and col-lagenase-3 gene expression in human chondrocytes. J Biol Chem. 1996;271:23577–23581. doi: 10.1074/jbc.271.38.23577. [DOI] [PubMed] [Google Scholar]
- Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 2000;43:801–811. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Vincenti MP, Coon CI, Mengshol JA, Yocum S, Mitchell P, Brinckerhoff CE. Cloning of the gene for interstitial collagenase-3 (matrix metalloproteinase-13) from rabbit synovial fibroblasts: differential expression with collagenase-1 (matrix metalloproteinase-1). Biochem J. 1998;331:341–346. doi: 10.1042/bj3310341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinckerhoff CE, Rutter JL, Benbow U. Interstitial collagenases as markers of tumor progression. Clin Cancer Res. 2000;6:4823–4830. [PubMed] [Google Scholar]
- Goldring MB. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000;43:1916–1926. doi: 10.1002/1529-0131(200009)43:9<1916::AID-ANR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS. c-Jun N-terminal kinase is required for met-alloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenti MP, Brinckerhoff CE. The potential of signal transduction inhibitors for the treatment of arthritis: Is it all just JNK? J Clin Invest. 2001;108:181–183. doi: 10.1172/JCI13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetlow LC, Adlam DJ, Woolley DE. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001;44:585–594. doi: 10.1002/1529-0131(200103)44:3<585::AID-ANR107>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Shlopov BV, Lie WR, Mainardi CL, Cole AA, Chubinskaya S, Hasty KA. Osteoarthritic lesions: involvement of three different collagenases. Arthritis Rheum. 1997;40:2065–2074. doi: 10.1002/art.1780401120. [DOI] [PubMed] [Google Scholar]
- Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, Geoghegan KF, Hambor JE. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest. 1996;97:761–768. doi: 10.1172/JCI118475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein GS, Paine MM, Littman BH. Gene expression (collagenase, tissue inhibitor of metalloproteinases, complement, and HLA-DR) in rheumatoid arthritis and osteoarthritis synovium. Quantitative analysis and effect of intraarticular corticos-teroids. Arthritis Rheum. 1991;34:1094–1105. doi: 10.1002/art.1780340905. [DOI] [PubMed] [Google Scholar]
- Clark IM, Powell LK, Ramsey S, Hazleman BL, Cawston TE. The measurement of collagenase, tissue inhibitor of metalloproteinases (TIMP), and collagenase-TIMP complex in synovial fluids from patients with osteoarthritis and rheumatoid arthritis. Arthritis Rheum. 1993;36:372–379. doi: 10.1002/art.1780360313. [DOI] [PubMed] [Google Scholar]
- Westhoff CS, Freudiger D, Petrow P, Seyfert C, Zacher J, Kriegsmann J, Pap T, Gay S, Stiehl P, Gromnica-Ihle E, Wernicke D. Characterization of collagenase 3 (matrix metalloproteinase 13) messenger RNA expression in the synovial membrane and synovial fibroblasts of patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:1517–1527. doi: 10.1002/1529-0131(199907)42:7<1517::AID-ANR27>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351:289–305. doi: 10.1042/0264-6021:3510289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Leppa S, Saffrich R, Ansorge W, Bohmann D. Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. Embo J. 1998;17:4404–4413. doi: 10.1093/emboj/17.15.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R, Ernst WH, Nordheim A. SAP1a is a nuclear target of signaling cascades involving ERKs. Oncogene. 1995;10:1209–1216. [PubMed] [Google Scholar]
- O'Hagan RC, Tozer RG, Symons M, McCormick F, Hassell JA. The activity of the Ets transcription factor PEA3 is regulated by two distinct MAPK cascades. Oncogene. 1996;13:1323–1333. [PubMed] [Google Scholar]
- Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Bondeson J, Brennan F, Foxwell B, Feldmann M. Effective adenoviral transfer of IkappaBalpha into human fibroblasts and chondrosarcoma cells reveals that the induction of matrix metalloproteinases and proinflammatory cytokines is nuclear factor-kappaB dependent. J Rheumatol. 2000;27:2078–2089. [PubMed] [Google Scholar]
- Barchowsky A, Frleta D, Vincenti MP. Integration of the NF-kappaB and mitogen-activated protein kinase/AP-1 pathways at the collagenase-1 promoter: divergence of IL-1 and TNF-dependent signal transduction in rabbit primary synovial fibroblasts. Cytokine. 2000;12:1469–1479. doi: 10.1006/cyto.2000.0743. [DOI] [PubMed] [Google Scholar]
- Vincenti MP, Coon CI, Brinckerhoff CE. Nuclear factor kappaB/p50 activates an element in the distal matrix metalloproteinase 1 promoter in interleukin-1beta-stimulated synovial fibroblasts. Arthritis Rheum. 1998;41:1987–1994. doi: 10.1002/1529-0131(199811)41:11<1987::AID-ART14>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- Heissmeyer V, Krappmann D, Hatada EN, Scheidereit C. Shared pathways of IkappaB kinase-induced SCF(betaTrCP)-mediated ubiquitination and degradation for the NF-kappaB precursor p105 and IkappaBalpha. Mol Cell Biol. 2001;21:1024–1035. doi: 10.1128/MCB.21.4.1024-1035.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch M, Lin L, Geleziunas R, Greene WC. The generation of nfkb2 p52: mechanism and efficiency. Oncogene. 1999;18:6201–6208. doi: 10.1038/sj/onc/1203022. [DOI] [PubMed] [Google Scholar]
- Heissmeyer V, Krappmann D, Wulczyn FG, Scheidereit C. NF-kappaB p105 is a target of IkappaB kinases and controls signal induction of Bcl-3-p50 complexes. Embo J. 1999;18:4766–4778. doi: 10.1093/emboj/18.17.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler-Heitbrock HW, Wedel A, Schraut W, Strobel M, Wendel-gass P, Sternsdorf T, Bauerle PA, Haas JG, Riethmuller G. Tolerance to lipopolysaccharide involves mobilization of nuclear factor kappa B with predominance of p50 homodimers. J Biol Chem. 1994;269:17001–17004. [PubMed] [Google Scholar]
- Franzoso G, Bours V, Azarenko V, Park S, Tomita-Yamaguchi M, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 can facilitate NF-kappa B-mediated transactivation by removing inhibiting p50 homodimers from select kappa B sites [published erratum appears in EMBO J 16:440]. Embo J. 1997;12:3893–3901. doi: 10.1002/j.1460-2075.1993.tb06067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbow U, Brinckerhoff CE. The AP-1 site and MMP gene regulation: what is all the fuss about? Matrix Biol. 1997;15:519–526. doi: 10.1016/s0945-053x(97)90026-3. [DOI] [PubMed] [Google Scholar]
- Chamberlain SH, Hemmer RM, Brinckerhoff CE. Novel phorbol ester response region in the collagenase promoter binds Fos and Jun. J Cell Biochem. 1993;52:337–351. doi: 10.1002/jcb.240520310. [DOI] [PubMed] [Google Scholar]
- White LA, Brinckerhoff CE. Two activator protein-1 elements in the matrix metalloproteinase-1 promoter have different effects on transcription and bind Jun D, c-Fos, and Fra-2. Matrix Biol. 1995;14:715–725. doi: 10.1016/s0945-053x(05)80014-9. [DOI] [PubMed] [Google Scholar]
- Han Z, Boyle DL, Aupperle KR, Bennett B, Manning AM, Firestein GS. Jun N-terminal kinase in rheumatoid arthritis. J Pharmacol Exp Ther. 1999;291:124–130. [PubMed] [Google Scholar]
- Rutter JL, Mitchell TI, Buttice G, Meyers J, Gusella JF, Ozelius LJ, Brinckerhoff CE. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter creates an Ets binding site and augments transcription. Cancer Res. 1998;58:5321–5325. [PubMed] [Google Scholar]
- Kanamori Y, Matsushima M, Minaguchi T, Kobayashi K, Sagae S, Kudo R, Terakawa N, Nakamura Y. Correlation between expression of the matrix metalloproteinase-1 gene in ovarian cancers and an insertion/deletion polymorphism in its promoter region. Cancer Res. 1999;59:4225–4227. [PubMed] [Google Scholar]
