

Abstract
To unravel the region of human eukaryotic release factor 1 (eRF1) that is close to stop codons within the ribosome, we used mRNAs containing a single photoactivatable 4-thiouridine (s4U) residue in the first position of stop or control sense codons. Accurate phasing of these mRNAs onto the ribosome was achieved by the addition of tRNAAsp. Under these conditions, eRF1 was shown to crosslink exclusively to mRNAs containing a stop or s4UGG codon. A procedure that yielded 32P-labeled eRF1 deprived of the mRNA chain was developed; analysis of the labeled peptides generated after specific cleavage of both wild-type and mutant eRF1s maps the crosslink in the tripeptide KSR (positions 63–65 of human eRF1) and points to K63 located in the conserved NIKS loop as the main crosslinking site. These data directly show the interaction of the N-terminal (N) domain of eRF1 with stop codons within the 40S ribosomal subunit and provide strong support for the positioning of the eRF1 middle (M) domain on the 60S subunit. Thus, the N and M domains mimic the tRNA anticodon and acceptor arms, respectively.
Keywords: eukaryotic ribosomes/human eRF1/NIKSR region/photocrosslinking/stop codons
Introduction
Termination of translation is the last critical step of protein synthesis, as it ensures the formation of normal-sized proteins. Release of the nascent polypeptide takes place within the ribosome when the A site is occupied by the stop codon (UGA, UAA or UAG) and by class-1 polypeptide chain release factors (RF1s) (reviewed in Nakamura and Ito, 1998; Kisselev and Buckingham, 2000; Poole and Tate, 2000; Bertram et al., 2001). In eukaryotes, eRF1 decodes all three stop codons, whereas, in prokaryotes, RF1 and RF2 decode UAG/UAA and UGA/UAA stop codons, respectively. In response to termination codons, RF1s trigger peptidyl-tRNA hydrolysis catalyzed by the peptidyl transferase center of the ribosome. It is believed (Nakamura et al., 2000) that RF1s decode stop codons via direct protein–mRNA interactions, as eRF1 strongly competes with suppressor tRNAs in vitro and in vivo (Weiss et al., 1984; Drugeon et al., 1997; Le Goff et al., 1997) and, at least in some cases, eRF1 was shown to be in close contact with the stop codon in the ribosome (Poole and Tate, 2000; Chavatte et al., 2001). All known RF1s contain a ubiquitous Gly-Gly-Gln (GGQ) motif, which was found to be essential for triggering peptidyl-tRNA hydrolysis and is thought to mimic the CCA end of tRNA (Frolova et al., 1999; Seit-Nebi et al., 2001). However, the molecular basis of stop codon recognition by RF1s remains a challenging problem.
In prokaryotes, a tripeptide was inferred by mutagenesis analysis to determine RF1 stop codon recognition. Thus, Pro-Ala-Thr in RF1 and Ser-Pro-Phe in RF2 were shown to be involved in the discrimination between the second and third purine bases of the stop signal (Ito et al., 2000; Nakamura et al., 2000). Yet, the recent crystal structure of RF2 (Vestergaard et al., 2001) failed to support the implication of these residues in the interaction with stop codons. To study RF2–mRNA contacts occurring in Escherichia coli ribosomes, Tate and colleagues performed site-directed crosslinking experiments (Tate et al., 1990; Brown and Tate, 1994; Poole et al., 1997) using mini-mRNAs in which the U residue of the stop signal was replaced by its photoactivatable analog, 4-thiouridine (s4U) (Favre et al., 1998). Both s4UAA and s4UGA (but not the non-cognate s4UGA) 36mer mRNAs appropriately programmed on E.coli ribosomes by tRNAAla2, were shown to crosslink to RF2. Further experiments, however, demonstrated that the nature of the phasing tRNA strongly influences the specificity of E.coli RF–mRNA crosslink formation (McCaughan et al., 1998). In a recent review (Poole and Tate, 2000), it was mentioned that a crosslink was identified in the structural domain D of RF2, near to, or within, the Ser-Pro-Phe motif.
eRF1s display low, if any, sequence similarity with prokaryotic RF1 and RF2 (Frolova et al., 1994). The domain organization of human eRF1 established on the basis of its crystal structure (Song et al., 2000) and from biochemical studies (Frolova et al., 2000) supports the ‘tRNA analog’ concept (Moffat and Tate, 1994). It has been suggested that the N-terminal (N) domain mimics the anticodon arm, the middle (M) domain is equivalent to the amino acid acceptor arm and the C-terminal (C) domain is responsible for interaction with the class-2 RF, eRF3 (Ebihara and Nakamura, 1999; Merkulova et al., 1999). Deletion of the C domain has no influence on in vitro eRF1 activity, and thus the active ‘core’ is composed of N and M domains (Frolova et al., 2000). For the Saccharomyces cerevisiae eRF1, mutation of certain amino acid residues of the N domain affected stop codon discrimination (Bertram et al., 2000). In some ciliate species, a variant nuclear genetic code is used, affecting the meaning of stop codons. For example, in hypotrich Euplotes, UAA and UAG are used as stop codons, whereas UGA codes for Cys. The fact that, in an in vitro release assay (performed with rabbit ribosomes), Euplotes aediculatus eRF1 responded to the UAA and UAG stop codons (but not to UGA) demonstrates that eRF1 itself bears stop codon recognition properties (Kervestin et al., 2001). Sequence alignment of eRF1s and the search for conserved residues within the N domain, which could vary following the reassignment of stop codons in ciliate eRF1, led to several hypotheses on eRF1 stop codon recognition (Inagaki and Doolittle, 2001; Lehman, 2001; Liang et al., 2001; Lozupone et al., 2001; Muramatsu et al., 2001; Inagaki et al., 2002). In vitro site-directed mutagenesis studies showed that the left part of the NIKS motif (positions 61 and 62) is important for the conservation of RF activity, whereas its right part (positions 63 and 64) and Arg residues in positions 65 and 68 are involved in ribosome binding (Frolova et al., 2002). The omnipotent decoding potential of the eRF1s of variant-code organisms was shown recently to be modulated by the TASNIKS sequence (Ito et al., 2002). All the above data focused on the N domain as containing the stop codons recognition site, but they failed to demonstrate that this domain directly contacts the stop codons.
