

Abstract
The essential membrane fusion apparatus in mammalian cells, the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, consists of four α-helices formed by three proteins: SNAP-25, syntaxin 1, and synaptobrevin 2. SNAP-25 contributes two helices to the complex and is targeted to the plasma membrane by palmitoylation of four cysteines in the linker region. It is alternatively spliced into two forms, SNAP-25a and SNAP-25b, differing by nine amino acids substitutions. When expressed in chromaffin cells from SNAP-25 null mice, the isoforms support different levels of secretion. Here, we investigated the basis of that different secretory phenotype. We found that two nonconservative substitutions in the N-terminal SNARE domain and not the different localization of one palmitoylated cysteine cause the functional difference between the isoforms. Biochemical and molecular dynamic simulation experiments revealed that the two substitutions do not regulate secretion by affecting the property of SNARE complex itself, but rather make the SNAP-25b-containing SNARE complex more available for the interaction with accessory factor(s).
INTRODUCTION
The identity of the proteinaceous machinery responsible for fusing intracellular membranes is rapidly being unraveled (Jahn et al., 2003). Among the molecular players the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins assume a special position, because they seem to form the essential fusion apparatus on which the other proteins work. Reconstituted SNARE proteins suffice to fuse vesicles in vitro (Weber et al., 1998), and many of the other proteins regulating membrane fusion (e.g., Sec1p/Munc18-proteins, synaptotagmins, and complexins) may be recruited to the fusion apparatus through binding to SNAREs. A simple model would be that the SNARE complex executes the membrane fusion reaction itself, whereas accessory proteins would provide the necessary regulation of the process. However, the situation seems more complicated, because the neuronal SNARE proteins that act in fast neuroexocytosis to release neurotransmitter in the synapse are present in alternative isoforms (Elferink et al., 1989; Archer et al., 1990; Bennett et al., 1992; Bark and Wilson, 1994). The question how these isoforms regulate secretion has not been resolved.
The core of the neuronal SNARE complex is a twisted coiled-coil structure composed of amphipathic helices contributed by the plasma membrane attached proteins syntaxin 1 and synaptosome-associated protein of 25 kDa (SNAP-25), and the vesicular synaptobrevin 2 (Sutton et al., 1998; Figure 1). SNAP-25 provides two of the four α-helices to the complex and is attached to the plasma membrane via palmitoylation of four cysteine residues in the linker region between the two SNARE domains. In contrast, both syntaxin 1 and synaptobrevin 2 have transmembrane domains and contribute to the SNARE complex with one α-helix each. The orientation of the four α-helices is parallel, so that the membrane anchors of syntaxin and synaptobrevin are located on the same side of the complex. This structure led to the suggestion that the SNARE complex when formed in trans would act as a molecular zipper, such that formation toward the transmembrane anchors brings the membranes into contact and eventually leads to fusion (Hanson et al., 1997). This model for SNARE action would predict that assembly of the SNARE complex might be rate limiting for secretion. Considering that fast chemical neurotransmission depends on the extremely tight temporal coupling (<0.5 ms) between the calcium trigger for exocytosis and neurotransmitter release, this raises the question whether residues in the SNARE domains are modified to enable physiological regulation of synaptic transmission or whether they are conserved so as not to compromise the basic fusogenic function of SNARE complexes.
Figure 1.

Alternative splicing of exon 5 introduces nine amino acid substitutions in SNAP-25. (A) The substitutions are located in the C-terminal end of the first SNARE motif and the first part of the linker and include a relocalization of one of the palmitoylated cysteines. The gray boxes and numbers show residues that are buried in the inside of the complex. (B) Crystal structure of the ternary SNARE complex (Sutton et al., 1998). The membrane anchors of syntaxin and synaptobrevin would attach at the right side. The linker between the two SNAP-25 SNARE domains (SN1 and SN2) was added using a drawing program. The structure was downloaded from PubMed (1SFC) and drawn using Swiss-Pdb viewer (Guex and Peitsch, 1997; http://www.expasy.org/spdbv/).
SNAP-25 is expressed as two isoforms that differ by nine amino acid substitutions. The substitutions cluster within the N-terminal region of the first SNARE domain and in the adjacent sequence and include a relocation of one of the four cysteine residues that are required for membrane association (Figure 1). The substitutions in the N-terminal SNARE domain of the two SNAP-25 isoforms include three charge changes (Figure 1). This is remarkable because in those syntaxin and synaptobrevin isoforms that have been shown to enter alternatively into the neuronal SNARE complex, substitutions in the SNARE domains are either nonexistent or conservative (syntaxin 1A and 1B, Bennett et al., 1992; synaptobrevin 1 and 2, Elferink et al., 1989; Archer et al., 1990; and synaptobrevin 2 and cellubrevin, McMahon et al., 1993; Borisovska et al., 2005). The two SNAP-25 isoforms are the product of developmentally regulated alternative splicing of duplicated but divergent copies of exon 5 (Bark and Wilson, 1994; Bark et al., 1995). In the embryonic brain, SNAP-25a is the prevalent isoform, whereas the expression of SNAP-25b increases robustly through postnatal brain development to become the predominant isoform in most, but not all, adult brain areas (Bark et al., 1995; Boschert et al., 1996). Impairment of this switch toward the SNAP-25b isoform in mice leads to premature mortality and a change in short-term plasticity in CA1 hippocampal synapses (Bark et al., 2004). In contrast, the SNAP-25a isoform remains the predominant species in adult rat and mouse adrenal chromaffin cells and in PC12 cells (Bark et al., 1995; Grant et al., 1999). Importantly, when expressed in chromaffin cells from SNAP-25 null mice, the two splice variants support different levels of secretion, due to differential regulation of the size of the releasable vesicle pools (Sørensen et al., 2003).
Here, we used a number of approaches to investigate the question how the two SNAP-25 isoforms differentially regulate secretion from chromaffin cells. The answer to this question has important implications for the understanding of how the SNARE complex can both regulate and trigger vesicle fusion.
MATERIALS AND METHODS
Chromaffin Cell Preparation, Mutagenesis, and Expression
SNAP-25 null embryos (E17-19) were recovered by Cesarean section and chromaffin cells prepared as described previously (Sørensen et al., 2003). Mutations were introduced into SNAP-25a- or SNAP-25b-containing pSFV1 plasmids (pSFV1 SNAP-25a-IRES-EGFP and pSFV1 SNAP-25b-IRES-EGFP) by using PCR mutagenesis, and all constructs were sequenced. Semliki Forest Virus (SFV) expressing SNAP-25 and enhanced green fluorescent protein (EGFP) were prepared as described previously (Ashery et al., 1999).
Plasma Membrane Sheets from Embryonic Mouse Chromaffin Cells: Generation and Immunofluorescence
Mouse chromaffin cells were plated on Ø 25-mm glass coverslips pretreated with 0.1 mg/ml poly-l-lysine (Sigma-Aldrich, St. Louis, MO) for 30 min. Plasma membrane sheets were generated 22–28 h after cell plating and 7 h after viral infection by placing the coverslip into 150 ml of ice-cold sonication buffer (120 mM potassium glutamate, 20 mM potassium acetate, 20 mM HEPES, 0.5 mM dithiothreitol [DTT], 2 mM ATP, 100 μM GTP, 4 mM MgCl2, 4 mM EGTA, 6 mM Ca2+-EGTA, [Ca2+]free = 300 nM, pH 7.2, and 310 mOsM/kg; bubbled with N2 for 30 min) in a round glass beaker with a final volume of 300 ml. The coverslip with the attached cells was centered 14 mm under the sonication tip (Ø 2.5 mm), and the cells were disrupted applying a single ultrasound pulse (Sonifier 450, power setting at 1.6–1.8 and a duty cycle of 100 ms; Branson, Danbury, CT). Apart from chromaffin cells, the primary culture contained a low number of other cell types (e.g., endothelial cells). However, the membrane sheets generated from these cells differed in size and had lower level of staining for syntaxin 1, so that they were easily recognized and excluded from analysis.
