

Abstract
Elevation of the intracellular cAMP concentration ([cAMP]i) regulates metabolism, cell proliferation, and differentiation and plays roles in memory formation and neoplastic growth. cAMP mediates its effects mainly through activation of protein kinase A (PKA) as well as Epac1 and Epac2, exchange factors activating the small GTPases Rap1 and Rap2. However, how cAMP utilizes these effectors to induce distinct biological responses is unknown. We here studied the specific roles of PKA and Epac in neuroendocrine PC12 cells. In these cells, elevation of [cAMP]i activates extracellular signal-regulated kinase (ERK) 1/2 and induces low-degree neurite outgrowth. The present study showed that specific stimulation of PKA triggered ERK1/2 activation that was considerably more transient than that observed upon simultaneous activation of both PKA and Epac. Unexpectedly, the PKA-specific cAMP analog induced cell proliferation rather than neurite outgrowth. The proliferative signaling pathway activated by the PKA-specific cAMP analog involved activation of the epidermal growth factor receptor and ERK1/2. Activation of Epac appeared to extend the duration of PKA-dependent ERK1/2 activation and converted cAMP from a proliferative into an anti-proliferative, neurite outgrowth-promoting signal. Thus, the present study showed that the outcome of cAMP signaling can depend heavily on the set of cAMP effectors activated.
INTRODUCTION
The normal development of organisms requires a tight coordination between growth and differentiation. Importantly, the regulation of this decision is altered in disease states, most notably in cancer, where growth signals overcome the physiological processes of differentiation. The differentiation of neuronal cells is important for the regular function of the brain. Thus, there is interest in understanding the molecular signaling mechanisms that regulate cell proliferation and differentiation.
In a number of cells, most notably of neuronal origin, Gs protein-coupled receptors (GsPCR) are key regulators of cell proliferation, differentiation, and survival and play important roles in numerous biological processes such as neoplasia, learning, and memory formation (Kandel, 2001; Spada and Lania, 2002; Wang et al., 2004). The signaling cascades downstream of GsPCRs to regulate these events include activation of adenylyl cyclase, elevation of the intracellular cAMP concentration ([cAMP]i), and modulation of the extracellular signal-regulated kinase (ERK) 1/2 pathway (Stork, 2003).
cAMP binds to and directly activates protein kinase A (PKA) as well as the cAMP binding guanine nucleotide exchange factors Epac1 and -2, which in turn stimulate the small GTPases Rap1 and Rap2. Together, PKA and Epacs appear to mediate the majority of effects of cAMP in mammalian cells (de Rooij et al., 1998; Kawasaki et al., 1998; Bos, 2003). Nevertheless, most work on cAMP-dependent pathways has been centered on the role of PKA. For instance, a role of PKA in memory formation has been verified in numerous in vitro and in vivo studies to the point that pharmacological activators of PKA are being developed for the treatment of memory performance (Kandel, 2001; Barad et al., 1998).
It is largely unknown how cAMP utilizes PKA and Epac to modulate cellular functions such as proliferation and differentiation. Furthermore, it is not known how the cells integrate the signaling pathways activated by PKA and Epac to achieve physiological outcomes. The cAMP-dependent responses may be mediated by activation of either one single cAMP effector or represent an integrated response of cAMP effectors. The present study aimed to dissect how selective activation of PKA or Epac modulates the signaling outcome of an elevated [cAMP]i in the neuroendocrine model cell line PC12. In these cells cAMP induces differentiation reflected by neurite outgrowth (Gunning et al., 1981). The data indicate that an elevated [cAMP]i utilized two different mechanisms to activate ERK1/2 and thereby fine tunes its signaling outcome. We unexpectedly found that specific stimulation of PKA triggered ERK1/2 activation to mediate cell proliferation rather than neurite outgrowth. Additional activation of Epac potentiated PKA-dependent ERK1/2 activation and switched cAMP from a proliferative into a differentiation signal. Our studies suggest that the signaling outcome of cAMP depended on the set of cAMP effectors activated.
MATERIALS AND METHODS
Reagents
Forskolin, pituitary adenylyl cyclase-activating peptide (PACAP) 38, human recombinant epidermal growth factor (EGF), anti-β-actin IgG were obtained from Sigma (St. Louis, MO). NGF was obtained from Promega (Madison, WI). Cell culture reagents were from Invitrogen (Grand Island, NY). 8-(4-Chlorphenylthio)-adenosine-3′,5′-cyclical monophosphate (CPT-cAMP), PD98059, PD153035, PD168393, H-89, and AG1478 were obtained from Calbiochem (San Diego, CA). N6-benzoyladenosine-3′,5′-cyclical monophosphate (6-Bnz-cAMP) and 8-(4-methoxyphenylthio)-2′-O-methyladenosine-3′,5′-cyclical monophosphate (2-Me-cAMP) were from BIOLOG (Bremen, Germany). The antibody recognizing dually-phosphorylated activated ERK1/2 was from Cell Signaling (Beverly, MA). Monoclonal anti-phosphotyrosine, polyclonal goat anti-EGFR, polyclonal anti-ERK1/2, monoclonal anti-ERK2, anti-Rap1, and horseradish peroxidase (HRP)-conjugated anti-goat were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-pY-1068-EGFR was from Biosource (Camarillo, CA). The monoclonal antibodies recognizing Myc- and HA-tags were from Roche (Mannheim, Germany). Enhanced chemiluminescence reagents were from Pierce (Rockford, IL).
Cell Culture
PC12 cells were grown in Dulbecco`s modified Eagle`s medium (DMEM) containing 10% horse serum and 5% fetal calf serum plus antibiotics. EGFR-overexpressing PC12 cells were kindly provided by P. Cohen (University of Dundee, United Kingdom; Traverse et al., 1994). PC12 cells defective in PKA activation were kindly provided by J. A. Wagner (Cornell University Medical College, New York; Ginty et al., 1991). Cells were serum-starved for at least 4 h before their exposure to stimuli in serum-free DMEM. Inhibitors (PD98059, PD153035, PD168393, H-89, and AG1478) were added to the cells 30–45 min before stimulation of the cells.
Detection of Neurite Outgrowth
Cells grown in 24-well dishes were exposed to the indicated reagents or vehicle for 24 h in serum-containing DMEM. Cells were visualized by phase-contrast microscopy and representative cells were photographed with a CCD camera. Images were prepared using Adobe Photoshop software (San Jose, CA).
[3H]Thymidine Incorporation
Cells, 2 × 104 cells/well, were seeded on 96-well plates, and the cells were switched to 2% serum and incubated with stimuli for the indicated time. Subsequently, 0.3 μCi [3H]thymidine was added to each well and the incubation was continued for 4 h. The cells were washed extensively, and the incorporated radioactivity was determined using a Wallac Trilux scintillation counter (PerkinElmer, Boston, MA).