We applied (Chavatte et al., 2001) to eukaryotes (rabbit ribosome, human eRF1) the same photocrosslinking strategy as previously used in the study of E.coli translation termination (Poole and Tate, 2000). The 42mer mRNA analogs used contain a GAC codon corresponding to tRNAAsp, followed by either s4UGA or the control sense s4UCA triplet. We demonstrated that, in non-phased ribosomes, both s4UGA and s4UCA mRNA analogs yielded very similar crosslinking patterns with rRNA and ribosomal proteins (rPs). In contrast, in phased ribosomes, they yielded different crosslinking patterns on the addition of eRF1. The formation of an eRF1–mRNA crosslink was detected only with s4UGA. It occurs with both full-length eRF1 and a truncated form lacking the C domain and requires the presence of tRNAAsp (Chavatte et al., 2001). Using perfluoroaryl-azido derivatized heptaribonucleotides containing a UCC codon for Phe followed by either a stop UAA or sense CAA codon, Bulygin et al. (2002) also observed that the formation of an eRF1–mini-mRNA crosslink on the irradiation of a mixture of 80S ribosome, mRNA analog, eRF1 and tRNAPhe was stop codon dependent.
In the present work, we compared the behavior of mRNA analogs bearing in the appropriate position one of the stop codons s4UGA, s4UAA and s4UAG or a closely related sense codon. The ribosome–mRNA complex was phased in the presence of tRNAAsp. To monitor the specificity of this interaction, mRNA constructs were used in which the s4UGA triplet was ‘frameshifted’ by one, two or three positions with respect to the Asp codon. The results demonstrate that correctly phased stop codons specifically crosslink with eRF1 within the ribosome. To locate on the eRF1 sequence the oligopeptide(s) that crossreact(s) with stop codons containing s4U, a procedure was developed that leads to highly purified 32P-labeled eRF1 depleted of mRNA. Specific cleavage of the polypeptide chain of wild-type 32P-labeled eRF1, as well as eRF1 mutants, followed by analysis of the labeled peptides, showed that the Lys-Ser-Arg tripeptide (positions 63–65) in the N domain of human eRF1 is crosslinked to the first position of stop codons.
Results
Specificity of eRF1–mRNA analog crosslink formation
Basically, the in vitro system consists of 80S ribosomes, tRNAAsp and mRNA analog. The 42mer to 45mer mRNA analogs are purine-rich to minimize the formation of secondary structure or self-association, which could restrict or prevent their binding to the ribosome and/or their template efficiency. In the middle of their sequence, they all contain (Figure 1A) a GAC codon for Asp, followed, on their 3′ side, either immediately or after a few residues, by a single specifically photoactivatable residue, s4U. The behavior of the ribosome programmed with a 5′-end 32P-labeled mRNA analog containing the GACs4UGA (UGA mRNA) sequence is represented in Figure 2, which shows the patterns of crosslinks obtained in the absence of tRNAAsp and eRF1 (non-phased ribosomes), in the presence of tRNAAsp (phased ribosomes) and in the presence of tRNAAsp and eRF1. The addition of tRNAAsp to the non-phased ribosomes increases the overall yield of crosslink formation (i.e. the fraction of added mRNA involved in crosslinks) from 5 to 50%, and this is mainly due to the enhancement of rRNA–mRNA crosslinks (apparent mass >100 kDa) and a 19 kDa rP–mRNA (34 kDa) crosslink (Chavatte et al., 2001). The transition from the non-phased to phased ribosome is achieved at 0.5 µM tRNAAsp, as no further changes in either yield or pattern of crosslink occur at higher tRNAAsp concentrations (data not shown). The addition of eRF1 to the phased ribosomes (Figure 2, lanes 3 and 4) results in significant quenching of all crosslinks (final yield close to 6%), with the exception of the 68–70 kDa one (lanes 3 and 4). The corresponding bands are weak (lane 1) or close to background (lane 2) in the absence of eRF1, but they are strongly stimulated in its presence (lanes 3 and 4). It has been unambiguously shown that this enhancement is due to the formation of the eRF1–mRNA crosslink (Chavatte et al., 2001). This finding strongly suggests that, on binding to the correctly programmed ribosomes, eRF1 is in contact with the first position of the stop codon (s4U), while preventing the s4U residue from crosslinking with neighboring rRNA and rP. Moreover, the final patterns are identical whether the eRF1 concentration is 2 or 6 µM (compare lanes 3 and 4).

Fig. 1. mRNA containing s4U. (A) mRNAs obtained by in vitro transcription in the presence of s4UTP (instead of UTP). All 42mer mRNAs contain a GAC codon (underlined) followed by a stop or sense codon (bold). As they differ only by the nature of the latter codon, they were named accordingly. In UGA+1, UGA+2 and UGA+3, the triplet was ‘frameshifted’ relative to the GAC codon by the insertion of one, two or three Gs, respectively. All these mRNAs were used after 5′-end 32P-labeling. (B) Scheme showing the synthesis of the internally labeled s4U*GA mRNA analog. This 42mer mRNA was assembled from two fragments: RNA(1), obtained by in vitro transcription; and RNA(2), synthesized chemically. Ligation was performed in the presence of complementary DNA template and T4 DNA ligase.
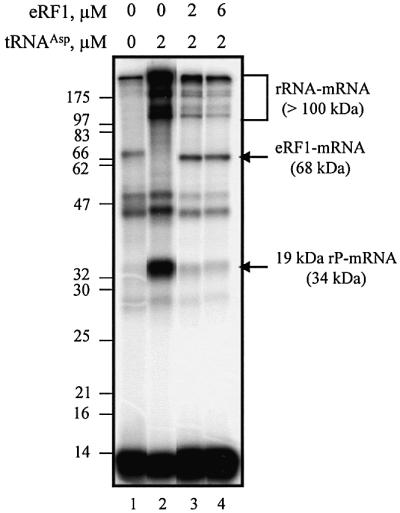
Fig. 2. Dependence of crosslink formation on s4UGA programmed ribosomes as a function of eRF1 concentration. Autoradiograph of the crosslink pattern obtained after irradiation of 0.1 µM 5′-end 32P-labeled s4UGA mRNA analog with 0.2 µM reassociated ribosomes and with or without 2 µM tRNAAsp and variable amounts of eRF1. The irradiated mixtures were analyzed by 10% SDS–PAGE. Lanes 1 and 2 are typical of non-phased and phased ribosome–mRNA complexes. Lanes 3 and 4 show the behavior of the phased ribosomes on the addition of different amounts of eRF1. Molecular mass markers are indicated (in kDa).