For immunolabeling, freshly prepared membrane sheets were fixed for 2 h at room temperature in phosphate-buffered saline (PBS) containing 4% paraformaldehyde. They were washed twice in PBS, incubated for 10 min with 50 mM NH4Cl in PBS to block free aldehyde groups, and then washed once more with PBS. Sheets were incubated for 2 h with primary antibodies raised against SNAP-25 (mouse monoclonal Cl 71.2, recognizing both SNAP-25a and b; Xu et al., 1999) and syntaxin 1 (rabbit polyclonal R31; Lang et al., 2001) diluted 1:100 in PBS containing 1% bovine serum albumin (PBS-bovine serum albumin). They were washed four times for 10 min each with PBS and then incubated for 1 h with secondary antibodies diluted 1:200 in PBS-bovine serum albumin (Cy3-coupled goat-anti-mouse and Cy5-coupled goat-anti-rabbit; Jackson ImmunoResearch Laboratories, West Grove, PA). Membrane sheets were washed four times in PBS and were then imaged in PBS containing 1-(4-trimethyl-amoniumphenyl)-6-phenyl-1,3,5-hexatriene (TMA-DPH; Invitrogen, Carlsbad, CA). TMA-DPH visualizes phospholipid membranes and therefore allows for the identification of membrane sheets. In addition, 0.2-μm TetraSpeck beads (Invitrogen) were added and allowed to adsorb to the glass coverslip, acting as a spatial reference to correct for vertical shifts that occur during filter changes.
Coverslips mounted in an open chamber were analyzed using a Zeiss Axiovert 100 TV fluorescence microscope with a 100× 1.4 numerical aperture plan achromate objective. Appropriate filter sets were used for TMA-DPH (BP 350/50, BS 395, and BP 420LP), Cy3 (BP 525/30, BS 550LP, and BP 575/30) and Cy5 (BP 620/60, BS 660LP, and BP 700/75). Throughout all experiments the focal position of the objective was controlled using a low-voltage piezo translator driver and a linear variable transformer displacement controller (Physik Instrumente, Waldbronn, Germany). Recordings were performed with a back-illuminated charge-coupled device camera (512 × 512-EEV chip, 24 × 24-μm pixel size; Princeton Instruments, Trenton, NJ) with a 2.5× Optovar magnifying lens. Images were acquired and analyzed using the program MetaMorph (Molecular Devices, Sunnyvale, CA).
For comparative quantitation of fluorescence intensity, membrane sheets were identified and selected in the TMA-DPH channel in an unbiased manner. Regions of interest (40 × 40 pixels corresponding to 3.7 × 3.7 μm) were placed onto the sheets and then transferred to the Cy3- and Cy5-channels with corrections being made to avoid obvious artifacts such as highly fluorescent contaminating particles that were occasionally seen. In the Cy3- and Cy5-channels, the average fluorescence intensity was determined and corrected for the local background measured in an area outside the membrane sheets.
From each animal, at least 10 membrane sheets were analyzed, and the mean value normalized to the mean of +/+ animals from the same litter. The animal means were used to calculate population mean and SEM (number of animals = 7–12 for each condition). For image representation, a linear lookup-table was applied using the autoscale-function. In some cases, the maximal value was decreased to make the finer structures (clusters) visible.
Correlative Features of Fluorescent Spots
To investigate correlative properties of fluorescent spots we calculated the normalized correlation coefficient between pairs of same-sized images (or regions of interests) detected in different channels. If f′ (x,y) and t′ (x,y) are the two images after subtraction of the respective means the normalized correlation coefficient is given by (Manders et al., 1992)
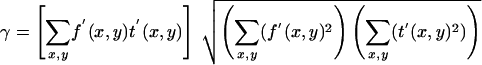 |
The calculations were carried out in a custom-written macro for Igor Prover 4.01 (Wavemetrics, Lake Oswego, Oregon). We used the fluorescence profile of TetraSpeck beads (Invitrogen) to align the images with another. To investigate the maximal degree of correlation that could be expected in our system between the two channels used for SNAP-25 and syntaxin 1 detection, given noise, image distortion, and so on, we calculated the correlation coefficient between images of artificial liposomes containing phosphatidylethanolamine-Oregon Green and Alexa594-synaptobrevin 2. This gave a correlation coefficient of 0.78 ± 0.02 (n = 4).
Electrophysiology and Electrochemistry
The cells were used 2–4 d after plating; 6–10 h after virus infection, whole-cell patch-clamp capacitance, amperometry, flash photolysis of caged calcium, and intracellular Ca2+ measurements were performed as described previously (Nagy et al., 2002). During the recordings, the mouse chromaffin cells were maintained in extracellular solution (145 mM NaCl, 2.8 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 2 mg/ml d-glucose, pH 7.20, 305 mOsM/kg). Capacitance and amperometric measurements were carried out in parallel to ensure that the fusion of catecholamine-containing vesicles was being monitored. Capacitance traces were fitted with a sum of exponential functions to separate pool sizes (noted as amplitudes of the exponentials) from the kinetics of fusion triggering (noted as time constants of the exponentials), as described previously (Nagy et al., 2002, 2004). Data are given as mean ± SEM, and the nonparametric Mann–Whitney U-test or the Kruskal–Wallis multiple comparison test were used to test statistical difference, which is indicated by *p < 0.05, **p < 0.01, and ***p < 0.001.
Protein Purification
The basic SNARE expression constructs in a pET28a vector (residues 1–206), the syntaxin 1A SNARE motif (residues 180–262), and synaptobrevin 2 (residues 1–96) have been described previously (Fasshauer and Margittai, 2004). For expression of SNAP-25b, the full-length gene was cloned into the pET28a vector. The recombinant SNARE proteins were isolated from Escherichia coli and purified by Ni2+-nitrilotriacetic acid affinity chromatography followed by ion exchange chromatography on an Äkta system (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) essentially as described previously (Fasshauer and Margittai, 2004). All ternary SNARE complexes were assembled overnight and purified using a Mono Q-column (GE Healthcare). Protein concentration was determined by absorption at 280 nm.
Circular Dichroism (CD) Spectroscopy Measurements
CD measurements were performed using a model J-720 instrument (Jasco, Tokyo, Japan). All experiments were carried out in 20 mM sodium phosphate, 2 M guanidine-HCl, pH 7.4, in the presence of 100 mM NaCl and 1 mM DTT. For thermal denaturation experiments, ∼10 μM purified ternary SNARE complexes were heated in Hellma quartz cuvettes with a pathlength of 0.1 cm. The ellipticity at 222 nm was recorded between 25 and 95°C at a temperature increment of 30°C/h.
Molecular Dynamics Simulations
Molecular dynamics simulations of the SNAP-25a-containing SNARE complex were started from the x-ray structure of the neuronal SNARE complex (chains A–D from PDB code: 1SFC; Sutton et al., 1998). The simulation system contained 3002 protein atoms, 27,785 SPC water molecules (Berendsen et al., 1981), and 15 sodium ions, resulting in a system size of 86,357 atoms. The “mutated structure” was generated using the molecular modeling suite WHATIF (Vriend, 1990). The MUTATE routine was used to replace H66 with Q and Q69 with K in the x-ray structure (1SFC), before the polar hydrogens were attached with the ADDHYD routine. Due to these mutations, the simulation system of the mutated structure contains 3003 protein atoms and 27,782 SPC water molecules. Fourteen sodium ions were added to keep the simulation system neutral.
Molecular dynamics simulations were carried out using the GROMACS simulation package (Lindahl et al., 2001). We used the GOMACS force field, which is the GROMOS 87 force field (van Gunsteren and Berendsen, 1987) with slight modifications (Van Buuren et al., 1993) and explicit hydrogens on the aromatic side chains. To allow an integration time step of 2 ns, covalent bond lengths were constrained using Lincs and Settle (Miyamoto and Kollman, 1992; Hess et al., 1997). Electrostatic interactions were calculated explicitly at a distance smaller than 1.0 nm. Long-range electrostatic interactions were calculated by particle-mesh Ewald summation (Darden et al., 1993). The protein and the solvent were coupled separately to an external temperature bath of 300 K (Berendsen et al., 1984) with a coupling constant of tau = 0.1 ps. The pressure was kept constant at 1 bar by weak coupling (tau = 1.0 ps) to a pressure bath (Berendsen et al., 1984).
