

Abstract
The dynamical nature of the binding of a substrate surrogate to lactate dehydrogenase is examined on the nanoseconds to milliseconds timescale by laser-induced temperature-jump relaxation spectroscopy. Fluorescence emission of the nicotinamide group of bound NADH is used to define the pathway and kinetics of substrate binding. Assignment of specific kinetic states and elucidation of their structures are accomplished using isotope edited infrared absorption spectroscopy. Such studies are poised to yield a detailed picture of the coupling of protein dynamics to function.
The key connection between protein structure and function is well recognized and established. It is also widely recognized that protein function is crucially dependent on the dynamical nature of proteins. For example, atomic motion is needed for proteins to bind ligands and to carry out enzymatic catalysis. Truly rigid structures, as suggested in static pictures of protein structure, would not be functional. Probing atomic motion in proteins is difficult, however, both theoretically as well as experimentally, and we know very little quantitively about this problem. New approaches are needed to study protein dynamics.
In this communication, we examine the atomic motion involved in the binding of substrate to an enzyme, lactate dehydrogenase (LDH), using a new and promising approach to define atomic motion in proteins over a large time range (10−10–10−2 s): laser-induced temperature-jump (T-jump) relaxation methods to perturb the reaction equilibrium and isotope-edited infrared (IR) spectroscopy to follow the accompanying structural relaxation dynamics. LDH is an efficient enzyme that catalyzes the direct transfer of a hydride ion from the pro-R face of the reduced nicotinamide group of NADH to the C2 carbon of pyruvate producing NAD+ and the alcohol lactate, accelerating the solution reaction by some 14 orders of magnitude (Fig. 1; (1,2)). Binding of the pyruvate substrate in LDH is ordered and follows the formation of the LDH/NADH binary complex. The unreactive substrate surrogate, oxamate, is employed in our study so that the observed kinetics are due solely to binding and not complicated by on-enzyme catalyzed chemical steps.
FIGURE 1 .

A cartoon of the active site of lactate dehydrogenase showing the relative arrangement of reacting groups (not to scale). The substrate pyruvate is shown; the −CH3 group is replaced by −NH2 to form oxamate. The hydride transfer is indicated by the bold arrow; hydrogen transfer by the light arrow.
The basic experimental design is to subject the chemical system:
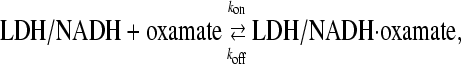 |
to a rapid change in temperature (induced by irradiating the protein solution with a pulse of near IR light; T-jumps of 20°C are typical). The relaxation of the system is monitored by a spectroscopic probe and the kinetic steps along with their microscopic rate constants are determined (3). Using the fluorescence of the nicotinamde ring of NADH as an indicator of structural transformation, we have determined (4) that oxamate binds in a multistep process (at 20°C):
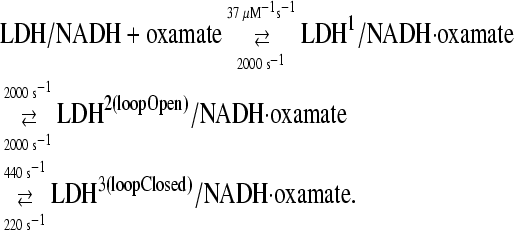 |
Although the NADH emission is an effective probe for following the kinetic steps, we turn to IR spectroscopy to provide specific structural attributes of the transient forms. One excellent marker for structure is the oxamate's C=O stretch frequency. Upon binding to the protein, this bond is highly polarized, with concomitant downshift in its stretch frequency, as the key catalytic groups of the active site, His-195 (located within the binding pocket) and Arg-109 (located on a 10-residue surface loop that closes over the binding pocket), form hydrogen bonds with the C=O moiety (5). These groups stabilize the polar transition state of the reaction by electrostatic interactions and are responsible for about half of the catalytic power of LDH (six orders of magnitude in rate enhancement).
In either static or kinetic isotope-edited IR spectroscopy, a difference measurement is made between the protein complex and an analog in which a specifically placed stable isotope has been incorporated. The isotope shifts the IR spectrum of just those modes that involve the labeled atom; subtraction of the two measurements results in a difference spectrum or transient containing the specifically affected modes. All other background components subtract out. In our studies, the carbon of the C=O group has been labeled with 13C, which downshifts the C=O stretch by 43 cm−1 (Fig. 2 A). When oxamate binds to LDH/NADH, two C=O stretches are observed, both downshifted from the solution position; this indicates that oxamate occupies two conformations in the ternary complex, a minor complex whose C=O stretch lies at 1622 cm−1 in the 13C2 ternary spectrum and a major complex at 1606 cm−1. There is evidence that the initial encounter complex (LDH1/NADH·oxamate in the scheme) represents an ensemble of bound oxamate conformations, each with a somewhat different C=O stretch, little shifted from solution (4). Tentatively, we then assign the 1622 cm−1 species to LDH2(loopOpen)/NADH·oxamate and the 1606 cm−1 form to LDH3(loopClosed)/NADH·oxamate.
FIGURE 2 .