To examine whether other stop codons are able to crosslink with eRF1 on phased ribosomes, 42mer mRNA analogs that contained the s4UAA or s4UAG codon in place of s4UGA (UAA and UAG mRNA analogs) were synthesized. As a control in these experiments, a set of sense codons related to the stop codons (UGG, UCA and UAC mRNA analogs) were used. As the pattern of crosslink with the rPs is sensitive to the ribosome preparation (Chavatte et al., 2001), the UGA mRNA analog was always tested in parallel as an internal reference. With the phased ribosomes, all these mRNA analogs yielded the same pattern of crosslinks as for the UGA mRNA (Figure 3A, lane 1). With 6 µM eRF1, the resulting patterns can be subdivided into three types: (i) when any of the stop codons is located at the A site, the eRF1–mRNA crosslinks are formed (Figure 3A, lanes 2–4), though with different efficiencies (Figure 3B), and the 19 kDa rP–mRNA or rRNA–mRNA crosslinks are severely quenched; (ii) when the s4UCA and s4UAC sense codons are located at the A site, no eRF1–mRNA crosslinks (the 68–70 kDa band corresponds closely to the background values observed in the absence of eRF1) are detected, and the formation of the rRNA–mRNA and 19 kDa rP–mRNA crosslinks is partly quenched; and (iii) the sense s4UGG triplet allows eRF1–mRNA crosslink formation (Figure 3A and B), but quenching of rRNA–mRNA and 19 kDa rP–mRNA crosslinks is less efficient than with stop codons.

Fig. 3. Codon dependence of eRF1–mRNA and 19 kDa rP–mRNA crosslink formation. (A) Autoradiograph of the crosslink patterns obtained with stop or sense codons containing 0.1 µM 42mer mRNA analogs in the presence of 0.2 µM reassociated ribosome, 6 µM eRF1 and 2 µM tRNAAsp (lanes 2–7). For comparison, a control without eRF1 is shown (lane 1). Molecular mass markers are indicated (in kDa) on the left. eRF1–mRNA and 19 kDa rP–mRNA crosslinks are indicated by arrows. (B) Codon-dependent formation of the eRF1–mRNA crosslink. The percentage of this crosslink over all of the crosslinks within the same lane was determined by quantification of the data in (A).
To probe more deeply the specificity of eRF1–mRNA crosslink formation, we examined the behavior of a set of mRNA analogs derived from the 42mer UGA mRNA by the insertion of one, two or three G residues between the GAC and the s4UGA triplets, yielding 43mer UGA+1, 44mer UGA+2 and 45mer UGA+3 mRNA analogs, respectively (Figure 1A). In the non-phased ribosomes, all these mRNA analogs yield patterns of crosslinks very similar to those obtained with the UGA mRNA analog, taking into account the respective masses of the mRNAs (data not shown). Incubation with tRNAAsp to phase the ribosomes increases the overall yield of crosslinks in every case (data not shown). It should place the Gs4UG triplet (UGA+1 mRNA), the GGs4U triplet (UGA+2 mRNA) and GGG triplet (UGA+3 mRNA analogs) within the ribo somal A site. Interestingly, the yields of crosslinks with rRNA and 29 and 32 kDa rPs remain unchanged (Figure 4, bands a–c). On the other hand, the formation of the crosslink with the 19 kDa rP (Figure 4, band d) is observed with UGA and UGA+1 mRNA, whereas the crosslink with the 13 kDa rP (Figure 4, band e) is observed almost exclusively with the UGA+1, UGA+2 and UGA+3 mRNA analogs. These data provide strong additional evidence for phasing of the mRNA analogs onto the ribosome on the addition of tRNAAsp. The addition of eRF1 to the phased ribosomes (Figure 4, right) markedly decreases the overall crosslinking yield only with the UGA mRNA analog (Figure 4, compare lanes 1 and 5) and much less with any mRNAs analogs containing G insertions. None of the ‘frameshifted’ mRNAs allow the formation of the eRF1–mRNA crosslink (Figure 4, lanes 6–8).

Fig. 4. Crosslink patterns obtained with ‘frameshifted’ mRNA analogs. 5′-end 32P-labeled frameshifted mRNA (0.1 µM) (Figure 1A) was mixed with 0.2 µM ribosomes and 2 µM tRNAAsp in the absence or presence of 6 µM eRF1. s4UGA mRNA (lanes 1 and 5) was used as a control. Molecular mass markers are indicated (in kDa). The nature of the crosslinks is indicated on the left: (a) >100 kDa rRNA–mRNA, (b) 32 kDa rP–mRNA, (c) 29 kDa rP–mRNA, (d) 19 kDa rP–mRNA and (e) 13 kDa rP–mRNA; the eRF1–mRNA crosslink is marked by an asterisk.