RESULTS
Plasma Membrane Localization of SNAP-25 Isoforms
SNAP-25 is targeted to the plasma membrane by a stretch of 36 amino acids (85–120) localized in the linker region between the two SNARE domains (Gonzalo et al., 1999). All four linker-cysteines are necessary for proper membrane localization because single-cysteine substitutions suffice to greatly diminish palmitoylation and membrane association (Veit et al., 1996; Lane and Liu, 1997). Because the position of one of these clustered cysteine residues differs between SNAP-25a and SNAP-25b, it has been suggested that the arrangement of potential fatty acylation sites may contribute to targeting the two SNAP-25 isoforms to different sites on the plasma membrane (Bark and Wilson, 1994; Bark et al., 1995). In general, targeting of SNARE proteins to distinct regions (e.g., lipid rafts) in the plasma membrane has been suggested to spatially control exocytosis (Chamberlain et al., 2001; Salaün et al., 2005). Thus, it is possible that the functional difference between SNAP-25a and SNAP-25b might simply reflect an altered targeting efficiency to exocytotic sites.
We addressed these questions by isolating plasma membrane sheets from SNAP-25 null cells overexpressing either SNAP-25 isoform using a SFV construct (see Materials and Methods). This technique allows examination of membrane proteins in their natural microenvironment defined by local lipid composition and bound proteins (Lang, 2003). Previously, by Western blot analysis we showed that the SNAP-25a/b SFV constructs express similar amounts of protein in bovine chromaffin cells (Sørensen et al., 2003); however, the preparation of mouse chromaffin cells does not yield enough protein for Western blot analysis. Thus, another advantage of the membrane sheet technique is that it allows the comparison of the amount of membrane-targeted protein in embryonic mouse chromaffin cells. Membrane-associated SNAP-25 was visualized by immunostaining, together with syntaxin 1, to assay the distribution and amount of the interacting SNARE protein that might be different upon ablation or overexpression of SNAP-25. In Figure 2, we present images of membrane sheets from wild-type (+/+) cells and knock-out cells (-/-) overexpressing either isoform. On overexpression, the amount of SNAP-25 increased to beyond wild-type levels (see below); however, to preserve spatial information the images C and D in Figure 2 were scaled independently of A and B (for quantitation, see Figure 3). In wild-type mouse chromaffin cells, both SNAP-25 and syntaxin 1 were clustered (Figure 2A), as shown previously in PC12 cells (Lang et al., 2001). In the plasma membrane of SNAP-25 null cells syntaxin 1 still formed clusters (Figure 2B), indicating that syntaxin 1 clusters do not depend on direct or indirect interactions between syntaxin 1 and SNAP-25. Importantly, in cells from SNAP-25 null mice overexpressing SNAP-25a (Figure 2C) or SNAP-25b (Figure 2D), a spotty pattern of SNAP-25 was observed, indistinguishable from that in wild-type cells. In overexpressing cells, this pattern was present on top of a stronger background level, which is not obvious at the image scaling chosen for presentation in Figure 2 (see Discussion). We compared the absolute fluorescence intensities per unit membrane area for both SNAP-25 isoforms and syntaxin 1 (Figure 3, A and B). The SNAP-25-specific signal was reduced by 33% in heterozygous (Snap-25 +/-) cells compared with wild-type (Snap-25 +/+) cells (p < 0.05 Tukey– Kramer multiple comparison test), showing a gene-dose effect. In null cells overexpressing either SNAP-25a or SNAP-25b, the immunoreactivity was much higher than in wild-type cells but not significantly different between isoforms (SNAP-25a, 13.7 ± 1.0-fold overexpression; SNAP-25b, 14.7 ± 2.2-fold overexpression, p > 0.05, Tukey–Kramer multiple comparison test). The levels of syntaxin 1 immunofluorescence were not strongly affected by ablation or overexpression of SNAP-25 isoforms (Figure 3B; p = 0.0393, ANOVA, posttests showed that the difference between heterozygotes and knockouts was just significant). These data show no difference in targeting efficiency between the SNAP-25 isoforms. In addition, the 14- to 15-fold increase compared with wild-type levels excludes that limited availability at the plasma membrane of either isoform causes the difference in secretion.
Figure 2.

SNAP-25 and syntaxin 1 on membrane sheets from embryonic mouse chromaffin cells. Plasma membrane sheets were generated from SNAP-25 +/+ chromaffin cells (A), SNAP-25 null chromaffin cells (B), SNAP-25 null chromaffin cells expressing SNAP-25a (C), and SNAP-25 null chromaffin cells expressing SNAP-25b (D). Sheets were immediately fixed with paraformaldehyde and immunostained for SNAP-25 and syntaxin 1. The samples were imaged in three channels: membranes were identified in the presence of TMA-DPH dye in the blue (not shown), SNAP-25 signal was detected in the red and syntaxin 1 in the long red channel. Overlay from SNAP-25 and syntaxin 1 indicates partial colocalization of two proteins. Note that the SNAP-25 images in A, C, and D were scaled independently of each other to preserve spatial information; however, the absolute immunofluorescence intensities in C and D were much higher than in A (see Figure 3). The colocalization was quantified using correlation analysis (see text).
Figure 3.

Quantification of SNAP-25 isoforms and syntaxin 1 in the plasma membrane of embryonic mouse chromaffin cells. The immunofluorescence of membrane sheets from 12 SNAP-25 null animals, 12 SNAP-25 WT(+/+) animals, 9 SNAP-25 heterozygous (+/-) animals, 9 SNAP-25 null animals expressing SNAP-25a and 7 SNAP-25 null animals expressing SNAP-25b were analyzed and plotted. A minimum of 10 sheets per animal were analyzed. The values indicate relative abundance (normalized to the mean of SNAP-25+/+ animals from the same litter) ± SEM of SNAP-25 (A) and syntaxin 1 (B) protein.
However, the differences might occur at the level of microdomain organization. When the colocalization of SNAP-25 and syntaxin 1 clusters was studied, findings ranged from only partial overlap (Lang et al., 2001; Ohara-Imaizumi et al., 2004) to nearly perfect colocalization (Rickman et al., 2004), but all studies suggest that the sites of overlap represent fusion sites. To assay for a putative difference in the sorting of the SNAP-25 isoforms into the syntaxin 1-cluster, we quantified the colocalization by calculating the correlation coefficient of the two images (see Materials and Methods). First, the degree of overlap was characterized in homozygous wild-type cells, resulting in a correlation coefficient of 0.25 ± 0.02 (n = 32 membranes analyzed), indicating that a significant fraction of SNAP-25 is within or close to syntaxin 1 clusters. The correlation coefficients were similar when heterozygous and SNAP-25 null cells overexpressing SNAP-25a or SNAP-25b were analyzed (heterozygous cells, 0.26 ± 0.02, n = 34; null cells overexpressing SNAP-25a, 0.25 ± 0.03, n = 24; and null cells overexpressing SNAP-25b, 0.25 ± 0.02, n = 31).
Finally, we conclude that the secretory difference between SNAP-25 isoforms cannot be explained by differential targeting efficiency to the plasma membrane or a change in microdomain organization as assayed by diffraction-limited light microscopy.