The isotope-edited FTIR spectra between [13C2]oxamate and [12C2]oxamate in solution and in the LDH/NADH·oxamate ternary complex at 20°C.
T-jump isotope-edited IR transient studies were carried out at 1606 cm−1 because this frequency is a marker band for the major population of the LDH/NADH·oxamate complex and because interfering protein IR background is minimal at this frequency. Fig. 3 shows the results along with T-jump fluorescence data under identical conditions. Two relaxation times are observed in both the IR and fluorescence traces. At the high concentrations used in this experiment, it is expected (4) that very small signals would be observed from the bimolecular formation of the encounter complex. Furthermore, previous measurements (4) have shown that most of the NADH fluorescence is highly quenched before the final step (loop closure) of substrate binding; thus the small positive amplitude process in the fluorescence transient (∼500 s−1) corresponds to the loop opening after the T-jump (i.e., the loss of the LDH3(loopClosed)/NADH·oxamate species in the reaction scheme). The faster relaxation process (∼7000 s−1) is therefore assigned to the loss of the LDH2(loopOpen)/ NADH·oxamate species.
FIGURE 3 .

Transient fluorescence at 450 nm (excitation 360 nm) (top) and IR isotope-edited absorption (13C2–12C2) at 1606 cm−1 (bottom) of LDH/NADH·oxamate solutions (600/620/700 μN) after laser T-jump to Tfinal = 38°C. Biexponential fits overlay the data and the fitting results are listed.
The IR data show that the hydrogen bonds between the protein and the substrate mimic C=O moiety are formed in separate events. Clearly, loss of the C=O–Arg-109 interaction dominates the slower relaxation process (∼500 s−1) because loop closure brings Arg-109 into the active site pocket in contact with substrate on this timescale (5,6) and the IR transient of Fig. 3 shows changes in the C=O stretch frequency with the same rate. The faster relaxation process (∼7000 s−1) must therefore correspond to the loss of the C=O–His-195 contact. Consistent with these assignments, the relative amplitudes of the two phases of the IR transient are inverted compared to the fluorescence amplitudes. Transient loss of IR absorbance at 1606 cm−1 primarily monitors the loss of the C=O–Arg-109 interaction (large amplitude slow phase; loop opening). The loss of the C=O–His-195 interaction is weakly detected at this frequency (small amplitude fast phase) because its characteristic IR band is at higher frequency (1622 cm−1), but is broad and upshifts strongly upon breaking the H-bonding interaction (5).
In summary, following the time-dependent perturbation of the oxamate C=O bond using isotope-edited IR spectroscopy has resolved the individual steps in the polarization of this bond, revealing how the catalytically competent Michaelis complex is formed. This approach yields unique insight into the coupling of protein dynamics (loop and other atomic motions) to substrate binding and activation.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org. This material contains details of methodology, including the production of 13C labeled oxamate.
Acknowledgments
We thank Herbert Wong for help with running and interpreting the NMR spectra and producing labeled oxamate.
This work was supported by the Institutes of General Medicine and Biomedical Imaging and Bioengineering of the National Institutes of Health (GM068036 and EB01958).
References
- 1.Holbrook, J. J., A. Liljas, S. J. Steindel, and M. G. Rossmann. 1975. Lactate Dehydrogenase. In The Enzymes. P. D. Boyer, editor. Academic Press: New York, NY. 191–293.
- 2.Burgner, J. W., and W. J. Ray. 1984. On the origin of lactate dehydrogenase induced rate effect. Biochemistry. 23:3636–3648. [DOI] [PubMed] [Google Scholar]
- 3.Callender, R., and R. B. Dyer. 2002. Probing protein dynamics using temperature jump relaxation spectroscopy. Curr. Opin. Struct. Biol. 12:628–633. [DOI] [PubMed] [Google Scholar]
- 4.McClendon, S., N. Zhadin, and R. Callender. 2005. The approach to the Michaelis complex in lactate dehydrogenase: the substrate binding pathway. Biophys. J. In press. [DOI] [PMC free article] [PubMed]
- 5.Deng, H., J. Zheng, A. Clarke, J. J. Holbrook, R. Callender, and J. W. Burgner. 1994. Source of catalysis in the lactate dehydrogenase system. Ground state interactions in the enzyme·substrate complex. Biochemistry. 33:2297–2305. [DOI] [PubMed] [Google Scholar]
- 6.Dunn, C. R., H. M. Wilks, D. J. Halsall, T. Atkinson, A. R. Clarke, H. Muirhead, and J. J. Holbrook. 1991. Design and synthesis of new enzymes based upon the lactate dehydrogenase framework. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 332: p. 177–185. [DOI] [PubMed] [Google Scholar]