Labeling of the crosslink on eRF1
The strategy used to map the site of attachment of the mRNA on eRF1 relies on the determination of the size of the 32P-labeled fragments of eRF1 obtained after specific cleavage of the polypeptide chain. This approach is reliable only if performed on a highly purified form of the stop codon–eRF1 crosslink. As the yield of this crosslink is low (close to 1% of added mRNA), it is likely to be contaminated with rP–mRNA and rRNA–mRNA crosslinked products (Figure 3A). To improve resolution and decrease the background level, we developed a procedure that allows further analysis of the 32P-labeled peptides derived from the eRF1–mRNA crosslink. For this purpose, an internally 32P-labeled 42mer s4U*GA mRNA was constructed, because the s4UGA codon was the most efficient in yielding the desired crosslink (Figure 3B). This mRNA analog was assembled by the ligation of the two oligoribonucleotides RNA(1) and RNA(2) in the presence of DNA ligase and a complementary DNA strand, as described previously (Yu, 1999). To improve correct hybridization, the RNA(1) sequence was identical to the corresponding 5′ part of the parent s4UGA mRNA analog, whereas the RNA(2) sequence was enriched in pyrimidine residues as compared with the 3′ part of the parent analog (Figure 1B). Prior to ligation, RNA(2) was 32P-labeled at its 5′ end. Thus, after ligation, the label was introduced 3′ of the s4U probe. Digestion with micrococcal nuclease of the crosslinked complexes obtained with this mRNA analog is expected to remove all mRNA and rRNA–mRNA contaminants while leaving a 32P-labeled residue attached to the crosslinks formed with proteins.
As ligated s4UGA mRNA differs from the UGA mRNA analog in the 3′ part of its sequence and in the position of the label (Figure 1), its behavior was first examined under previously defined conditions (Figure 2). After irradiation of the s4U*GA ribosome–mRNA–tRNAAsp ternary complex in the presence of eRF1, one aliquot was treated as usual and the other was digested with micrococcal nuclease prior to SDS–PAGE analysis. The data obtained in a typical experiment are shown in Figure 5. First, they show that the internally labeled mRNA behaves exactly as its 5′ end-labeled analog (compare lane 1 in Figure 5 with lanes 3 and 4 in Figure 2). Secondly, they demonstrate that the nuclease treatment greatly improves the separation of 32P-labeled proteins on the gel. In particular, the 68 kDa band previously shown to correspond either to an rP–mRNA crosslink (in the absence of eRF1) or to, essentially, an eRF1–mRNA crosslink (when eRF1 was added) is now resolved after nuclease treatment into 52 and 55 kDa bands (Figure 5, lane 2). To unambiguously identify the eRF1 band, we took advantage of the presence of a His-tag fused to the N-terminus of eRF1. An aliquot of phased ribosomes containing eRF1 was loaded onto an Ni-NTA column after irradiation and nuclease treatment. The fraction eluted in the presence of 150 mM imidazole was then concentrated and analyzed on the gel (Figure 5, lane 3), demonstrating that the 55 kDa band corresponds to 32P-labeled eRF1 (eRF1p*). Thus, under the conditions used, ligated s4U*GA mRNA digestion is complete and the procedure makes it possible to isolate purified eRF1p*.
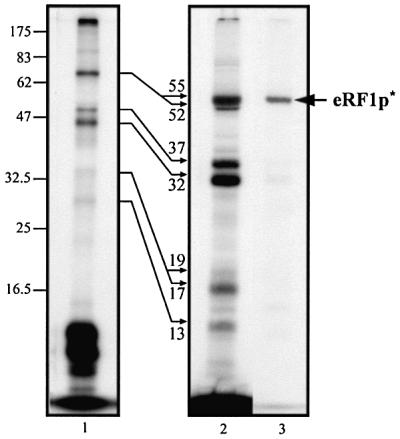
Fig. 5. Crosslink patterns obtained with internally labeled s4U*GA mRNA analog and phased ribosomes in the presence of eRF1: lane 1, without nuclease treatment; lane 2, after microccocal digestion of tRNAs. Reaction conditions: 0.1 µM mRNA, 0.2 µM ribosome and, where indicated, 2 µM tRNAAsp and 6 µM eRF1. Autoradiogram after 12.5% SDS–PAGE. To localize the His6-tagged eRF1p* product (arrow), an aliquot of the mixture obtained after irradiation of the complete mixture was nuclease treated and passed through an Ni-NTA column. The fraction eluted with 150 mM imidazole was analyzed in parallel (lane 3). Molecular masses of the rPp* and eRF1p* obtained after nuclease digestion are indicated (in kDa) close to the thin arrows.
Mapping of the crosslink on the eRF1 protein
To map the crosslink, eRF1p* was either purified by the Ni-NTA column, or the band corresponding to eRF1 (Figure 5, lane 2) was cut from a dried gel. None of these preparations revealed significant contamination by rPp* crosslinks, as shown for the sample purified by the Ni-NTA column (Figure 5, lane 3). Treatment of eRF1p* with CNBr, which cleaves the polypeptides after Met residues (nine sites available in wild-type human eRF1), and subsequent analysis by SDS–PAGE (Figure 6A, lane 1) revealed two major labeled polypeptides of 15.5 and 17.5 kDa (apparent masses) in a 2:1 ratio. From the positions of Met residues along the eRF1 sequence (Figure 6B), it can be inferred that the 15.5 kDa polypeptide is the 52–195 fragment, whereas the 17.5 kDa polypeptide corresponds to the 35–195 fragment. This interpretation was verified as follows. (i) eRF1p* was subjected to limited CNBr cleavage, leaving intact up to 70% of the initial protein. The polypeptides containing the His-tag were selected using an Ni-NTA column and analyzed by SDS–PAGE; the shortest labeled fragment migrated as an ∼25 kDa band, corresponding to the N-terminal fragment up to position 195 (theoretical mass ∼24 kDa). (ii) Treatment of free eRF1 by CNBr generated the expected peptides (80% yield) plus fragments resulting from incomplete cleavage, as shown by mass spectrometry and gel analysis followed by silver staining (data not shown).

Fig. 6. Mapping the crosslinking site on wild-type eRF1. (A) Determination of the size of the 32P-labeled eRF1p* fragments after specific treatments. In lanes 1, 2 and 4, eRF1p* was treated by CNBr (Met↓X), protease V8 (Glu↓X and Asp↓X) and protease Arg-C (Arg↓X), respectively. Samples were analyzed by either 12.5% Tris–glycine (lane 1) or 16.5 % Tris–tricine (lanes 2–5) SDS–PAGE. Digestion of 15 µg of eRF1 with either V8 (lane 3) or Arg-C (lane 5) proteases followed by visualization by silver staining. Asterisks indicate the positions of V8 and Arg-C on the gel. Arrows indicate the position of full-length eRF1. (B) Schematic representation of the sites of cleavage of full-length human eRF1by CNBr or V8 and Arg-C proteases; sites are represented as vertical bars. The size of 32P-labeled peptides in agreement with the data of (A) are shown in gray (fragments resulting from complete cleavage) or as black bars (incomplete cleavage) for CNBr and V8. The data obtained with Arg-C show that the 9.3 kDa peptide, positions 82–166 (crossed black bar), is not labeled. Compilation of the data is shown as a black box (positions 55–81 of human eRF1).