Immunoprecipitation and Immunoblotting
For the experiments serum-starved cells were incubated with the indicated agents at 37°C. At specified times, the incubation was stopped by the addition of lysis buffer (50 mM HEPES, pH 7.0, 100 mM NaCl, 0.2 mM MgSO4, 0.5 mM Na3VO4, 1% Triton X-100, 10 μg/ml leupeptin, 10 μg/ml aprotinin). The EGFR was immunoprecipitated by the addition of anti-EGFR antibody. After incubation for 3 h at 4°C with gentle agitation, protein G-Sepharose was added and the incubation was continued for 2 h. The immunoprecipitates were washed three times in lysis buffer, resuspended in 2× SDS sample buffer, boiled for 4 min, and separated on SDS polyacrylamide gels under reducing conditions. Gel-resolved proteins were electrotransferred to nitrocellulose sheets, and immunological detection of the proteins was performed as recently described (Piiper et al., 2000, 2003a). Antigen-antibody complexes were visualized using HRP-conjugated antibodies and the enhanced chemiluminescence system.
cDNAs and Transfection
Myc-tagged ERK2 was generated by inserting the cDNA of ERK2 (kindly provided by A. Cuenda, University of Dundee, Scotland) into pCMVMyc (Clontech, Mountain View, CA). For transient transfection cells grown in six-well plates were cotransfected with 3 μg of a plasmid encoding hemagglutinin (HA)-tagged Rap1GAP, a GTPase-activating protein of Rap1 (kindly provided by J. Bos, University of Utrecht, The Netherlands), and 1 μg of the Myc-ERK2 plasmid using Lipofectamine 2000 according to the instructions of the manufacturer (Invitrogen). 40 h after transfection, the cells were serum-starved, stimulated with CPT-cAMP for the indicated duration. After cell lysis, Myc-tagged ERK2 was immunoprecipitated and phosphorylation of ERK2 was determined by anti-phospho-ERK1/2 immunoblotting (Piiper et al., 2000).
Reproducibility of the Results
Results are representative of at least three experiments performed on different occasions.
RESULTS
The PKA-selective cAMP Analog 6-Bnz-cAMP Elicits Short-lived ERK1/2 Activation
While cAMP inhibits growth factor-induced ERK1/2 activation (Cook and McCormick, 1993; Wu et al., 1993; Burgering et al., 1993), it stimulates ERK1/2 in several cell types, most notably in neuroendocrine cells (Stork, 2003). In these cells, an additional pathway involving the 95-kDa isoform of B-Raf complexed to 14-3-3 appears to mediate the stimulatory effect of cAMP on ERK1/2 (Vossler et al., 1997; Qiu et al., 2000; Fujita et al., 2002). The neuroendocrine cell line PC12 is an excellent model to study the mechanism of cAMP-induced ERK1/2 activation and how ERK1/2 activation governs subsequent cell proliferation and differentiation (Marshall, 1995). To investigate the roles of PKA and Epacs in cAMP-induced ERK1/2 activation, we examined the effects of cAMP analogs that specifically activate either PKA or Epac, respectively, and thus allow to distinguish their potentially distinct functions on ERK1/2 activation in PC12 cells (Enserink et al., 2002; Christensen et al., 2003). Activation of PKA was determined by immunoblotting of cellular extracts with an antibody that recognizes the PKA phosphorylation motif RRXpS/pT, whereas Epac activation was monitored by detecting GTP-loading of the Epac downstream effector Rap1. 8-(4-chlorphenylthio)-adenosine 3′,5′-cyclic monophosphate (CPT-cAMP), which activates both PKA and Epac, and the PKA-specific cAMP analog 6-Bnz-cAMP increased PKA substrate motif phosphorylation, whereas the Epac-selective cAMP analog 2-Me-cAMP or EGF had no effect (Supplementary Figure 1, A and B). Conversely, CPT-cAMP and 2-Me-cAMP, but not 6-Bnz-cAMP, increased GTP-loading of Rap1 (Supplementary Figure 1C). These data confirm that 6-Bnz-cAMP and 2-Me-cAMP specifically activated PKA and Epac, respectively, in our experimental model. ERK1/2 activation was detected by immunoblotting of cellular lysates with an antibody that specifically recognizes the dually phosphorylated activated form of the enzyme. 2-Me-cAMP increased ERK1/2 phosphorylation only slightly (Figure 1, A and B). This increase was, however, significant, because the effect was observed in different clones of PC12 cells and repeatedly was present in all our experiments. 6-Bnz-cAMP elicited rapid, but short-lived ERK1/2 phosphorylation that was more pronounced than the effect of 2-Me-cAMP (Figure 1, A and C). Treatment of the cells with CPT-cAMP caused phosphorylation of ERK1/2 similar to 6-Bnz-cAMP, but the response was stronger and more sustained as it remained high after the 15–30 min of treatment (Figure 1A). Combined incubation of the cells with both 6-Bnz-cAMP and 2-Me-cAMP mimicked CPT-cAMP-induced ERK1/2 phosphorylation (Figure 1B). These data suggest that stimulation of PKA is sufficient to induce ERK1/2 activation, whereas activation of Epac may play a role in the sustained signaling of cAMP toward ERK1/2.
Figure 1.

The PKA- and the Epac-selective cAMP analogs each have specific effects on ERK1/2 phosphorylation, whereas their combined effects resemble that of CPT-cAMP. Parental cells (A–C) or cells defective in PKA activation (C, bottom panel) were exposed to 6-Bnz-cAMP (100 μM), 2-Me-cAMP (100 μM), 6-Bnz-cAMP plus 2-Me-cAMP or CPT-cAMP (100 μM) in the presence or absence of H-89 (10 μM) for the indicated time periods at 37°C in serum-free DMEM. The incubation was terminated by replacement of the medium with lysis buffer. Lysates were analyzed by anti-phospho-ERK1/2 immunoblotting. Antigen-antibody complexes were visualized by HRP-conjugated antibodies and the enhanced chemiluminescence system. Equal loading of the lanes was controlled by reprobing of the blots with anti-ERK1/2 or anti-actin.
To further examine the role of PKA in cAMP-induced ERK1/2 activation, we studied the effect of H-89, a specific PKA inhibitor, on CPT-cAMP-, 6-Bnz-cAMP-, and 2-Me-cAMP-induced ERK1/2 phosphorylation. Complete inhibition of CPT-cAMP- or 6-Bnz-cAMP-induced phosphorylation of the PKA substrate motif required 10 μM H-89 (Supplementary Figure 1, A and B). H-89 completely inhibited 6-Bnz-cAMP-induced ERK1/2 phosphorylation (Figure 1C) and partially inhibited the response to CPT-cAMP (Supplementary Figure 1B) or combined incubation of the cells with 6-Bnz-cAMP and 2-Me-cAMP, whereas the effects of EGF and 2-Me-cAMP remained unchanged. The inhibitory effect of H-89 on CPT-cAMP-induced PKA substrate phosphorylation correlated with its inhibitory effect on ERK1/2 activation (Supplementary Figure 1B). These data further support the involvement of PKA in cAMP-induced ERK1/2 activation.
Finally, we studied the effect of 6-Bnz-cAMP on ERK1/2 activation in cells expressing cAMP-insensitive regulatory subunit of PKA that inhibits cAMP-induced activation of PKA (Ginty et al., 1991). In these cells, 6-Bnz-cAMP-induced ERK1/2 activation was clearly reduced (Figure 1C). These data provide additional evidence that PKA indeed mediates the effects of 6-Bnz-cAMP on ERK1/2 activity.