Identification of the Critical Amino Acid Substitutions Defining Neurosecretion Properties of SNAP-25a and SNAP-25b
To identify the amino acid substitutions responsible for the physiological differences in secretion mediated by SNAP-25 isoforms, we generated chimeric constructs and assayed their ability to rescue exocytosis in SNAP-25 null chromaffin cells. Secretion was assayed by simultaneous whole cell patch-clamp recordings to measure capacitance increase resulting from vesicular fusion (Figure 4A, middle) and amperometry (Figure 4A, bottom), which detects the release of oxidizable vesicle contents (adrenaline and noradrenaline). Viral-infected SNAP-25-expressing cells were recognized by the coexpression of green fluorescent protein (see Materials and Methods), and exocytosis was triggered by flash photolysis of caged Ca2+ (Figure 4A, at arrow). Because different preparations of chromaffin cells vary in secretory competence, we adopted the strategy of dividing the chromaffin cells obtained from each SNAP-25 null embryo between several coverslips and performed rescue experiments with both mutated and control constructs on cells from the same animal on each experimental day. Only experiments done in parallel were compared statistically. Because we did not measure a difference in membrane targeting between the two isoforms, we reasoned that most likely the critical amino acid substitutions would be present in the SNARE domain, rather than in the linker. Hence, we first assayed the three nonconservative substitutions (from SNAP-25a to SNAP-25b; Figure 1) N65D, H66Q, and Q69K in the N-terminal SNARE domain, which could affect properties and function of the SNARE core complex.
Figure 4.

A group of three amino acids is sufficient to switch SNAP-25a to SNAP-25b phenotype. (A) Mean [Ca2+]i (top, error bars represent SEM), capacitance change (middle), and amperometric current (bottom) were measured simultaneously after a step-like elevation of [Ca2+]i caused by flash photolysis of caged Ca2+ (flash at arrow). The traces are averages of many experiments, so the individual fusion events (spikes) are not recognizable in the amperometric signal. Left, secretion after the first stimulation; right, secretion in response to the second stimulation (left). Shown are means of 38 SNAP-25 null cells expressing SNAP25b cells (black) and 38 cells overexpressing SNAP-25aN65D/H66Q/Q69K for 6–8 h (gray). There was no difference in preflash [Ca2+]i between two groups (our unpublished data). The data for SNAP-25a overexpression were taken from another series of experiments and are shown here for comparison. Secretion from cells transfected with SNAP-25aN65D/H66Q/Q69K and SNAP-25b was similar. (B) Amplitudes of exponential fits to individual responses. The amplitudes (mean ± SEM) of the fast and the slow burst component and the rate of sustained component were similar in both stimuli (dark bars, SNAP-25b; gray bars, SNAP-25aN65D/H66Q/Q69K).
As reported previously, expression of SNAP-25b in SNAP-25-deficient chromaffin cells resulted in a twofold increase in the size of the exocytotic burst, as determined by the secretion 0–1 s after flash photolysis of caged Ca2+, compared with that supported by SNAP-25a overexpression (Figure 4A; note that the SNAP-25a trace is taken from a separate experimental series, and hence the quantification of these experiments is not presented in Figure 4B; also see Sørensen et al., 2003). The exocytotic burst phase represents the fusion of the two primed vesicle pools (the readily releasable and the slowly releasable pools, denoted RRP and SRP, respectively), whereas the sustained phase represents slower priming of new vesicles, followed by fusion as long as the intracellular calcium concentration ([Ca2+]i) stays high. By fitting of a sum of exponential functions to the capacitance trace, we can separate the size of the two releasable pools from their fusion time constants (Nagy et al., 2002, 2004). The time constants for fusion from the two releasable pools were not different between SNAP-25a- and SNAP-25b-expressing cells (not shown; Sørensen et al., 2003). In addition, the rate of sustained secretion was independent of the isoform of SNAP-25 expressed (1–5 s after Ca2+ release; Figure 4A and Sørensen et al., 2003). However, it should be noted that by comparison with SNAP-25 null cells, it was shown that SNAP-25 expression is necessary for both the normal fusion rate constants and the rate of the sustained component (Sørensen et al., 2003), indicating that SNAP-25 participates in all phases of release from chromaffin cells. However, the difference between SNAP-25 isoform is only evident on the size of the fast and slow exocytotic burst component, indicating that they only differ in their regulation of the size of the releasable vesicle pools.
Expression of a SNAP-25a variant in which the three amino acids in positions 65, 66, and 69 (Figure 1) were switched to SNAP-25b residues—N65D/H66Q/Q69K—resulted in a SNAP-25b-like secretory phenotype (Figure 4A). Kinetic analysis showed that the size of both burst components and the rate of the sustained component were indistinguishable from SNAP-25b overexpression (Figure 4B). In addition, providing a second flash stimulation ∼100 s after the initial stimulus resulted in almost the same level of secretion as were recorded in SNAP-25b-expressing cells (Figure 4, A and B). The small reduction in secretion driven by the N65D/H66Q/Q69K construct during the second flash stimulation was not statistically significant (Figure 4B). This experiment tested the ability to refill depleted vesicle pools and confirmed that this component of secretion was also indistinguishable between cells expressing the triple SNAP-25a mutation and SNAP-25b. Thus, the three amino acid substitutions N65D/H66Q/Q69K were sufficient to provide SNAP-25a with the properties of SNAP-25b in mediating secretion from these chromaffin cells.
We next tested the effect of single mutations N65D, Q69K, and H66Q imposed on the SNAP-25a protein sequence background (Figure 5, A–D). The N65D mutation resulted in a level of secretion that was indistinguishable from SNAP-25a (Figure 5, A and B, red traces; compare with the trace for SNAP-25a in Figure 4). In contrast, the Q69K mutation resulted in an intermediate level of secretion, with an exocytotic burst that was significantly larger than in SNAP-25a N65D but smaller than for the N65D/H66Q/Q69K triple mutation (Figure 5, A and B). Kinetic analysis confirmed that the Q69K had intermediate sized RRP and SRP (Figure 5B). Note, that the sustained rate of release was not changed by any of these mutations (Figure 5B, bottom) –or by any of those that were studied in the following experiments. This is consistent with the difference between the SNAP-25a and SNAP-25b phenotype, where the burst size was changed, but the sustained rate was invariant (Figure 4; Sørensen et al., 2003). The H66Q single mutation resulted in a SNAP-25a-like phenotype (Figure 5C, compare with trace for SNAP-25a in Figure 4). Therefore, the Q69K substitution was necessary for the stronger secretory phenotype produced by SNAP-25b, but in itself seemed not to be sufficient. Finally, we tested the effect of combining the Q69K with the H66Q substitution on secretion. Expression of the double substitution resulted in sizes of the fast and slow burst components that were indistinguishable from SNAP-25b (Figure 5, C and D). Together, these data indicate that the substitutions H66Q/Q69K define critical residues that distinguish the secretory properties attributed to SNAP-25 isoforms in chromaffin cells.
Figure 5.

H66Q/Q69K: the minimal mutation in SNAP-25a that gives SNAP-25b phenotype. (A) Response to the first stimulation in SNAP-25 null cells expressing SNAP25aN65D (red, 23 cells), SNAP25aQ69K (blue, 30 cells) and SNAP25aN65D/H66Q/Q69K (black, 40 cells). For explanation, see the legend to Figure 4. (B) Size of the burst (fast + slow burst) component and the rate of sustained secretion. The secretory phenotype of SNAP25aQ69K is intermediate between SNAP25aN65D and the triple mutation. No difference in the rate of the sustained component was detected. (C) Response to the first flash stimulation in SNAP-25 null cells expressing SNAP25aH66Q/Q69K (green, 30 cells), SNAP25aH66Q (gray, 16 cells) and SNAP25b (black, 23 cells). (D) The amplitudes of the burst components and the rate of sustained release in cells expressing SNAP25aH66Q/Q69K were indistinguishable from cells expressing SNAP-25b.