Proteolytic fragmentation of eRF1p* was also performed with either endoproteinase V8, which cuts after Glu and Asp residues (64 sites available in human eRF1), or Arg-C, which cuts after Arg residues (18 sites available) (Figure 6B). Digestion of free unlabeled eRF1 was examined in parallel. SDS–PAGE analysis showed that protease V8 efficiently cuts free eRF1 (Figure 6A, lane 3), which is no more detectable on the gel, and yielded 6.7–5.3 kDa fragments. Mass spectra confirmed their presence, together with a number of smaller fragments (data not shown). In the eRF1p* V8 digest, both 6.7 and 5.3 kDa fragments plus other minor fragments were strongly labeled (Figure 6A, lane 2), suggesting that the label was attached between positions 56 and 104 (Figure 6B). Arg-C digestion of eRF1p* yielded mainly 4, 4.5 and 5.5 kDa polypeptides, excluding the possibility that the label is attached between positions 82 and 166, which would have generated a polypeptide of 9.3 kDa and longer fragments. Taken together, the data imply that the site of crosslinking maps between positions 55 and 81 in the N domain of eRF1 (Figure 6B).
To refine the position of the crosslink, a strategy based on the CNBr treatment of eRF1 mutants bearing substitution of a single amino acid for Met in the region of interest (positions 55–81) was used. Cleavage on the N-terminal side of the labeled crosslink will generate a large labeled fragment (13–15 kDa), whereas cleavage on its C-terminal side will yield fast-migrating labeled peptides (1–2.5 kDa). Mutations (S60M, V66M and G73M) were performed in non-conserved positions of eRF1. All the corresponding mutant proteins retain full crosslinking ability but variable RF activity (Table I). CNBr cleavage yields a large labeled fragment (∼15 kDa) with S60M, whereas it generates fast-migrating polypeptides with V66M and G73M (Figure 7A), showing that the crosslink maps between positions 61 and 66.
Table I. Percentage crosslinking yields (obtained with internally labeled s4U*GA mRNA analog) and RF activity of Met mutants relative to wild-type eRF1.
| Wild type | S60M | I62M | K63M | S64M | V66M | G73M | |
|---|---|---|---|---|---|---|---|
| Crosslink |
100 |
97 |
95 |
43 |
85 |
98 |
80 |
| RF activity with UAA |
100 |
100 |
7 |
61 |
100 |
52 |
nd |
| RF activity with UAG |
100 |
100 |
5 |
67 |
100 |
30 |
nd |
| RF activity with UGA | 100 | 100 | 28 | 66 | 100 | 39 | nd |
Values are the averages of at least three independent experiments and are given with ±10% (crosslinks) and ±11% (RF activity) precision.
nd, not determined.

Fig. 7. Mapping of the crosslink with mutant eRF1s. (A) Patterns of CNBr induced cleavage fragments obtained with S60M, V66M and G73M (left) and S60M, I62M, K63M and S64M (right). Numbers 1–4 and 5–8 refer to fragments shown in (B) and (C), respectively. Samples were analyzed by 12.5% Tris–glycine SDS–PAGE, and wild-type eRF1p* was used as a control. (B) Expected size of 32P-labeled fragments (shown in gray) for S60M, V66M and G73M, indicating that the crosslink maps in the NIKS region. (C) Size of fragments expected from CNBr induced cleavage of the 52–195 polypeptide on the substitution of S60, I62, K63 or S64 by a Met residue.
For further refinement of the crosslink positions, I62M, K63M and S64M eRF1 mutants were constructed, some of which are affected in either their RF activity or crosslinking ability (Table I). I62M is significantly impaired in its RF activity but crosslinks well. On CNBr treatment, it generates two large labeled polypeptides (Figure 7). The smaller 15.5 kDa fragment migrates faster than the 52–195 fragment obtained with wild-type eRF1 and thus corresponds to a 63–195 labeled polypeptide. It should be noticed that the cleavage efficiency at position 62 is reduced ∼2-fold. K63M crosslinks poorly and, on CNBr cleavage, yields a pattern identical to the one obtained with I62M. On the other hand, S64M generates a fast-migrating labeled peptide with a reduced yield (compare its behavior with that of V66M or G73M). Further analysis of the data above (see Discussion) narrowed the site of crosslink to the KSR peptide (positions 63–65 of human eRF1).
Discussion
Phasing of mRNA onto ribosomes
Basically, our assay includes factor-free rabbit ribosomes and a 42mer to 45mer mRNA analog containing a single photoactivatable s4U residue but is devoid of an energy source. A 2-fold molar excess of ribosomes over mRNA was used in order to select ribosomes prone to efficient mRNA binding. The characteristic change of the patterns of crosslinks observed on the addition of tRNAAsp to 42mer ribosome–mRNA mixtures (Figure 2) is due to mRNA phasing onto the ribosome. It is well established that, in the presence of its cognate codon, a tRNA exhibits a much higher affinity for the ribosomal P site than for the A or E sites (Graifer et al., 1992 and references therein). Therefore, in the phased state of the ribosome–mRNA– tRNAAsp complex, tRNAAsp and its GAC codon are located in the P site, automatically placing the s4UNN codon of 42mer mRNAs in the A site. This is in agreement with our finding that all 42mer mRNAs yield the same crosslinking patterns both qualitatively and quantitatively (data not shown). ‘Frameshifted’ s4UGA mRNAs (Figure 1A) provide additional evidence for correct phasing of the mRNA on the ribosome. Indeed, the left part of Figure 4 shows that the s4U residue crossreacts with the 19 kDa rP, yielding the 34 kDa crosslink, only when located immediately 3′ to the mRNA decoding site or 3′-shifted by one residue (UGA mRNA and UGA+1 mRNA, Figure 1A). Conversely, it crossreacts with the 13 kDa rP, yielding the 28 kDa crosslink, when it is 3′-shifted by one, two or three residues (UGA+1, UGA+2 and UGA+3 mRNA analogs, respectively).