Inhibition of Rap1 Abolishes the Sustained Phase of CPT-cAMP-induced ERK1/2 Activation
Epacs stimulate the small GTPases Rap1 and Rap2 independently from PKA (de Rooij et al., 1998; Kawasaki et al., 1998; Bos, 2003). To investigate if Rap1 participates in the sustained phase of cAMP-dependent ERK1/2 activation, we tested the effect of inhibition of Rap1 activation by overexpression of Rap1GAP on CPT-cAMP-induced ERK phosphorylation. To this end, Rap1GAP was cotransfected with Myc-tagged ERK2, and ERK2 activation was detected after anti-Myc immunoprecipitation by anti-phospho-ERK1/2 immunoblotting. Overexpression of Rap1GAP inhibited the sustained phase of CPT-cAMP-induced ERK2 phosphorylation, whereas the initial phase remained unchanged (Figure 2). Together with the data of Figure 1, these data suggest a role of Epac/Rap1 in the sustained phase of cAMP-induced ERK activation, whereas activation of Epac/Rap1 appears to be dispensable for the initial PKA-dependent phase of cAMP-induced ERK1/2 activation.
Figure 2.
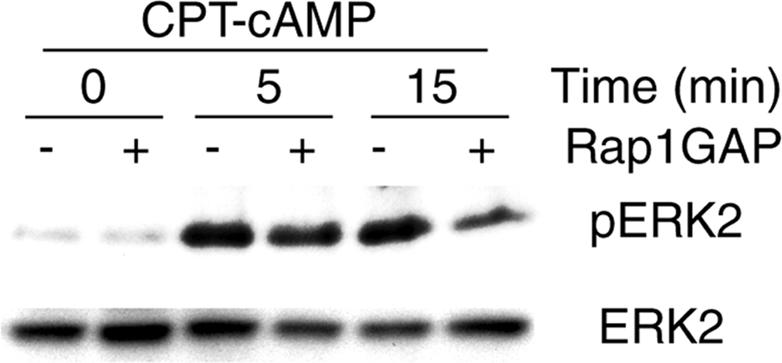
Activation of Rap1 is required for the sustained phase of CPT-cAMP-induced ERK phosphorylation. Parental PC12 cells were cotransfected with HA-Rap1GAP and Myc-ERK2. After 40 h, the cells were switched to serum-free medium. After additional 5 h, the cells were exposed to CPT-cAMP (100 μM) for the indicated duration at 37°C in serum-free DMEM. Lysates were immunoprecipitated with anti-Myc, and the immunoprecipitates were analyzed by anti-phospho-ERK1/2 immunoblotting. Antigen-antibody complexes were visualized by HRP-conjugated antibodies and the enhanced chemiluminescence system. Total ERK2 in the cell lysates was verified by anti-ERK2 immunoblotting.
The PKA-selective cAMP Analog 6-Bnz-cAMP Elicits Proliferation of PC12 Cells, Whereas cAMP Analogs Causing Simultaneous Activation of Both PKA and Epac Support Neurite Outgrowth
In PC12 cells, activation of the ERK1/2 signaling cascade can lead either to proliferation or neurite outgrowth. In these cells, the decision whether growth factor receptor activation elicits proliferation or neurite outgrowth through the ERK1/2 signaling pathway appears to depend on the set of adaptor proteins activated by the receptor tyrosine kinase and the kinetics of ERK1/2 activation (Traverse et al., 1992; Qui and Green, 1992; Marshall, 1995; York et al., 1998). Elevation of [cAMP]i does not elicit proliferation of PC12 cells and is a modest inducer of neurite outgrowth (Gunning et al., 1981; Frodin et al., 1994; Young et al., 1994). Therefore, we were surprised to observe that treatment of the cells with 6-Bnz-cAMP, which specifically activates PKA without affecting Epac in PC12 cells (Christensen et al., 2003), drastically affected the cell number in our experiments. To quantify cell proliferation, PC12 cells were incubated for various duration with the three different cAMP analogs or EGF followed by addition of [3H]thymidine for the last 4 h and detection of the radioactivity incorporated into the cells. As illustrated in Figure 3A, the PKA-specific cAMP analog 6-Bnz-cAMP strongly increased cell proliferation, whereas the Epac-activating cAMP analogs 2-Me-cAMP and CPT-cAMP did not elicit this response. Similar data were obtained in several different PC12 cell clones or when cell proliferation was determined by the 3-(4,5-dimethyldiazol-2-yl)-2,5 diphenyl tetrazolium bromide (MTT) assay (Supplementary Figure 2). Cell cycle analyses revealed that 6-Bnz-cAMP increased the portion of cells in the S-phase (Supplementary Table 1), thus further confirming the proliferative effect of 6-Bnz-cAMP in PC12 cells.
Figure 3.

6-Bnz-cAMP causes cell proliferation, whereas neurite outgrowth requires stimulation with cAMP analogs capable of activating both Epac and PKA. (A–C) PC12 cells grown in 96-well plates were exposed to 100 μM (A) or the indicated concentration (B and C) of 6-Bnz-cAMP, 2-Me-cAMP, CPT-cAMP, and/or EGF in 2% serum-containing DMEM for 70 h (B and C) or the indicated time (A). During the last 4 h of the incubation 0.2 μCi [3H]thymidine per well was added. The incorporation of radioactivity into the cells was determined by scintillation counting. The data are means ± SE. Asterisks (*) indicate significant differences to the condition without stimuli. The statistical significance was calculated by ANOVA (* p < 0.05). (D and E) Parental (D) or EGFR overexpressing PC12 cells (E) were incubated with the indicated concentration of cAMP analogs and/or EGF (20 ng/ml). After 48 h of incubation, phase-contrast images of representative cells were taken with a CCD camera.
PKA binds cAMP with considerably higher affinity than Epac (Bos, 2003), raising the possibility that at low CPT-cAMP concentrations CPT-cAMP might selectively activate PKA similar to 6-Bnz-cAMP and stimulate cell proliferation. However, CPT-cAMP did not increase cell proliferation at any concentration tested (10 nM to 100 μM; Figure 3B). This suggested that the selective activation of PKA rather than the concentration of the cAMP analogs was crucial for the proliferative effect of cAMP in PC12 cells.
To confirm the involvement of PKA in 6-Bnz-cAMP-induced PC12 cell proliferation, we examined the effect of 6-Bnz-cAMP on cell proliferation in the presence of H-89 and in PC12 cells defective in PKA activation. 6-Bnz-cAMP failed to elicit cell proliferation in the presence of H-89 (Supplementary Figure 2A), and 6-Bnz-cAMP-induced cell proliferation was inhibited in PKA-defective cells, whereas the effect of EGF was not significantly altered (Supplementary Figure 2B). These data indicate that PKA is essential for the proliferative response of 6-Bnz-cAMP in PC12 cells.