To confirm that these residues are sufficient to explain the difference in secretory phenotype between isoforms, we constructed the complementary substitution mutations in SNAP-25b. As shown in Figure 6, secretion rescued by the mutation Q66H/K69Q in SNAP-25b was almost indistinguishable from that obtained with SNAP-25a (Figure 6A, gray and black traces, respectively). Kinetic analysis confirmed that there was no statistical significant difference in the size of either burst component between cells expressing SNAP-25a and SNAP-25b Q66H/K69Q (Figure 6C, dark and gray bars). In fact, the single mutation K69Q in SNAP-25b was enough to secure the reversion to the SNAP-25a phenotype, as judged by the overall secretion (Figure 6B, dark and gray traces) and kinetic analysis of the burst sizes (Figure 6, B and C, dark and white). The fact that the Q69K mutation in the SNAP-25a background leads to an intermediate phenotype (Figure 5A), whereas the SNAP-25b K69Q mutation is indistinguishable from SNAP-25a, indicates that the two positions 66 and 69 have nonadditive effects on secretion. Given the complexity of protein–protein interaction such effects are not surprising.
Figure 6.

Q66H/K69Q mutation in SNAP-25b results in SNAP-25a-like phenotype. (A) The opposite experiment to the one shown in Figure 5. Here, we mutated the positions 66 and 69 in SNAP-25b into the residues found in SNAP-25a. Response to a first flash stimulation in SNAP-25 null cells expressing SNAP25bQ66H/K69Q (gray, 26 cells) in comparison with SNAP25a (black, 26 cells). The data for SNAP-25b overexpression were taken from another series of experiments and are shown here for comparison. (B) Response to a first flash stimulation in SNAP-25 null cells expressing SNAP25bK69Q (gray, 26 cells) in comparison with SNAP25a (black, 26 cells). Again, data for SNAP-25b overexpression were taken from another experimental series (C). Size of fast and slow burst and rate of the sustained component. Dark bars, SNAP-25a; gray bars, SNAP-25b Q66H/K69Q; and white bars, SNAP-25b K69Q. No significant changes were found between these three constructs (e.g., for the sustained component; p = 0.13, Kruskal–Wallis test).
In conclusion, by systematically swapping nonconservative amino acid differences between the isoforms, we find that the residue substitutions Q66H and K69Q, in SNAP-25a and SNAP-25b, respectively, are necessary and sufficient to account for the difference in secretory phenotype supported by these two isoforms in mouse chromaffin cells.
A Neighboring Residue, Asp-70, Defines a Hydrophilic Stretch of Amino Acids Regulating Secretion
One possible explanation for the difference in secretory phenotype between SNAP-25 isoforms is that an accessory factor binds to the surface of the helical coiled-coil structure of the SNARE complex around positions 66 and 69 (see Discussion). If this is the case, then this factor may bind to a longer stretch than just these two residues. Alternatively, the difference in secretory phenotype might be explained by an interaction of the residues at positions 66 and 69 with neighboring residues in the SNARE complex, leading to different properties of the SNAP-25a- and SNAP-25b-containing SNARE core complexes. In both cases, structural neighbors of the amino acids at positions 66 and 69 might participate in the difference in secretory phenotype.
To identify other amino acids that could be involved, we mutated the neighboring aspartate D70, which is present in both SNAP-25a and SNAP-25b (Figure 1; also see Figure 9), and measured secretion after rescue of null cells. Mutation of D70 to alanine (D70A) in SNAP-25a did not compromise secretion, as shown in the overall secretion and following kinetic analysis of secretory components (Figure 7A). Strikingly, however, expression of the D70A SNAP-25b mutant resulted in a clear decrease in secretion compared with cells expressing native SNAP-25b (Figure 7B). The resulting secretion seemed indistinguishable from the level of vesicular fusion supported by SNAP-25a (Figure 7B). Moreover, we found no significant differences between the burst sizes between SNAP-25a and SNAP-25b D70A (Figure 7B, bottom). These data suggest that the neighboring amino acid D70 in SNAP-25 cooperates with K69 and Q66 in SNAP-25b to induce a stronger secretory phenotype for the SNAP-25b isoform.
Figure 9.

Structural arrangement of the SNARE complex around the SNAP-25 residues 66 and 69. A. Structure of the SNAP-25a-containing ternary SNARE complex (Sutton et al., 1998) after MD simulation for 20 ns (as explained in the text). (B) Structure of the ternary SNARE complex after introduction of Q66 and K69 in the SNAP-25a-containing ternary SNARE complex and MD simulation for 20 ns. Note that the structural arrangements shown here are “snapshots” taken from a longer simulation and do not represent a preferred or typical arrangement of the side chains.
Figure 7.

A neighboring aspartate (D70) present in both isoforms is necessary for the SNAP-25b-like phenotype. (A) Response to a first flash stimulation in SNAP-25 null cells expressing SNAP25aD70A (gray, 22 cells) in comparison with SNAP25a (black, 21 cells). Bottom, size of fast and slow burst and rate of sustained component. (B) Response to a first flash stimulation in SNAP-25 null cells expressing SNAP25bD70A (gray, 35 cells) in comparison with SNAP25a (black, 41 cells). The data for SNAP-25b overexpression were taken from another series of experiments and are shown here for comparison. Bottom, sizes of fast and slow burst of release, and rate of sustained component. No significant changes were found between SNAP-25a and SNAP-25bD70A, showing that the aspartate at position 70 is necessary for the SNAP-25b secretory phenotype but not for the SNAP-25a-like secretory phenotype.
Biochemical Properties of the Two Splice Variants of SNAP-25
We next investigated whether different biochemical properties of the SNARE complex formed with either SNAP-25 splice variant could account for the different secretory phenotypes. The pathway of SNARE complex formation in vitro involves a transient interaction between the N-terminal ends of the SNARE domains of syntaxin 1 and SNAP-25, which results in the formation of a 1:1 syntaxin 1:SNAP-25 precomplex (Fasshauer and Margittai, 2004) and serves as an acceptor for synaptobrevin. Therefore, mutations or changes in the N-terminal ends of the SNAP-25 SNARE domains can lead to slowdown of assembly, whereas C-terminal mutations do not (Fasshauer and Margittai, 2004; our unpublished data). The amino acid substitutions between the SNAP-25a and SNAP-25b isoforms are placed in the C-terminal end of the first SNARE motif (Figure 1); therefore, no difference in in vitro assembly rates can be expected.
The stability of the SNARE complex can be assayed by CD spectroscopy, which makes use of the fact that the α-helical structure of the SNARE domains is induced during complex formation, whereas uncomplexed SNARE domains are unstructured (Fasshauer et al., 1997). To assess the consequences of the SNAP-25 isoform for the stability of the SNARE complex, we performed thermal denaturation experiments on in vitro assembled ternary SNARE core complexes. All analyzed complexes unfolded in a single, cooperative reaction (Figure 8). Interestingly, SNAP-25b-containing complexes melted at a slightly higher temperature (4–5°C) compared with those constituted with SNAP-25a, indicating that SNAP-25b increases the stability of the ternary SNARE complex. We next tested the stability of the SNAP-25a H66Q/Q69K double substitution that leads to a SNAP-25b-like secretory phenotype (see above). As shown in Figure 8, replacement of these residues in SNAP-25a resulted in a complex with the same stability as SNAP-25b, indicating that both the difference in stability and secretory phenotype can be attributed to these two amino acids. In contrast, complexes formed with SNAP-25a containing the Q69K single mutation did not affect the melting temperature, suggesting that this substitution cannot in isolation contribute to the differential stability of the isoforms. Finally, we tested the SNAP-25b mutation D70A. Remarkably, complexes containing SNAP-25b with the D70A substitution retained the higher melting temperature and thus stability characteristic of SNAP-25b, even though this single substitution was sufficient to provide a SNAP-25a-like secretory phenotype to SNAP-25b (Figure 7). These experiments suggest that despite a slight difference in stability of complexes containing either SNAP-25b or SNAP-25a, this change is probably not causal for the difference in secretory phenotype.
Figure 8.