eRF1 binding to programmed ribosomal A site and eRF1–mRNA crosslink formation
The addition of eRF1 to s4UGA stop codon programmed ribosomes triggers quenching of all previously observed crosslinks (Figure 3A, lanes 2–4) and the formation of a new 68 kDa band previously identified as an eRF1–mRNA crosslink (Chavatte et al., 2001). Obviously, the quenching effect reflects the occupancy of the A site by eRF1, and this is particularly clear for the 34 kDa crosslink (Figure 2). When induced by the addition of 5 µM eRF1, this quenching effect appears to depend solely on the nature of the triplet present in the A site. It is weak but significant (∼30%) for the sense codons s4UCA, s4UAC (Figure 3) or Gs4UG (Figure 4), higher (∼50%) for s4UGG and reaches ∼70% for the stop codons (Figure 3, lanes 2–4). As shown in Figure 2 (lanes 3 and 4) for UGA mRNA, a 2 µM concentration of eRF1 is sufficient to trigger maximum quenching. These data indicate that stop codons promote strong eRF1 binding to the ribosomal A site as compared with sense codons, whereas the near-cognate s4UGG triplet adopts an intermediate behavior (a detailed analysis of eRF1 binding isotherms will be published elsewhere). All three stop codons and the near-cognate s4UGG triplet allow the formation of the eRF1–mRNA crosslink, whereas only background values are detected with the s4UCA and s4UAC codons. The observation that sense codons (except UGG) are unable to promote the formation of the 68 kDa crosslink is confirmed by the data obtained with ‘frameshifted’ mRNAs (Figure 4). It can thus be concluded that the s4U-purine-purine triplet programmed A site allows both efficient binding of eRF1 to this site and the formation of the eRF1–mRNA crosslink. Conversion of a single purine residue into pyrimidine markedly decreases the affinity of eRF1 for the A site and prevents the formation of this crosslink. Under in vitro conditions, the RF activity of eRF1 was shown to be promoted by stop codons but not by UGG or other sense codons (Frolova et al., 1994). In this context, the ability of the s4UGG triplet to induce an eRF1–mRNA crosslink possibly reflects the lack of sensitivity of the activity assay. Remarkably, UGG stands out as a hot-spot in RF2-dependent termination of protein synthesis in a pro karyotic system (Freistroffer et al., 2000), and our data suggest it should behave similarly in the corresponding eRF1-dependent eukaryotic system. In any case, the highly stringent conditions required to observe the formation of an eRF1–mRNA crosslink justifies our attempt to map the crosslink on eRF1.
Identification of the eRF1 crosslinked peptide
Mapping was achieved on wild-type eRF1 in two steps. First, eRF1p* (eRF1 32P-labeled at the site of the crosslink) was freed from contaminating labeled rPs by SDS–PAGE (Figure 5), or His6-tagged eRF1p* was directly purified from the irradiated incubation mixture (after nuclease digestion) by affinity chromatography (Figure 5, lane 3). In the second step, purified eRF1p* was specifically cleaved and the size of the resulting fragments was determined (Figure 6A). Taken together, the data show that the crosslink is attached to a fragment that encompasses positions 55–81 (Figure 6B) of eRF1. To narrow the positioning of the crosslink in this region of eRF1, a set of eRF1 mutants was prepared, where a pre-selected amino acid residue between positions 60 and 73 was substituted for Met. We took advantage of the fact that, on CNBr treatment, the large 52–195 fragment will be asymmetrically cut (Figure 7), thus generating small (0.9–2.4 kDa) and large (≥13.5 kDa) fragments. If the fast-migrating fragment is labeled, then the crosslink is attached on the N-terminal side of the new cleavage site. S60M, V66M and G73M behave similarly to wild-type eRF1 with respect to their crosslinking ability (Table I). CNBr treatment leads to efficient cleavage at the introduced Met residue (Figure 7A), generating either a large (∼15 kDa, band 2 for S60M) fragment or fast-migrating peptides (bands 3 and 4 for V66M and G73M, respectively). These data unambiguously map the crosslink in the highly conserved NIKSR pentapeptide (positions 61–65 of human eRF1). Residue V66 can be excluded from this target site, as the formation of a crosslink involving M66 of V66M would impede CNBr induced cleavage at this position, which is not observed (Figure 7A).
We further attempted to refine the mapping using I62M, K63M and S64M. Their RF activities can be anticipated from earlier data (Frolova et al., 2002) and show no apparent correlation with their crosslinking abilities (Table I). After CNBr treatment, I62M and K63M generate very similar patterns (Figure 7), indicating that cleavage occurs at the inserted Met, yielding 14.7 kDa (band 5, I62M) and 14.6 kDa (band 6, K63M) labeled fragments. On the other hand, S64M yields a large 14.5 kDa fragment (band 7) and a fast-migrating one (band 8). These data should be interpreted with caution, as the cleavage yields at inserted Met residues are 2-fold lower than observed with either wild-type eRF1 or S60M and V66M (Figure 7A), which could be due to partial oxidation of the introduced Met by a short-lived O2 singlet (1O2) generated during the irradiation step by the closely positioned s4U residue (reviewed in Favre, 1990). Cleavage might also be prevented if the considered Met is involved in the crosslink. Taking the above considerations into account, it can be concluded that I62 does not crossreact with s4U, as no fast-migrating peptide could be detected with K63M (Figure 7A). On the other hand, a labeled fragment of 14.5 kDa is observed with S64M, which suggests that some 32P label may be attached to R65. Taken together, these data restrict the site of crosslink to the KSR tripeptide (positions 63–65 of human eRF1). K63M protein is the only one exhibiting a markedly decreased crosslinking ability (Table I), suggesting that the anomalous behavior of K63M is due essentially to the low reactivity of M63, which favors s4U crosslinking with S64 and R65. This strongly points to K63 as the main site of attachment of the s4U residue in wild-type eRF1.