The finding that 6-Bnz-cAMP-induced cell proliferation was surprising because it was known that elevation of [cAMP]i does not cause proliferation of PC12 cells. cAMP even inhibits the proliferative effect of EGF and converts EGF from a proliferative into a differentiation stimulus (Yao et al., 1995; Mark and Storm, 1997). To investigate which of the cAMP effectors (PKA and Epac) mediates the anti-proliferative effect of cAMP in PC12 cells, we tested the effects of 2-Me-cAMP on [3H]thymidine incorporation in response to EGF or 6-Bnz-cAMP. As shown in Figure 3B, 2-Me-cAMP inhibited 6-Bnz-cAMP-induced [3H]thymidine incorporation, indicating that activation of Epac suppressed the proliferative effect of 6-Bnz-cAMP. 2-Me-cAMP also inhibited EGF-induced [3H]thymidine incorporation similarly to CPT-cAMP, whereas 6-Bnz-cAMP had no effect (Figure 3C). 6-Bnz-cAMP did not increase EGF-induced [3H]thymidine incorporation, probably because cell proliferation was already maximally stimulated by EGF. These results suggested that activation of Epac, but not of PKA, inhibited EGF-induced cell proliferation.
Elevation of [cAMP]i can induce low-degree neurite outgrowth on its own (Gunning et al., 1981) and potentiates growth factor-induced differentiation (Yao et al., 1995; Mark and Storm, 1997). To investigate if the anti-proliferative effect of Epac activation by 2-Me-cAMP is accompanied by neurite outgrowth, PC12 cells were exposed to the three different cAMP analogs in the presence or absence of EGF. 6-Bnz-cAMP, 2-Me-cAMP, or combined stimulation with these two cAMP analogs failed to induce neurite outgrowth (Figure 3D). Even combined stimulation of the cells with EGF and 6-Bnz-cAMP or 2-Me-cAMP did not induce neurite outgrowth (Figure 3D), whereas combined stimulation with CPT-cAMP and EGF or with 6-Bnz-cAMP, 2-Me-cAMP, and EGF elicited neurite outgrowth. These results indicated that individual stimulation of PKA or Epac was not sufficient to induce neurite outgrowth and that cAMP-induced neurite outgrowth required activation of both PKA and Epac.
To further study the roles of Epac and PKA in cAMP-induced neurite outgrowth, we investigated the effects of 6-Bnz-cAMP, 2-Me-cAMP, CPT-cAMP, and EGF on neurite outgrowth in EGFR-overexpressing PC12 cells. EGFR-overexpressing PC12 cells behave different from parental PC12 cells in that individual stimulation with EGF or CPT-cAMP is sufficient to cause neurite outgrowth (Traverse et al., 1994). CPT-cAMP or EGF elicited neurite outgrowth in EGFR-overexpressing cells, whereas 6-Bnz-cAMP or 2-Me-cAMP did not (Figure 3E). These data support the notion that cAMP-induced differentiation of PC12 cells requires activation of both PKA and Epac.
Involvement of the MEK/ERK1/2 Signaling Pathway in 6-Bnz-cAMP-induced ERK1/2 Activation and Proliferation of PC12 Cells
Our finding that specific activation of PKA in PC12 cells triggered cell proliferation prompted us to study the signaling pathways involved in this response. To investigate if 6-Bnz-cAMP-induced cell proliferation depends on activation of the ERK1/2 signaling pathway, the major intracellular signaling cascade regulating cell proliferation, we studied the effect of PD98059, a compound that specifically inhibits MEK1/2, the immediate upstream kinases of ERK1/2, on 6-Bnz-cAMP-induced cell proliferation. PD98059 strongly reduced 6-Bnz-cAMP-induced ERK1/2 activation (Figure 4A) and [3H]thymidine incorporation (Figure 4B), indicating that 6-Bnz-cAMP-induced PKA-dependent proliferation occurs through stimulation of the MEK/ERK1/2 pathway.
Figure 4.

Involvement of the MEK/ERK1/2 signaling pathway in PKA-dependent ERK1/2 activation and proliferation of PC12 cells. (A) Cells were exposed to 6-Bnz-cAMP (100 μM) or EGF (10 ng/ml) in the presence or absence of PD98059 (5 μM) for the indicated time periods at 37°C in serum-free DMEM. ERK1/2 activation was determined by anti-phospho-ERK1/2 immunoblotting. Equal amounts of ERK1/2 of the samples were controlled by immunoblotting with anti-ERK1/2. (B) PC12 cells grown in 96-well plates were exposed to 6-Bnz-cAMP or EGF in the presence or absence of PD98059 in 2% serum-containing DMEM for 70 h. During the last 4 h of the incubation 0.2 μCi [3H]thymidine per well was added. The incorporation of radioactivity into the cells was determined by scintillation counting. The proliferation data are means ± SE. Asterisks (*) indicate significant differences. Statistical significance was calculated by ANOVA (* p < 0.05).
Involvement of the EGFR in PKA-dependent ERK1/2 Activation and Proliferation of PC12 Cells
MEK inhibition blunted 6-Bnz-cAMP-induced ERK1/2 activation and cell proliferation (Figure 4), indicating that the MEK/ERK signaling pathway mediated 6-Bnz-cAMP-induced cell proliferation. Although a number of studies indicate that PKA participates in cAMP-induced ERK1/2 activation, it is unclear how PKA activates ERK1/2. To investigate if the 6-Bnz-cAMP-induced ERK1/2 activation depends on the EGFR, we studied the effect of two different EGFR tyrosine kinase inhibitors (AG1478, PD153035) on ERK1/2 phosphorylation in response to 6-Bnz-cAMP. As illustrated in Figure 5, A and B, each of the two EGFR tyrosine kinase inhibitors reduced 6-Bnz-cAMP-induced ERK1/2 phosphorylation, indicating a role of the EGFR in 6-Bnz-cAMP-induced ERK1/2 activation. The proliferative effect of 6-Bnz-cAMP was also reduced by EGFR tyrosine kinase blockade (Figure 5C), suggesting that the mitogenic action of PKA activation is EGFR tyrosine kinase-dependent. To obtain further evidence for a role of the EGFR in PKA-dependent ERK1/2 activation and proliferation in PC12 cells, we investigated the effects of 6-Bnz-cAMP and EGF on tyrosine phosphorylation of the EGFR. As illustrated in Figure 5D, 6-Bnz-cAMP caused rapid transient tyrosine phosphorylation of the EGFR similar to CPT-cAMP (Piiper et al., 2002), although to a lower degree than a maximal effective concentration of EGF. 6-Bnz-cAMP-induced ERK1/2 activation was slightly more sustained in EGFR overexpressing cells and correlated with 6-Bnz-cAMP-induced tyrosine phosphorylation of the EGFR. These data confirm a role of the EGFR in 6-Bnz-cAMP-induced mitogenic signaling. The EGFR tyrosine kinase inhibitor PD153035 ablated 6-Bnz-cAMP-induced tyrosine phosphorylation of the EGFR (Figure 5D), indicating that 6-Bnz-cAMP-induced tyrosine phosphorylation of the EGFR occurred through receptor autophosphorylation. In contrast, 2-Me-cAMP had no significant effect on EGFR tyrosine phosphorylation (data not shown).
Figure 5.