The thermal stability of the ternary SNARE complex is slightly higher in the presence of SNAP-25b. Thermal melts of purified ternary SNARE complexes containing different SNAP-25 variants or point mutations. Thermal stability of ternary SNARE complexes formed overnight was assayed by CD spectroscopy in the presence of 2 M guanidine hydrochloride. Complexes formed with SNAP-25b unfolded at a slightly higher temperature (4–5°C) than SNAP-25a-containing complexes. The double mutation H66Q/Q69K in SNAP-25a was sufficient to get a complex of higher stability, whereas the single mutation Q69K did not change stability. The D70A mutation in SNAP-25b led to a complex with SNAP-25b-like stability, but a SNAP-25a-like secretory phenotype (Figure 7B). This finding shows that the difference in stability is not causing the difference in secretory phenotype.
Structural Implications of the Replacement of Two Amino Acids in the SNARE Domains
To identify potential molecular interactions within the SNARE core complex involving the side chains of the two critical amino acid positions, we inspected the crystal structure of the SNARE core complex. The original crystal structure (Sutton et al., 1998) and a later refined structure (Ernst and Brunger, 2003) both used the SNAP-25a isoform (note that in those publications this was denoted as SNAP-25b). By replacing H66 with Q and Q69 with K, we created a model structure to explore the possibilities for interaction with neighboring residues (Figure 9). Next, we performed molecular dynamics (MD) simulations of the wild-type (WT) crystal structure and the mutated structure. In both cases, the crystal structure was solvated in a box of water molecules (Materials and methods) and simulated for 20 ns. The results show that in the WT (SNAP-25a-containing) crystal structure the side chains of the residues H66 and Q69 displayed more interactions (H-bonds) with neighboring residues than when mutated to the (SNAP-25b) residues Q66 and K69. In the SNAP-25a structure, the two nitrogen atoms (Nδ1 and Nε2) in the imidazole ring of H66 participated in H-bonds 85 and 46% of the time, respectively, whereas the Q66 was hydrogen bound only 36% of the time. Similarly, at position 69, the Q residue of SNAP-25a participated in H-bonds 37% of the time, whereas K69 showed hydrogen bonds only 29% of the time. These simulations show that the substitution of histidine to glutamine at position 66 and glutamine to lysine at position 69 leads to decreased interaction of the side chains with neighboring amino acids within the SNARE complex. Together with the experimental findings, these computer simulations are consistent with the idea that the difference in secretory phenotype induced by the two amino acid substitutions is most likely caused by differential interaction with accessory factors on the surface of the coiled-coil structure of the SNARE complex.
DISCUSSION
Although there is considerable evidence that SNARE protein complexes participate in nearly all intracellular membrane fusion events, different opinions exist as to which step in the multiple-step process of membrane fusion is catalyzed by SNAREs. Similarly, the question of the physiological importance of different SNARE isoforms and how they may participate either in different membrane trafficking pathways or in the same vesicle fusion events has yet to be resolved. We have investigated these questions by comparing the two splice variants of SNAP-25, SNAP-25a and SNAP-25b, which can both support secretion when expressed in SNAP-25 null mutant chromaffin cells. In spite of only differing by nine amino acid substitutions, expression of the two splice variants results in a two- to threefold difference in the size of the exocytotic burst, indicating that they have a different ability to support the primed pool of vesicles. Here, we identify the two key amino acid residues that lead to the physiological differences in secretion. Biochemical and computer-assisted simulation analysis lead us to propose that the function of these residues, whose side chains face the external surface of the coiled-coil SNARE structure, is to interact with accessory proteins that may influence the stabilization or formation of the primed vesicle state.
Results from the plasma membrane sheet assay suggest that both SNAP-25 isoforms are targeted to the plasma membrane with the same efficiency, are available in excess, and colocalize to the same extent with syntaxin 1-clusters. Concerning the latter conclusion, it should be noted that the antibody used for SNAP-25 detection is less sensitive for SNAP-25 in a complex with syntaxin 1 (Lang et al., 2002), leading to a reduction of the signal-to-noise ratio for the detection of this pool compared with free SNAP-25. Nevertheless, the presence of significant positive correlations between the SNAP-25 and syntaxin 1 signals show that the proteins are targeted to the same subdomains in the plasma membrane. It might be surprising that a 14- to 15-fold increase of SNAP-25 leaves the correlation coefficient with syntaxin 1 unchanged compared with wild-type cells. However, it should be noted that correlation analysis was done on mean-subtracted images (see Materials and Methods). Therefore, the finding is consistent with two scenarios. First, independently of the SNAP-25 level always the same fraction of SNAP-25 ends up in clusters, or second, the mechanism that causes clustering has a limited capacity, and after saturation additional SNAP-25 is uniformly distributed in the membrane. Linescan analysis of the membrane sheets showed that after overexpression, the amplitude of the spotty signals is increased but that the background fluorescence level is increased even more (our unpublished data). Therefore, we suggest that the mechanism that causes clustering has a limited capacity, which, however, is not exhausted in control cells.
Although evidence has suggested that in some other cells coexpression with syntaxin 1 is required to retain SNAP-25 on intracellular membranes (Rowe et al., 1999; Vogel et al., 2000; Washbourne et al., 2001), more recent studies have supported the view that targeting of SNAP-25 to the plasma membrane in neuronal cells does not require syntaxin 1 (Loranger and Linder, 2002). This model is in agreement with our finding that membrane associated SNAP-25 can be increased from nothing to >10-fold wild-type levels within 8 h without changing the plasma membrane syntaxin 1 level. The observation that the positioning of the four cysteines is rearranged between the two SNAP-25 isoforms had been suggested to provide a mechanism that might contribute to differences in membrane localization or targeting (Bark and Wilson, 1994). Our data show no discernible difference between the patterns of membrane-associated clusters formed by the two SNAP-25 isoforms. Furthermore, our search for the functionally relevant substitutions showed that the relocation of the cysteine does not contribute to the difference in secretory phenotype between SNAP-25 isoforms in chromaffin cells. Nevertheless, previous studies using tagged proteins expressed in nerve growth factor-differentiated PC12 cells indicated that SNAP-25b localized more to varicosities and terminals, whereas SNAP-25a showed a more diffuse localization (Bark et al., 1995). Thus, this question should be reexamined in polarized neurons.
Our data demonstrate that the major functional distinction between the two SNAP-25 isoforms in neurosecretion is their ability to support the pool of releasable vesicles. By systematic mutagenesis, we identified the two nonconservative substitutions in the N-terminal SNARE domain (from SNAP-25a to SNAP-25b)—H66Q and Q69K—as being both necessary and sufficient for the functional difference between the isoforms. Thermal denaturation studies showed that SNAP-25b-containing complexes were somewhat (4–5°C) more stable than the SNAP-25a-containing complexes, which were used for structural studies (Sutton et al., 1998) and that the difference in stability could be attributed to the same two amino acid substitutions. Moreover, further experiments demonstrated that a neighboring conserved aspartate (D70) also is required to obtain the stronger SNAP-25b secretory phenotype. Mutation of D70 to alanine in SNAP-25b did not change the stability of the SNARE complex, even though the ability to support secretion was reduced to the level of SNAP-25a. Therefore, we conclude that the change in complex stability is most likely not causal for the functional difference between SNAP-25a and SNAP-25b.
The idea that the two substitutions H66Q and Q69K do not regulate secretion through a change in the property of the SNARE complex itself was corroborated by molecular dynamics simulations, which showed that Q66 and K69 participate less frequently in intracomplex interactions than the SNAP-25a residues H66 and Q69. It should be noted that this result is not in conflict with the higher thermostability of the SNAP-25b-containing complex measured biochemically. The thermostability of the SNARE complex does not depend strongly on hydrogen-bonding on the surface of the complex but more on the hydrophobic interactions in the interior of the complex, on the tendency of the SNARE domains to adopt different secondary structures, on the stability of partly unfolded states, and so on, which could all be changed in subtle ways by the H66Q and Q69K substitutions.