An excited s4U residue crossreacts efficiently only if it is able to come into contact (3–4 Å) with reactive acceptor groups with a favorable orientation (Favre, 1990; Favre et al., 1998). Thus, the photocrosslinking procedure used does not necessarily detect all residues susceptible to interaction with s4U. In this context, the low yield of formation of the eRF1–mRNA crosslink (∼1% of total mRNA but 6–15% of the crosslinks formed in the presence of eRF1) is most likely to be due to the sequestration of s4U when bound to eRF1, thus preventing favorable contacts with neighbor groups. This view is supported by the severe quenching of crosslinks observed on the occupancy of the A site by eRF1 (Figures 2 and 3A). Thus, it can be safely concluded that residues of the NIKSR loop, and particularly K63, participate in the recognition of the invariant U of stop codons.
Crosslinking data and hypotheses on eRF1 stop codon recognition
The resemblance between eRF1 and tRNA is based primarily on the functional similarity between both macromolecules. In particular, both occupy the correctly programmed ribosomal A site, recognizing a specific set of codons (stop and sense codons, respectively), and both are known to act at the ribosome peptidyl transferase center to trigger either release of the polypeptide or peptide bond formation. This resemblance is also supported by the similar shapes of eRF1 and tRNA (Figure 8A and B) and of tRNA and the prokaryotic ribosome recycling factor (Selmer et al., 1999; Kim et al., 2000; Toyoda et al., 2000; Fujiwara et al., 2001; Yoshida et al., 2001). As eRF1 consists of three domains (Song et al., 2000), the major problem still remains to assign the N and M domains relative to the anticodon and acceptor arms of the tRNA, as this cannot be determined solely on the basis of X-ray data. Our finding that the first position of stop codons specifically crossreacts with the KSR tripeptide (positions 63–65 of human eRF1; Figure 7) provides the first direct evidence that the N domain does mimic the anticodon arm, as was proposed, but not proven, previously (Bertram et al., 2000; Song et al., 2000; Ito et al., 2002). This tripeptide is located in a short loop surrounded by two helices (α2 and α3), which reinforces the topological similarity with the anticodon loop located at the end of a double helix (Figure 8). Our data also strongly support the assignment of the M domain as equivalent to the acceptor arm of tRNA (Song et al., 2000), based on the observation that this domain contains a highly conserved and functionally important GGQ motif, probably involved in triggering peptidyl-tRNA hydrolysis at the peptidyl transferase center of the ribosome (Frolova et al., 1999; Seit-Nebi et al., 2001).

Fig. 8. Comparison of the tRNA (A) and eRF1 (B) crystallographic structures. The similarity of the structures is shown by a side view (left) and a front view (right). Regions displaying similar roles (anticodon versus KS and CCA versus GGQ) are colored yellow. Molecular structures are shown by their water accessible surfaces, and the N domain is dark blue. (C) Enlarged view of the N domain of eRF1 showing the relative positions of the site of crosslink (yellow) and of residues E55, S123 and Y125 (orange), previously proposed to contact the base moiety of the invariant U residue of stop codons (Bertram et al., 2000; Muramatsu et al., 2001, Inagaki et al., 2002). Pink sticks show amino acids connecting the N and M domains.
Several models describing the interaction of stop codons with the N domain of eRF1 have been proposed on the basis of either mutagenesis studies (Bertram et al., 2000) or eRF1 sequence alignments (Nakamura et al., 2000; Muramatsu et al., 2001; Inagaki et al., 2002). In the cavity-binding model of Bertram et al. (2000), the invariant U in the first position of stop codons is expected to contact Met51 and Ser123, whereas, in the anticodon-mimicry model of Muramatsu et al. (2001), it was positioned close to Glu55. Analysis of the primary structure of the eRF1 family, combined with the estimation of the evolutionary rates at amino acid sites, allowed Inagaki et al. (2002) to infer that the invariant U might bind to a pocket defined by Glu55, Val71, Tyr125 and Cys127. As shown in Figure 8C, the proposed sites of contact are far removed from the KSR region in the crystallographic structure of eRF1, and thus none of these models appear consistent with our data.
In contrast, our data strongly suggest that K63 and, possibly, neighbor residues of the NIKSR loop are involved in the binding and recognition of the first position of stop codons, and this is consistent with other findings. (i) Amino acid residues of this loop are either strictly conserved, such as I62, or among the slowest evolving residues of eRF1s (Lozupone et al., 2001; Inagaki et al., 2002). It makes sense that the invariant U of stop codons binds to a relatively well conserved region of eRF1. (ii) N61 and I62 appear to be important for eRF1 release activity, as mutations at these sites impedes it but do not provide critical discrimination among the three stop codons (Frolova et al., 2002; Table I). (iii) The decoding capacity of eRF1s can be modulated by the interaction of the TAS (KAT) tripeptides located immediately upstream of the NIKS region (Ito et al., 2002). In the crystallographic structure of eRF1, the distance between the NIKS and GGQ motifs is larger than between the corresponding anticodon and CCA region of tRNA (100 versus 75 Å), as shown in Figure 8. This implies a conformational change of eRF1 when bound to a stop codon programmed A site in order to allow the GGQ motif to contact the peptidyl transferase center on the large subunit. The identification of the eRF1 residues contacting the second and third positions of stop codons, which could be readily achieved using a photoactivatable G analog (s6G) and applying the methodology developed here, is required before this model is fully consistent.
Materials and methods
Synthesis of s4U modified mRNAs
Oligonucleotides used as mRNAs (and presented in Figure 1A) were synthesized by in vitro transcription of synthetic DNA templates (Genset, Paris, France) with T7 RNA polymerase, as described previously (Chavatte et al., 2001). The nucleotide triphosphate mixture was composed of ATP, GTP, CTP and s4UTP (Amersham Pharmacia Biotech). UTP was replaced by s4UTP, allowing the incorporation of the s4U probe in the single position available. These mRNAs were 5′ end-labeled with [γ-32P]ATP (ICN) by polynucleotide kinase. They were purified by 15% PAGE, eluted and ethanol precipitated.