6-Bnz-cAMP elicits tyrosine phosphorylation of the EGFR and thereby ERK1/2 activation and cell proliferation. (A–D) Parental (A–C) or EGFR overexpressing PC12 cells (D) were exposed to 6-Bnz-cAMP (100 μM), CPT-cAMP (100 μM) or EGF (10 ng/ml) in the presence or absence of AG1478 (250 nM; A) or PD153035 (500 nM; B) for the indicated time periods at 37°C in serum-free DMEM. (A and B) Lysates were analyzed by anti-phospho-ERK1/2 and anti-ERK2 immunoblotting. In the majority of experiments, 6-Bnz-cAMP-induced ERK1/2 activation was maximal 5 min after beginning of the incubation in most experiments, but could occasionally be maximal even as early as 3 min after beginning of the incubation. (C) Cells grown in 96-well plates were exposed to CPT-cAMP, 6-Bnz-cAMP, 2-Me-cAMP (100 μM each), or EGF (10 ng/ml) in the presence or absence of PD168393 (250 nM) in 2% serum-containing DMEM for 70 h. During the last 4 h of the incubation 0.2 μCi [3H]thymidine per well was added. The incorporation of radioactivity into the cells was determined by scintillation counting. The proliferation data are means ± SE. Asterisks (*) indicate significant differences according to ANOVA (* p < 0.05). (D) The EGFR was immunoprecipitated and tyrosine phosphorylation of the EGFR was detected by anti-phosphotyrosine immunoblotting. Equal loading of the lanes was controlled by reprobing of the blot with anti-EGFR.
Participation of the EGFR in the Early Response to cAMP
A close examination of the effect of AG1478 on the time course of ERK1/2 activation in response to the adenylyl cyclase activator forskolin as well as CPT-cAMP revealed that AG1478 completely blocked the initial phase of ERK1/2 activation elicited by these agents, whereas the sustained phase was much less sensitive to EGFR tyrosine kinase inhibition (Figure 6, A and B). AG1478 completely blocked EGF-induced ERK1/2 activation (Figure 6C), but had no effect on NGF-induced ERK1/2 activation (Figure 6B), confirming the specificity of this substance for the EGFR tyrosine kinase. These data support the notion that the initial phase of cAMP-induced ERK1/2 activation is strongly EGFR-dependent, whereas the sustained phase is less EGFR-dependent.
Figure 6.

EGFR tyrosine kinase inhibition blocks the initial phase of cAMP-induced ERK1/2 activation, whereas the sustained phase is less EGFR-dependent. Parental (A–C and E) or EGFR overexpressing PC12 cells (D and F) were exposed to forskolin (20 μM), CPT-cAMP (100 μM), PACAP (10 nM), EGF (10 ng/ml), or NGF (10 ng/ml) in the presence or absence of AG1478 (250 nM) or H-89 (10 μM) for the indicated time periods at 37°C in serum-free DMEM. Cell lysates were analyzed by anti-phospho-ERK1/2 or anti-phospho-EGFR immunoblotting. Equal loading of the lanes was controlled by immunoblotting of the same samples anti-ERK1/2 or anti-actin as indicated. The band migrating above ERK1 is probably identical to ERK1b, a splice variant of ERK1 (Yung et al., 2000).
To investigate if the EGFR plays a similar role in ERK1/2 activation in response to a physiological stimulus that activates ERK1/2 through stimulation of adenylyl cyclase and subsequent elevation of [cAMP]i, we studied the effects of AG1478 and H-89 on ERK1/2 activation in response to the pituitary neuropeptide hormone PACAP, which stimulates ERK1/2 by PKA- and phospholipase C-dependent pathway (Bouschet et al., 2003). As shown in Figure 6D, inhibition of PKA by H-89 reduced PACAP-induced ERK1/2 phosphorylation. Inhibition of the EGFR by AG1478 (Figure 6E) abolished initial PACAP-induced ERK1/2 phosphorylation, whereas the sustained phase was less inhibited by AG1478, similar to CPT-cAMP- or forskolin-induced ERK1/2 activation. In agreement with a role of the EGFR in PACAP signaling, PACAP caused an increase in tyrosine phosphorylation of the EGFR (Figure 6D and 6F), and the PACAP-induced tyrosine phosphorylation of the EGFR correlated well with phosphorylation of ERK1/2, thus supporting a role of the EGFR in PACAP-induced ERK1/2 activation. Both H-89 (Figure 6D) and AG1478 (Figure 6F) partially inhibited PACAP-induced tyrosine phosphorylation of the EGFR, suggesting that PKA as well as the EGFR intrinsic tyrosine kinase activity participate in PACAP-induced EGFR tyrosine phosphorylation. Together, these data suggest that the initial phase of PACAP-induced ERK1/2 activation is mediated by transactivation of the EGFR, whereas the sustained phase is less EGFR-dependent. Furthermore, these data support the notion that a physiological stimulator (PACAP) also used the signaling intermediates, time course and the EGFR that we had characterized using cAMP analogs.
DISCUSSION
cAMP was the first second messenger to be discovered and is known to be involved in metabolism, cell proliferation, and differentiation. In particular, cAMP signaling has an important role in neuronal cells, where it is a key mediator of memory formation. PKA and Epac appear to mediate most of the effects of the second messenger [cAMP]i (Bos, 2003). Although other studies show involvement of either Epac or PKA in a given signaling pathway or biological response of cAMP, the data of the present study indicate that the biological outcome of an elevated [cAMP]i depends on the set of cAMP effectors activated: stimulation with the PKA-selective cAMP analog, which does not activate Epac, caused cell proliferation, whereas additional activation of Epac prevented PKA-induced proliferation and promoted neurite outgrowth.
Our finding that a compound that selectively activates PKA (Christensen et al., 2003) increased proliferation of PC12 cells was unexpected, because earlier studies have shown that elevation of [cAMP]i by forskolin or membranepermeant cAMP analogs are modest inducers of differentiation and have no significant effect on proliferation on their own and even prevent EGF-induced proliferation of PC12 cells (Mark and Storm, 1997). The present study indicates that additional activation of Epac overturned the proliferative effect of cAMP mediated by selective activation of PKA by 6-Bnz-cAMP, i.e., cAMP can elicit cell proliferation only in the absence of Epac activation. Earlier studies have shown that inhibition of proliferation, e.g., by rapamycin, also converts EGF from a proliferative into an anti-proliferative stimulus in PC12 cells, suggesting that the differentiation elicited by EGF and cAMP may be due to the anti-proliferative action of combined stimulation with cAMP and EGF (Mark and Storm, 1997). However, although the Epac-specific cAMP analog inhibited 6-Bnz-cAMP- or EGF-induced cell proliferation, significant neurite outgrowth required combined stimulation of the cells with EGF and activators of both Epac and PKA. Although we failed to observe significant neurite outgrowth upon stimulation of the cells with 6-Bnz-cAMP plus 2-Me-cAMP, a recent study reported modest neurite outgrowth in response to stimulation of the cells with five times higher concentrations of 6-Bnz-cAMP plus 2-Me-cAMP in the absence of EGF (Christensen et al., 2003), supporting the contention that Epac activation converts cAMP from a proliferative stimulus into a differentiating signal.