We thus suggest that the side chains of these two polar amino acids are available for interaction with accessory factor(s). Although the identity of such putative binding partner(s) is unknown, our findings suggest that the factor would bind to a stretch of amino acids at the N terminus of the coiled-coil SNARE structure. Two scenarios can be suggested: either this factor binds to only one of the two complexes (SNAP-25a or SNAP-25b containing), or it binds to both complexes, but with different affinities, which in turn regulates the size of the releasable vesicle pools. Because usually protein–protein interactions involve binding to multiple residues, but only two substitutions are necessary to explain the difference between isoforms, the latter scenario seems most likely. Interestingly, complexin proteins bind to the groove between syntaxin 1 and synaptobrevin 2 (Chen et al., 2002), whereas Q66, K69 and D70 face the C-terminal SNAP-25 helix and syntaxin 1. Synaptotagmin 1 has been shown to bind to charged amino acids in the C-terminal end of the SNARE complex; however, the residues responsible for binding are found in the SN2 domain of SNAP-25 (refer to Figure 1; Zhang et al., 2002). Furthermore, increasing the amount of synaptotagmin 1 in mouse chromaffin cells leads to a secretory phenotype, which is not consistent with the difference between SNAP-25 isoforms (our unpublished data). Finally, the accessory factor proposed here might, in fact, be the phospholipids themselves. Such an interaction may cause the SNARE complex to assume a more flat position on the plasma membrane, which could stabilize the primed vesicle state, but this notion remains speculative. The identification of the polar stretch of amino acids Q66, K69, and D70 should make it possible to identify the binding partner(s) by biochemical interaction experiments.
The effect of incorporating different SNAP-25 isoforms into the SNARE core complex is to regulate the size of the exocytotic burst, which represents the number of release-ready vesicles. Importantly, this is achieved without changing the rate of release from the releasable pools (either the slowly releasable pool or the readily releasable pool; our unpublished data; Sørensen et al., 2003). This adds to our previous findings that alterations of protein kinase C or protein kinase A phosphorylation sites in SNAP-25 also modify upstream priming reactions, without affecting the fusion rate from the primed vesicle pools (Nagy et al., 2002, 2004). Together, these results establish a function of SNAREs in regulating the priming reaction, which confers release competence to the vesicles. However, other studies have involved the SNARE complex in exocytosis triggering and fusion pore formation (Gil et al., 2002, Sørensen et al., 2003; Han et al., 2004; Borisovska et al., 2005; our unpublished data). Collectively, these observations suggest that although the C-terminal end of the coiled-coil domain of the SNARE complex is involved in both vesicle priming and fusion triggering, these functions can be separately encoded within this small subdomain with certain residues being involved exclusively in one or the other reaction. Specifically, residues facing the surface of the SNARE bundle may regulate priming by binding to accessory factors, whereas residues facing the inside of the bundle will affect the assembly speed of the ternary complex and therefore may regulate the fusion rate, even though this has so far not been shown. The first of these steps (defining vesicle priming) may correspond to assembly of a precomplex between syntaxin 1 and SNAP-25, which in a later step serves as an acceptor for synaptobrevin 2 (An and Almers, 2004; Fasshauer and Margittai, 2004).
Thus, the SNAREs are not only required for the last steps in exocytosis (exocytosis triggering and fusion pore formation) but also for an upstream step (vesicle priming), and they can regulate the upstream reaction without compromising the fidelity and speed of the downstream fusion step. This arrangement allows regulation of the extent of exocytosis without compromising the exocytotic event itself, which is exactly what is required for the neuronal fusion apparatus.
Acknowledgments
We are grateful to Erwin Neher for ongoing discussions and for commenting on the manuscript. We thank Ina Herfort and Dirk Reuter for expert technical assistance. This work was supported Deutsche Forschungsgemeinschaft Grants SFB523/TP4, SO 708/1-1 (to J.B.S.) and GRK521 (to G. N.) and by National Institutes of Health Grant MH-48989 (to M.C.W.). I. M. is a Ph.D. student of the International Ph.D./M.D.-Ph.D. Program in the Neurosciences of the International Max Planck Research School and holds a Boehringer-Ingelheim Fond Fellowship.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-07-0595) on September 29, 2005.
Abbreviations used: SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; SNAP-25, synaptosome-associated protein of 25 kDa.
References
- An, S. J., and Almers, W. (2004). Tracking SNARE complex formation in live endocrine cells. Science 306, 1042-1046. [DOI] [PubMed] [Google Scholar]
- Archer, B. T., Özcelik, T., Jahn, R., Francke, U., and Südhof, T. C. (1990). Structures and chromosomal localizations of two human genes encoding synaptobrevins 1 and 2. J. Biol. Chem. 265, 17267-17273. [PubMed] [Google Scholar]
- Ashery, U., Betz, A., Xu, T., Brose, N., and Rettig, J. (1999). An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J. Cell Biol. 78, 525-532. [DOI] [PubMed] [Google Scholar]
- Bark, C., Bellinger, F. P., Kaushal, A., Mathews, J. R., Partridge, L. D., and Wilson, M. C. (2004). Developmentally regulated switch in alternatively spliced SNAP-25 isoforms alters facilitation of synaptic transmission. J. Neurosci. 24, 8796-8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark, I. C., Hahn, K. M., Ryabinin, A. E., and Wilson, M. C. (1995). Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc. Natl. Acad. Sci. USA 92, 1510-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark, I. C., and Wilson, M. C. (1994). Human cDNA clones encoding two different isoforms of the nerve terminal protein SNAP-25. Gene 139, 291-292. [DOI] [PubMed] [Google Scholar]
- Bennett, M. K., Calakos, N., and Scheller, R. H. (1992). Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257, 255-259. [DOI] [PubMed] [Google Scholar]
- Berendsen, H.J.C., Postma, J. P., DiNola, A., and Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684-3690. [Google Scholar]
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W. F., and Hermans, J. (1981). Interaction models for water in relation to protein hydration. In: Intermolecular Forces, ed. B. Pullman, Dordrecht, The Netherlands: D. Reidel Publishing Co., 331-342.