- Nishioka Y, Kobayashi K, Sagae S, Ishioka S, Nishikawa A, Matsushima M, Kanamori Y, Minaguchi T, Nakamura Y, Tokino T, Kudo R. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter in endometrial carcinomas. Jpn J Cancer Res. 2000;91:612–615. doi: 10.1111/j.1349-7006.2000.tb00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Enomoto-Iwamoto M, Iwamoto M, Nomura S, Himeno M, Kitamura Y, Kishimoto T, Komori T. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J Biol Chem. 2000;275:8695–8702. doi: 10.1074/jbc.275.12.8695. [DOI] [PubMed] [Google Scholar]
- Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, Ochi T, Endo N, Kita-mura Y, Kishimoto T, Komori T. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn. 1999;214:279–290. doi: 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.3.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Hess J, Porte D, Munz C, Angel P. AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast-specific element 2/AP-1 composite element. J Biol Chem. 2001;276:20029–20038. doi: 10.1074/jbc.M010601200. [DOI] [PubMed] [Google Scholar]
- Selvamurugan N, Chou WY, Pearman AT, Pulumati MR, Partridge NC. Parathyroid hormone regulates the rat collagenase-3 promoter in osteoblastic cells through the cooperative interaction of the activator protein-1 site and the runt domain binding sequence. J Biol Chem. 1998;273:10647–10657. doi: 10.1074/jbc.273.17.10647. [DOI] [PubMed] [Google Scholar]
- Porte D, Tuckermann J, Becker M, Baumann B, Teurich S, Higgins T, Owen MJ, Schorpp-Kistner M, Angel P. Both AP-1 and Cbfa1-like factors are required for the induction of interstitial collagenase by parathyroid hormone. Oncogene. 1999;18:667–678. doi: 10.1038/sj/onc/1202333. [DOI] [PubMed] [Google Scholar]
- Brown PD. Ongoing trials with matrix metalloproteinase inhibitors. Expert Opin Invest Drugs. 2000;9:2167–2177. doi: 10.1517/13543784.9.9.2167. [DOI] [PubMed] [Google Scholar]
- Funder JW. Glucocorticoid and mineralocorticoid receptors: biology and clinical relevance. Annu Rev Med. 1997;48:231–240. doi: 10.1146/annurev.med.48.1.231. [DOI] [PubMed] [Google Scholar]
- Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science. 1990;249:1266–1272. doi: 10.1126/science.2119054. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS., Jr Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol Cell Biol. 1995;15:943–953. doi: 10.1128/mcb.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA. 1994;91:752–756. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Williams-Skipp C, Scheinman RI. Mapping of glucocorticoid receptor DNA binding domain surfaces contributing to transrepression of NF-kappa B and induction of apoptosis. J Biol Chem. 2001;276:2329–2332. doi: 10.1074/jbc.C000526200. [DOI] [PubMed] [Google Scholar]
- Pan L, Eckhoff C, Brinckerhoff CE. Suppression of collagenase gene expression by all-trans and 9-cis retinoic acid is ligand dependent and requires both RARs and RXRs. J Cell Biochem. 1995;57:575–589. doi: 10.1002/jcb.240570402. [DOI] [PubMed] [Google Scholar]
- Schroen DJ, Brinckerhoff CE. Inhibition of rabbit collagenase (matrix metalloproteinase-1; MMP-1) transcription by retinoid receptors: evidence for binding of RARs/RXRs to the -77 AP-1 site through interactions with c-Jun. J Cell Physiol. 1996;169:320–332. doi: 10.1002/(SICI)1097-4652(199611)169:2<320::AID-JCP11>3.3.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Yang-Yen HF, Zhang XK, Graupner G, Tzukerman M, Sakamoto B, Karin M, Pfahl M. Antagonism between retinoic acid receptors and AP-1: implications for tumor promotion and inflammation. New Biol. 1991;3:1206–1219. [PubMed] [Google Scholar]
- Schule R, Rangarajan P, Yang N, Kliewer S, Ransone LJ, Bolado J, Verma IM, Evans RM. Retinoic acid is a negative regulator of AP-1-responsive genes. Proc Natl Acad Sci USA. 1991;88:6092–6096. doi: 10.1073/pnas.88.14.6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroen DJ, Brinckerhoff CE. Nuclear hormone receptors inhibit matrix metalloproteinase (MMP) gene expression through diverse mechanisms. Gene Expr. 1996;6:197–207. [PMC free article] [PubMed] [Google Scholar]