The finding that MEK inhibition abolished 6-Bnz-cAMP-induced cell proliferation and ERK1/2 activation indicated that the mitogenic effect of 6-Bnz-cAMP is mediated through activation of the ERK1/2 pathway, the major mitogenic signaling cascade. A number of studies suggest that PKA participates in cAMP-induced ERK1/2 activation, although the mechanism how cAMP induces ERK1/2 activation remained unclear. PKA may stimulate the ERK1/2 pathway by phosphorylating and activating Rap1 and subsequent activation of B-Raf (Altschuler et al., 1995; Vossler et al., 1997). In the present study, however, the PKA-selective cAMP analog 6-Bnz-cAMP activated ERK1/2 in the absence of any detectable Rap1 activation, indicating that Rap1 is not essential for PKA-induced ERK1/2 activation in our PC12 cells. The strong inhibitory effect of dominant-negative Ras on cAMP-induced ERK1/2 activation in several cell lines, including PC12 cells, suggests Ras might be involved in PKA-induced ERK1/2 activation (Erhardt et al., 1995; Iida et al., 2001; Enserink et al., 2002; Piiper et al., 2002; Norum et al., 2003; Bouschet et al., 2003).
Recently, several studies have shown an essential role of the EGFR in cAMP-induced ERK1/2 activation in PC12 and other cell types (Piiper et al., 2002, 2003b; Bertelsen et al., 2004; Piiper and Zeuzem, 2004; Sales et al., 2004), indicating that cAMP-induced ERK1/2 activation can proceed through transactivation of the EGFR. The results of the present study are in agreement with those showing that EGFR tyrosine kinase inhibitors reduce EGFR tyrosine phosphorylation and ERK1/2 activation in response to CPT-cAMP or forskolin (Piiper et al., 2002) and suggest that these effects were due to activation of PKA. This is concluded from our findings that 6-Bnz-cAMP increased tyrosine phosphorylation of the EGFR and that EGFR tyrosine kinase inhibition blocked 6-Bnz-cAMP-induced ERK1/2 phosphorylation and cell proliferation. The finding of the present study that EGFR tyrosine kinase inhibition blocked 6-Bnz-cAMP-induced EGFR tyrosine phosphorylation suggests that PKA-dependent EGFR tyrosine phosphorylation occurs through EGFR autophosphorylation. However, details on the mechanism how PKA causes transactivation of the EGFR remain to be elucidated. Our finding that transactivation of the EGFR mediates only the initial part of cAMP-induced ERK1/2 activation, whereas the sustained responses were considerably less sensitive to EGFR tyrosine kinase inhibitors, is compatible with the notion that the initial phase of cAMP-induced ERK1/2 activation depends on activation of PKA/EGFR, whereas the sustained phase requires additional activation of Epac.
The mechanism how stimulation of Epac overturned the proliferative effect of 6-Bnz-cAMP and EGF and instead promoted differentiation is not yet clear. The time course of ERK1/2 activation appears to be critical for the biological readout of growth factors such as EGF and NGF, i.e., short-lasting ERK1/2 activation and prolonged ERK1/2 activation are accompanied by proliferation and differentiation, respectively (Traverse et al., 1992, 1994; Marshall, 1995). In support of a critical role of the kinetics of ERK1/2 activation for the biological outcome of cAMP, we observed that ERK1/2 activation in response to the proliferative cAMP analog 6-Bnz-cAMP was more transient than those of CPT-cAMP or 2-Me-cAMP. Thus, the more rapid time course of ERK1/2 activation by 6-Bnz-cAMP correlated with cell proliferation and the more sustained ERK1/2 activation by CPT-cAMP with neurite outgrowth similar as observed for EGF and NGF, respectively. Cytoplasmic-to-nuclear movement of ERK1/2 has been suggested to be important for the long-term consequences of ERK1/2 activation in multiple cellular processes, such as cell growth, differentiation, and neuronal plasticity (Marshall, 1995). Sustained ERK1/2 activation has been linked to an increased nuclear translocation of ERK in PC12 cells (Traverse et al., 1992; Marshall, 1995). Thus, it is possible that the more sustained ERK1/2 activation upon stimulation of Epac/Rap1 could increase the nuclear translocation of ERK1/2 and thereby alter gene expression and the biological readout.
In the present study, the Epac-specific cAMP analog 2-Me-cAMP elicited a significant but low-degree ERK1/2 activation. Furthermore, CPT-cAMP-induced ERK1/2 activation was incompletely blocked by H-89 or in PKA-defective cells. These data suggest that cAMP can stimulate ERK1/2 in the absence of PKA activation in PC12 cells. The low levels of ERK1/2 phosphorylation may explain why other studies found no effect of this Epac-selective cAMP analog on ERK1/2 activity in PC12 cells (Enserink et al., 2002; Johnson-Farley et al., 2005). In agreement with a role of Epac in cAMP-induced ERK1/2 activation in PC12 cells, overexpression of Epac1 enhances ERK1/2 activation upon stimulation of Gs-coupled 5-HT7A receptors in these cells (Johnson-Farley et al., 2005).
Epacs are guanine nucleotide exchange factors on Rap1 and Rap2 and may thus exert their effects on ERK1/2 through activation of Rap1 or Rap2 (Chen et al., 2004; Keiper et al., 2004). It has been reported that cAMP-induced ERK1/2 activation involves PKA-dependent activation of Rap1, which in turn activates B-Raf in PC12 cells (Vossler et al., 1997). The data of the present study supported the contention that cAMP-induced ERK1/2 activation involves stimulation of Rap1 in PC12 cells. However, we and others have found that cAMP-induced activation of Rap1 proceeds through activation of Epacs independently from PKA in PC12 cells (Supplementary Figure 1C and Enserink et al., 2002). Furthermore, our findings suggested a role of Rap1 in the sustained phase of cAMP-induced ERK1/2 activation. This is in agreement with recent studies, which have shown that activation of Rap1 is required for the sustained phase of NGF- and PACAP-induced ERK1/2 activation (York et al., 1998; Bouschet et al., 2003). Thus, Rap1 might be essential for the sustained phase of ERK1/2 activation in response to a variety of different stimuli.
The present study also provides evidence for an involvement of the EGFR in ERK1/2 activation in response to PACAP, a neuropeptide agonist on GsPCRs. PACAP stimulates adenylyl cyclase, elevates [cAMP]i, and exerts a number of growth factor-like effects that depend on the cell type and the developmental stage. These include regulation of cell proliferation, survival, and neurite outgrowth (Waschek, 2002). PACAP also plays important roles in repair upon neuronal injury. In PC12 cells, PACAP signals through Pac1 receptors, activates ERK1/2 and induces neurite outgrowth (Deutsch and Sun, 1992; Waschek, 2002). Our experiments showed that PACAP caused tyrosine phosphorylation of the EGFR and that EGFR tyrosine kinase inhibition suppressed specifically the initial phase of PACAP-induced ERK1/2 activation. Thus, the EGFR transactivation pathway described for the initial activation of ERK1/2 by the second-messenger cAMP might be relevant for a GsPCR, which signals via [cAMP]i, because a neuropeptide activating the cAMP pathway in PC12 cells also triggered EGFR transactivation.