- Borisovska, M., Zhao, Y., Tsytsyura, Y., Glyvuk, N., Takamori, S., Matti, U., Rettig, J., Südhof, T. C., and Bruns, D. (2005). v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J. 24, 2114-2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschert, U., O'Shaugnessy, C., Dickinson, R., Tessari, M., Bendotti, C., Catsicas, S., and Pich, E. M. (1996). Developmental and plasticity-related differential expression of two SNAP-25 isoforms in the rat brain. J. Comp. Neurol. 367, 177-193. [DOI] [PubMed] [Google Scholar]
- Chamberlain, L. H., Burgoyne, R. D., and Gould, G. W. (2001). SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proc. Natl. Acad. Sci. USA 98, 5619-5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X., Tomchick, D. R., Kovrigin, E., Arac, D., Machius, M., Südhof, T. C., and Rizo. J. (2002) Three-dimensional structure of the complexin/SNARE complex. Neuron 33, 397-409. [DOI] [PubMed] [Google Scholar]
- Darden, T., York, D., and Pedersen, L. (1993). Particle mesh Ewald - an N log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089-10092. [Google Scholar]
- Elferink, L. A., Trimble, W. S., and Scheller, R. H. (1989). Two vesicle-associated membrane protein genes are differentially expressed in the rat central nervous system. J. Biol. Chem. 264, 11061-11064. [PubMed] [Google Scholar]
- Ernst, J. A., and Brunger, A. T. (2003). High resolution structure, stability and synaptotagmin binding of a truncated neuronal SNARE complex. J. Biol. Chem. 278, 8630-8636. [DOI] [PubMed] [Google Scholar]
- Fasshauer, D., Bruns, D., Shen, B., Jahn, R., and Brünger, A. T. (1997). A structural change occurs upon binding of syntaxin to SNAP-25. J. Biol. Chem. 272, 4582-4590. [DOI] [PubMed] [Google Scholar]
- Fasshauer, D., and Margittai, M. (2004). A transient N-terminal interaction of SNAP-25 and syntaxin nucleates SNARE assembly. J. Biol. Chem. 279, 7613-7621. [DOI] [PubMed] [Google Scholar]
- Gil, A., Guitérrez, L. M., Carrasco-Serrano, C. Alonso, M. T., Viniegra, S., and Criado, M. (2002). Modifications in the C terminus of the synaptosome-associated protein of 25 kDa (SNAP-25) and in the complementary region of synaptobrevin affect the final steps of exocytosis. J. Biol. Chem. 277, 9904-9910. [DOI] [PubMed] [Google Scholar]
- Gonzalo, S., Greentree, W. K., and Linder, M. E. (1999). SNAP-25 is targeted to the plasma membrane through a novel membrane-binding domain. J. Biol. Chem. 274, 21313-21318. [DOI] [PubMed] [Google Scholar]
- Grant, N. J., Hepp, R., Krause, W., Aunis, D., Oehme, P., and Langley, K. (1999). Differential expression of SNAP-25 isoforms and SNAP-23 in the adrenal gland. J. Neurochem. 72, 363-372. [DOI] [PubMed] [Google Scholar]
- Guex, N., and Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-Pdb-Viewer: an environment for comparative protein modeling. Electrophoresis 18, 2714-2723. [DOI] [PubMed] [Google Scholar]
- Han, X., Wang, C. T. Bai, J. Chapman, E. R. and Jackson. M. B. (2004) Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304, 289-292. [DOI] [PubMed] [Google Scholar]
- Hanson, P. I., Roth, R., Morisaki, H., Jahn, R., and Heuser, J. E. (1997). Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell 90, 523-535. [DOI] [PubMed] [Google Scholar]
- Hess, B., Bekker, H., Berendsen, H.J.C., and Fraaije, J.G.E.M. (1997). LINCS: a linear constraint solver for molecular simulations. J. Comp. Chem. 18, 1463-1472. [Google Scholar]
- Jahn, R., Lang, T., and Sudhof, T. C. (2003). Membrane fusion. Cell 112, 519-533. [DOI] [PubMed] [Google Scholar]
- Lane, S. R., and Liu, Y. (1997). Characterization of the palmitoylation domain of SNAP-25. J. Neurochem. 69, 1864-1869. [DOI] [PubMed] [Google Scholar]
- Lang, T. (2003). Imaging SNAREs at work in “unroofed” cells–approaches that may be of general interest for functional studies on membrane proteins. Biochem. Soc. Trans. 31, 861-864. [DOI] [PubMed] [Google Scholar]
- Lang, T., Bruns, D., Wenzel, D., Riedel, D., Holroyd, P., Thiele, C., and Jahn, R. (2001). SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 20, 2202-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, T., Margittai, M., Holzler, H., and Jahn, R. (2002). SNAREs in native plasma membranes are active and readily form core complexes with endogenous and exogenous SNAREs. J. Cell Biol. 158, 751-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl, E., Hess, B., and van der Spoel, D. (2001). GROMACS 3. 0, a package for molecular simulation and trajectory. J. Mol. Model. 7, 306-317. [Google Scholar]
- Loranger, S. S., and Linder, M. E. (2002). SNAP-25 traffics to the plasma membrane by a syntaxin-independent mechanism. J. Biol. Chem. 277, 34303-34309. [DOI] [PubMed] [Google Scholar]
- Manders, E.M.M., Stap, J., Brakenhoff, G. J., Van Driel, R., and Aten, J. A. (1992). Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 103, 857-862. [DOI] [PubMed] [Google Scholar]
- McMahon, H. T., Ushkaryov, Y. A., Edelmann, L., Link, E., Binz, T., Niemann, H., Jahn, R., and Südhof, T. C. (1993). Cellubrevin is a ubiquitous tetanustoxin substrate homologous to a putative synaptic vesicle fusion protein. Nature 364, 346-349. [DOI] [PubMed] [Google Scholar]
- Miyamoto, S., and Kollman, P. A. (1992). SETTLE: an analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comp. Chem. 13, 952-962. [Google Scholar]
- Nagy, G., Matti, U., Nehring, R. B., Binz, T., Rettig, J., Neher, E., and Sørensen. J. B. (2002) PKC-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J. Neurosci. 22, 9278-9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy, G., Reim, K., Matti, U., Brose, N., Binz, T., Rettig, J., Neher, E., and Sørensen, J. B. (2004). Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 41, 351-365. [DOI] [PubMed] [Google Scholar]
- Ohara-Imaizumi, M., Nishiwaki, C., Nakamichi, Y., Kikuta, T., Nagai, S., and Nagamatsu, S. (2004). Correlation of syntaxin-1 and SNAP-25 clusters with docking and fusion of insulin granules analysed by total internal reflection fluorescence microscopy. Diabetologia 47, 2200-2207. [DOI] [PubMed] [Google Scholar]
- Rickman, C., Meunier, F. A., Binz, T., and Davletov, B. (2004). High affinity interaction of syntaxin and SNAP-25 on the plasma membrane is abolished by botulinum toxin E. J. Biol. Chem. 279, 644-651. [DOI] [PubMed] [Google Scholar]
- Rowe, J., Corradi, N., Malosio, M. L., Taverna, E., Halban, P., Meldolesi, J., and Rosa, P. (1999). Blockade of membrane transport and disassembly of the Golgi complex by expression of syntaxin 1A in neurosecretion-incompetent cells: prevention by rbSEC1. J. Cell Sci. 112, 1865-1877. [DOI] [PubMed] [Google Scholar]
- Salaün, C., Gould, G. W., and Chamberlain, L. H. (2005). Lipid raft association of SNARE proteins regulates exocytosis in PC12 cells. J. Biol. Chem. 280, 19449-19453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen, J. B., Nagy, G., Varoqueaux, F., Nehring, R. B., Brose, N., Wilson, M. C., and Neher, E. (2003). Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114, 75-86. [DOI] [PubMed] [Google Scholar]
- Sutton, R. B., Fasshauer, D., Jahn, R., and Brunger, A. T. (1998). Crystal structure of a SNARE complex involved in synaptic exocytosis at 2. 4 Å resolution. Nature 395, 347-353. [DOI] [PubMed] [Google Scholar]
- Van Buuren, A. R., Marrink, S.-J., and Berendsen, H.J.C. (1993). A molecular dynamics study of the decane/water interface. J. Phys. Chem. 97, 9206-9212. [Google Scholar]
- van Gunsteren, W. F., and Berendsen, H.J.C. (1987). GROMOS manual. BIOMOS, biomolecular Software, Laboratory of Physical Chemistry, University of Groningen, The Netherlands.
- Veit, M., Söllner, T. H., and Rothman, J. E. (1996). Multiple palmitoylation of synaptotagmin and the t-SNARE SNAP-25. FEBS Lett. 385, 119-123. [DOI] [PubMed] [Google Scholar]
- Vogel, K., Cabaniols, J.-P., and Roche, P. A. (2000). Targeting of SNAP-25 to membranes is mediated by its association with the target SNARE syntaxin. J. Biol. Chem. 275, 2959-2965. [DOI] [PubMed] [Google Scholar]
- Vriend, G. (1990). WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8, 52-56. [DOI] [PubMed] [Google Scholar]
- Xu, T., Rammner, B., Margittai, M., Artalejo, A. R., Neher, E., and Jahn, R. (1999). Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell 99, 713-722. [DOI] [PubMed] [Google Scholar]
- Washbourne, P., Cansino, V., Mathews, J. R., Graham, M., Burgoyne, R. D., and Wilson, M. C. (2001). Cysteine residues of SNAP-25 are required for SNARE disassembly and exocytosis, but not for membrane targeting. Biochem. J. 357, 625-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, T., Zemelman, B. V., McNew, J. A., Westermann, B., Gmachl, M., Parlati, F., Söllner, T. H., and Rothman, J. E. (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92, 759-772. [DOI] [PubMed] [Google Scholar]
- Zhang, X., Kim-Miller, M. J., Fukuda, M., Kowalchyk, J. A., and Martin, T. F. (2002). Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron 34, 599-611. [DOI] [PubMed] [Google Scholar]