- Kuwabara K, Shudo K, Hori Y. Novel synthetic retinoic acid inhibits rat collagen arthritis and differentially affects serum immunoglobulin subclass levels. FEBS Lett. 1996;378:153–156. doi: 10.1016/0014-5793(95)01440-3. [DOI] [PubMed] [Google Scholar]
- Mix KS, Mengshol JA, Benbow U, Vincenti MP, Sporn MB, Brinckerhoff CE. A synthetic triterpenoid selectively inhibits the induction of matrix metalloproteinases 1 and 13 by inflammatory cytokines. Arthritis Rheum. 2001;44:1096–1104. doi: 10.1002/1529-0131(200105)44:5<1096::AID-ANR190>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Suh N, Wang Y, Honda T, Gribble GW, Dmitrovsky E, Hickey WF, Maue RA, Place AE, Porter DM, Spinella MJ, Williams CR, Wu G, Dannenberg AJ, Flanders KC, Letterio JJ, Mangelsdorf DJ, Nathan CF, Nguyen L, Porter WW, Ren RF, Roberts AB, Roche NS, Sub-baramaiah K, Sporn MB. A novel synthetic oleanane triter-penoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res. 1999;59:336–341. [PubMed] [Google Scholar]
- Wang Y, Porter WW, Suh N, Honda T, Gribble GW, Leesnitzer LM, Plunket KD, Mangelsdorf DJ, Blanchard SG, Willson TM, Sporn MB. A synthetic triterpenoid, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), is a ligand for the peroxisome proliferator-activated receptor gamma. Mol Endocrinol. 2000;14:1550–1556. doi: 10.1210/mend.14.10.0545. [DOI] [PubMed] [Google Scholar]
- Fahmi H, Di Battista JA, Pelletier JP, Mineau F, Ranger P, Martel-Pelletier J. Peroxisome proliferator–activated receptor gamma activators inhibit interleukin-1beta-induced nitric oxide and matrix metalloproteinase 13 production in human chondrocytes. Arthritis Rheum. 2001;44:595–607. doi: 10.1002/1529-0131(200103)44:3<595::AID-ANR108>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Ridley SH, Sarsfield SJ, Lee JC, Bigg HF, Cawston TE, Taylor DJ, DeWitt DL, Saklatvala J. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J Immunol. 1997;158:3165–3173. [PubMed] [Google Scholar]
- Badger AM, Bradbeer JN, Votta B, Lee JC, Adams JL, Griswold DE. Pharmacological profile of SB 20 a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J Pharmacol Exp Ther. 3580;279:1453–1461. [PubMed] [Google Scholar]
- Jackson JR, Bolognese B, Hillegass L, Kassis S, Adams J, Griswold DE, Winkler JD. Pharmacological effects of SB 22 a selective inhibitor of P38 mitogen-activated protein kinase, in angiogenesis and chronic inflammatory disease models. J Pharmacol Exp Ther. 0025;284:687–692. [PubMed] [Google Scholar]
- Badger AM, Griswold DE, Kapadia R, Blake S, Swift BA, Hoffman SJ, Stroup GB, Webb E, Rieman DJ, Gowen M, Boehm JC, Adams JL, Lee JC. Disease-modifying activity of SB 24 a selective inhibitor of p38 mitogen-activated protein kinase, in rat adjuvant-induced arthritis. Arthritis Rheum. 2235;43:175–183. doi: 10.1002/1529-0131(200001)43:1<175::AID-ANR22>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Manning AM. Signal transduction and transcription factors in rheumatic disease. Arthritis Rheum. 1999;42:609–621. doi: 10.1002/1529-0131(199904)42:4<609::AID-ANR3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson J, Foxwell B, Brennan F, Feldmann M. Defining therapeutic targets by using adenovirus: blocking NF-kappaB inhibits both inflammatory and destructive mechanisms in rheumatoid synovium but spares anti-inflammatory mediators. Proc Natl Acad Sci USA. 1999;96:5668–5673. doi: 10.1073/pnas.96.10.5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IK, Gerondakis S, O'Donnell K, Wicks IP. Distinct roles for the NF-kappaB1 (p50) and c-Rel transcription factors in inflammatory arthritis. J Clin Invest. 2000;105:1799–1806. doi: 10.1172/JCI8298. [DOI] [PMC free article] [PubMed] [Google Scholar]