In summary, using a well-characterized cellular model system, we uncovered novel mechanisms of signal integration by cAMP effectors. It was particularly interesting to note that activation of single cAMP effectors can trigger opposite physiological outcomes. The integration of cAMP signals at the ERK1/2 level is consistent with previously identified transient and sustained ERK1/2 activation as key mediators of the physiological outcome (Qui and Green, 1992; Traverse et al., 1994; Marshall, 1995; York et al., 1998). Finally, the use of a PKA-specific cAMP analog suggests that PKA is the cAMP effector that triggers transactivation of EGFR and transient activation of ERK1/2. Previous years have been dedicated to identifying linear signaling pathways. The present work appears as an example of the relevance of the integration of different signals and transactivation of signaling pathways in the physiological response to stimuli.
Supplementary Material
Acknowledgments
We thank P. Cohen (University of Dundee, Scotland) for providing EGFR-overexpressing PC12 cells. PC12 cells defective in PKA activation were kindly provided by J. A. Wagner (Cornell University Medical College, New York). The cDNA encoding for HA-tagged Rap1GAP was kindly provided by J. Bos(University Medical Center Utrecht, The Netherlands). The cDNA encoding ERK2 was a gift of A. Cuenda (University of Dundee, Scotland). We are grateful to B. Collins (University of Dundee, Scotland) for critically reading the manuscript, I. Dikic and W. Müller-Esterl (University of Frankfurt/M., Germany) for support, and H. Diehl for excellent technical assistance. We thank A. Wadle (University of Saarland, Germany) for advice concerning cell cycle analyses. This work was supported by grants from the Deutsche Forschungsgemeinschaft (Pi 258/7–1), the Wilhelm Sander Foundation (2003.119.1), and the University of Saarland (Anschubfinanzierung 2004).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–05–0432) on October 5, 2005.
Abbreviations used: 2-Me-cAMP, 8-(4-methoxyphenylthio)-2′-O-methyl-adenosine-3′,5′-cyclic AMP; 6-Bnz-cAMP, N6-benzoyladenosine-3′,5′-cyclic monophosphate; [cAMP]i, intracellular cAMP concentration; CPT-cAMP, 8-(4-chlorphenylthio)-adenosine-3′,5′-cyclic monophosphate; EGF, epidermal growth factor; EGFR, EGF receptor; ERK, extracellular signal-regulated kinase; GsPCR, Gs protein-coupled receptors; HRP, horseradish peroxidase; NGF, nerve growth factor; PACAP, pituitary adenylyl cyclase-activating peptide; PKA, protein kinase A; Rap1GAP, GTPase-activating protein of Rap1.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Altschuler, D. L., Peterson, S. N., Ostrowski, M. C., and Lapetina, E. G. (1995). Cyclic AMP-dependent activation of Rap1b. J. Biol. Chem. 270, 10373–10376. [DOI] [PubMed] [Google Scholar]
- Barad, M., Bourtchouladze, R., Winder, D. G., Golan, H., and Kandel, E. (1998). Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc. Natl. Acad. Sci. USA 95, 15020–15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen, L. S., Barrett, K. E., and Keely, S. J. (2004). Gs protein-coupled receptor agonists induce transactivation of the epidermal growth factor receptor in T84 cells: implications for epithelial secretory responses. J. Biol. Chem. 279, 6271–6279. [DOI] [PubMed] [Google Scholar]
- Bos, J. L. (2003). Epac: a new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell. Biol. 4, 733–738. [DOI] [PubMed] [Google Scholar]
- Bouschet, T., Perez, V., Fernandez, C., Bockaert, J., Eychene, A., and Journot, L. (2003). Stimulation of the ERK pathway by GTP-loaded Rap1 requires the concomitant activation of Ras, protein kinase C (PKC), and protein kinase A in neuronal cells. J. Biol. Chem. 278, 4778–4785. [DOI] [PubMed] [Google Scholar]
- Burgering, B.M.T., Pronk, G. J., van Weeren, P. C., Chardin, P., and Bos, J. L. (1993). cAMP antagonizes p21ras-directed activation of extracellular signal-regulated kinase 2 and phosphorylation of mSos nucleotide exchange factor. EMBO J. 12, 4211–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C., Koh, A. J., Datta, N. S., Zhang, J., Keller, E. T., Xiao, G., Franceschi, R. T., D'Silva, N. J., and McCauley, L. K. (2004). Impact of the mitogen-activated protein kinase pathway on parathyroid hormone-related protein actions in osteoblasts. J. Biol. Chem. 279, 29121–29129. [DOI] [PubMed] [Google Scholar]
- Christensen, A. E. et al. (2003). cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J. Biol. Chem. 278, 35394–35402. [DOI] [PubMed] [Google Scholar]
- Cook, S. J., and McCormick, F. (1993). Inhibition by cAMP of Ras-dependent activation of Raf. Science 262, 1069–1072. [DOI] [PubMed] [Google Scholar]
- de Rooij, J., Zwartkruis, F. J., Verheijen, M. H., Cool, R. H., Nijman, S. M., Wittinghofer, A., and Bos, J. L. (1998). Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477. [DOI] [PubMed] [Google Scholar]
- Deutsch, P. J., and Sun, Y. (1992). The 38-amino acid form of pituitary adenylate cyclase-activating polypeptide stimulates dual signaling cascades in PC12 cells and promotes neurite outgrowth. J. Biol. Chem. 267, 5108–5113. [PubMed] [Google Scholar]
- Enserink, J. M., Christensen, A. E., de Rooij, J., Van Triest, M., Schwede, F., Genieser, H. G., Doskeland, S. O., Blank, J. L., and Bos, J. L. (2002). A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 4, 901–906. [DOI] [PubMed] [Google Scholar]
- Erhardt, P., Troppmair, J., Rapp, U. R., and Cooper, G. M. (1995). Differential regulation of Raf-1 and B-Raf and Ras-dependent activation of mitogen-activated protein kinase by cyclic AMP in PC12 cells. Mol. Cell. Biol. 15, 5524–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin, M., Peraldi, P., and Van Obberghen, E. (1994). Cyclic AMP activates the mitogen-activated protein kinase cascade in PC12 cells. J. Biol. Chem. 269, 6207–6214. [PubMed] [Google Scholar]
- Fujita, T., Meguro, T., Fukuyama, R., Nakamuta, H., and Koita, M. (2002). New signaling pathway for parathyroid hormone and cyclic AMP action on extracellular-regulated kinase and cell proliferation in bone cells. Checkpoint of modulation by cyclic AMP. J. Biol. Chem. 277, 22191–22200. [DOI] [PubMed] [Google Scholar]
- Ginty, D. D., Glowacka, D., DeFranco, C., and Wagner, J. A. (1991). Nerve growth factor-induced neuronal differentiation after dominant repression of both type I and type II cAMP-dependent protein kinase activities. J. Biol. Chem. 266, 15325–15333. [PubMed] [Google Scholar]
- Gunning, P. W., Landreth, G. E., Bothwell, M. A., and Shooter, E. M. (1981). Differential and synergistic actions of nerve growth factor and cyclic AMP in PC12 cells. J. Cell Biol. 89, 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida, N., Namikawa, K., Kiyama, H., Ueno, H., Nakamura, S., and Hattori, S. (2001). Requirement of Ras for the activation of mitogen-activated protein kinase by calcium influx, cAMP, and neurotrophin in hippocampal neurons. J. Neurosci. 21, 6459–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Farley, N. N., Kertesy, S. B., Dubyak, G. R., and Cowen, D. S. (2005). Enhanced activation of Akt and extracellular-regulated kinase pathways by simultaneous occupancy of Gq-coupled 5-HT2A receptors and Gs-coupled 5-HT7A receptors in PC12 cells. J. Neurochem. 92, 72–82. [DOI] [PubMed] [Google Scholar]
- Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. [DOI] [PubMed] [Google Scholar]
- Kawasaki, H., Springett, G. M., Mochizuki, N., Toki, S., Nakaya, M., Matsuda, M., Housman, D. E., and Graybiel, A. M. (1998). A family of cAMP-binding proteins that directly activate Rap1. Science 282, 2275–2279. [DOI] [PubMed] [Google Scholar]
- Keiper, M. et al. (2004). Epac- and Ca2+-controlled activation of Ras and extracellular signal-regulated kinases by Gs-coupled receptors. J. Biol. Chem. 279, 46497–46508. [DOI] [PubMed] [Google Scholar]
- Mark, M., and Storm, D. R. (1997). Coupling of epidermal growth factor (EGF) with the antiproliferative activity of cAMP induces neuronal differentiation. J. Biol. Chem. 272, 17238–17244. [DOI] [PubMed] [Google Scholar]
- Marshall, C. J. (1995). Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179–185. [DOI] [PubMed] [Google Scholar]
- Norum, J. H., Hart, K., and Levy, F. O. (2003). Ras-dependent ERK activation by the human G(s)-coupled serotonin receptors 5-HT4(b) and 5-HT7(a). J. Biol. Chem. 278, 3098–3104. [DOI] [PubMed] [Google Scholar]
- Piiper, A., Gebhardt, R., Kronenberger, B., Giannini, C. D., Elez, R., and Zeuzem, S. (2000). Pertussis toxin inhibits cholecystokinin- and epidermal growth factor-induced mitogen-activated protein kinase activation by disinhibition of the cAMP signaling pathway and inhibition of c-Raf-1. Mol. Pharmacol. 58, 608–613. [DOI] [PubMed] [Google Scholar]
- Piiper, A., Dikic, I., Lutz, M. P., Leser, J., Cramer, H., Kronenberger, B., Elez, R., Müller-Esterl, W., and Zeuzem, S. (2002). Cyclic AMP induces transactivation of the receptors for epidermal growth factor and nerve growth factor, thereby modulating activation of MAP kinase, Akt, and neurite outgrowth in PC12 cells. J. Biol. Chem. 277, 43623–43630. [DOI] [PubMed] [Google Scholar]
- Piiper, A., Elez, R., You, S. J., Kronenberger, B., Loitsch, S. M., and Zeuzem, S. (2003a). Cholecystokinin stimulates extracellular signal-regulated kinase through activation of the epidermal growth factor receptor, Yes, and PKC. Signal amplification at the level of Raf by activation of protein kinase Cepsilon. J. Biol. Chem. 278, 7065–7072. [DOI] [PubMed] [Google Scholar]
- Piiper, A., Lutz, M. P., Cramer, H., Elez, R., Kronenberger, B., Dikic, I., Müller-Esterl, W., and Zeuzem, S. (2003b). Protein kinase A mediates cAMP-induced tyrosine phosphorylation of the epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 301, 848–854. [DOI] [PubMed] [Google Scholar]
- Piiper, A., and Zeuzem, S. (2004). Receptor tyrosine kinases are signaling intermediates of G protein-coupled receptors. Curr. Pharm. Design 10, 3539–3545. [DOI] [PubMed] [Google Scholar]
- Qiu, W., Zhuang, S., von Lintig, F. C., Boss, G. R., and Pilz, R. B. (2000). Cell type-specific regulation of B-Raf kinase by cAMP and 14-3-3 proteins. J. Biol. Chem. 275, 31921–31929. [DOI] [PubMed] [Google Scholar]
- Qui, M. S., and Green, S. H. (1992). PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron 9, 705–717. [DOI] [PubMed] [Google Scholar]
- Sales, K. J., Maudsley, S., and Jabbour, H. N. (2004). Elevated prostaglandin EP2 receptor in endometrial adenocarcinoma cells promotes vascular endothelial growth factor expression via cyclic 3′,5′-adenosine monophosphate-mediated transactivation of the epidermal growth factor receptor and extracellular signal-regulated kinase 1/2 signaling pathways. Mol. Endocrinol. 18, 1533–1545. [DOI] [PubMed] [Google Scholar]
- Spada, A., and Lania, A. (2002). Growth factors and human pituitary adenomas. Mol. Cell. Endocrinol. 197, 63–68. [DOI] [PubMed] [Google Scholar]
- Stork, P. J. (2003). Does Rap1 deserve a bad Rap? Trends Biol. Sci. 28, 267–275. [DOI] [PubMed] [Google Scholar]
- Traverse, S., Gomez, N. Paterson, H., Marshall, C., and Cohen, P. (1992). Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Biochem. J. 288, 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse, S., Seedorf, K., Paterson, H., Marshall, C. J., Cohen, P., and Ullrich, A. (1994). EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr. Biol. 4, 694–701. [DOI] [PubMed] [Google Scholar]
- Vossler, M. R., Yao, H., York, R. D., Pan, M. G., Rim, C. S., and Stork, P. J. (1997). cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89, 73–82. [DOI] [PubMed] [Google Scholar]
- Wang, H., Ferguson, G. D., Pineda, V. V., Cundiff, P. E., and Storm, D. R. (2004). Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat. Neurosci. 7, 635–642. [DOI] [PubMed] [Google Scholar]
- Waschek, J. A. (2002). Multiple actions of pituitary adenylyl cyclase activating peptide in nervous system development and regeneration. Dev. Biol. 24, 14–23. [DOI] [PubMed] [Google Scholar]
- Wu, J., Dent, P., Jelinek, T., Wolfman, A., Weber, M. J., and Sturgill, T. W. (1993). Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′-monophosphate. Science 262, 1066–1069. [DOI] [PubMed] [Google Scholar]
- Yao, H., Labbuda, K., Rim, C., Capodieci, P., Loda, M., and Stork, P. J. (1995). Cyclic adenosine monophosphate can convert epidermal growth factor into a differentiating factor in neuronal cells. J. Biol. Chem. 270, 20748–20753. [DOI] [PubMed] [Google Scholar]
- York, R. D., Yao, H., Dillon, T., Ellig, C. L., Eckert, S. P., McCleskey, E. W., and Stork, P. J. (1998). Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392, 622–626. [DOI] [PubMed] [Google Scholar]
- Young, S. W., Dickens, M., and Tavaré, J. M. (1994). Differentiation of PC12 cells in response to a cAMP analogue is accompanied by sustained activation of mitogen-activated protein kinase. Comparison with the effects of insulin, growth factors and phorbol esters. FEBS Lett. 338, 212–216. [DOI] [PubMed] [Google Scholar]
- Yung, Y., Yao, Z., Hanoch, T., and Seger, R. (2000). ERK1b, a 46-kDa ERK isoform that is differentially regulated by MEK. J. Biol. Chem. 275, 15799–15808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.