The internally labeled s4U*GA mRNA was assembled by the ligation of the two fragments RNA(1) and RNA(2) following a previously described procedure (Sontheimer, 1994; Yu, 1999). RNA(1) synthesized by in vitro transcription (see above) contained a single s4U residue at its 3′ end. RNA(2) (Eurogentec) was 32P-labeled at its 5′ end with T4 polynucleotide kinase. Both oligonucleotides were hybridized to the same DNA template (Figure 1C), and phosphodiester bond formation was catalyzed by T4 DNA ligase. Thus, 40 pmol of RNA(1), 30 pmol of 32P-labeled RNA(2) and 40 pmol of DNA template were incubated overnight at 37°C in 50 mM Tris–HCl pH 7.6, 10 mM MgCl2, 1 mM ATP, 1 mM DTT and 5% (w/v) polyethylene glycol-8000 (final volume 30 µl), together with 15 U of T4 DNA ligase, allowing the formation of 15–20 pmol of ligated s4U*GA mRNA. The 42mer mRNA was then purified by 15% PAGE.
Ribosomes and yeast tRNAAsp
Isolation and purification of 80S reticulocyte ribosomes was performed as described previously (Frolova et al., 1998). Standard 80S ribosomes washed by 0.5 M KCl (‘high salt-washed’ ribosomes) were purified by centrifugation through a 20% sucrose cushion and further purified by dissociation into subunits and subsequent reassociation (reassociated ribosomal subunits). Yeast tRNAAsp gene transcript was obtained by in vitro transcription (16 h at 37°C) of a linearized plasmid (derived from pUC19) in which the T7 RNA polymerase promoter region was directly connected to the downstream tRNA sequence (Frugier et al., 1993).
Cloning and mutagenesis of human eRF1
The full-length cDNA encoding human eRF1 carrying His-tag at the N-terminus was cloned in pQE30 vector (Qiagen), as described previously (Frolova et al., 1998, 2000). For site-directed mutagenesis, the full-length cDNA encoding human eRF1 with the C-terminal His-tag was cloned into pET23b(+) vector (Novagen), as described previously (Seit-Nebi et al., 2001; Frolova et al., 2002). The mutagenesis procedure generating eRF1 mutants was performed according to the PCR-based ‘megaprimer’ method (Sarkar and Sommmer, 1990), as described previously (Frolova et al., 2002). The primers used for the generation of eRF1 mutants are the following: S60M, 5′-ACTGCAATGAACA TTAAGTCACGA-3′; I62M, 5′-CTCGTGACTTCATGTTAGATGC-3′; K63M, 5′-GTTTACTCGTGACATAATGTTAGATG-3′; S64M, 5′-GG TTTACTCGCATCTTAATGTTAG-3′; V66M, 5′-TCACGAATGAAC CGCCTTTCAGTCC-3′; and G73M, 5′-GTCCTGATGGCCATTACAT CTGTAC-3′.
Expression and purification of human eRF1 and assay for RF activity
Wild-type human eRF1 and its mutants were expressed in E.coli strain BL21(DE3), purified using Ni-NTA resin (Superflow, Qiagen), and RF activity was measured as described previously (Frolova et al., 1994, 2000).
Crosslinking procedures
The standard in vitro system was composed of 0.1 µM 32P-labeled mRNA, 2 µM tRNAAsp , 0.2 µM high-salt-washed or reassociated (when mentioned in figure legends) 80S ribosomes and variable amounts of eRF1 (≤6 µM) in a final volume of 10 µl. The irradiation buffer contained 50 mM Tris–HCl pH 7.4, 100 mM KCl, 10 mM MgCl2 and 1 mM DTT. Each sample was incubated for 1 h at 37°C, placed into a siliconized glass capillary tube and irradiated for 30 min at 4°C. The light source was an HBO 150 W superpressure mercury lamp placed 5 cm from the sample and providing near-ultraviolet light (>320 nm), because shorter wavelengths were removed using an MTO J320A filter. These conditions allowed the completion of the crosslinking reactions. The irradiated sample was diluted in dye buffer and then resolved by 10% SDS–PAGE. The gel was then dried and analyzed on a PhosphorImager (Molecular Dynamics). Pre-stained molecular weight markers were systematically analyzed on a parallel lane of the gel. Crosslinks were counted using Image-Quant software.
eRF1 mapping
Ligated s4U*GA mRNA was prepared just before the irradiation procedure in order to prevent radiolysis. s4U*GA (0.1 µM) mRNA was mixed with 0.2 µM ribosomes, 2 µM tRNAAsp and 5 µM eRF1 in a final volume of 50 µl of irradiation buffer. Crosslinking was performed as described above. The irradiated sample was then diluted in nuclease buffer containing 50 mM Tris–HCl pH 8.8 and 10 mM CaCl2. Micrococcal nuclease (1 U, MBI Fermentas) was added, allowing complete RNA digestion after 2 h at 37°C. Two procedures were used to separate eRF1 from other labeled proteins (and RNA fragments). (i) The nuclease activity was stopped by the addition of dye buffer containing EDTA, and the mixture was subjected to 12.5% SDS–PAGE. The gel was dried and analyzed by autoradiography, and the band corresponding to eRF1p* was excised. (ii) The mixture containing His6-tagged eRF1 was loaded onto an Ni-NTA column (Superflow). The column was first washed with 200 vol. of IMAC 10 (20 mM Tris–HCl pH 8, 0.2 M KCl, 10% glycerol and 10 mM imidazole). His6-tagged eRF1 was then eluted with IMAC 150 (20 mM Tris–HCl pH 8, 0.2 M KCl, 10% glycerol and 150 mM imidazole). The corresponding fractions were pooled and precipitated with ethanol (final concentration 66% v/v).
The purified eRF1p* was digested directly in the gel slice or in solution by overnight reaction with CNBr (a few crystals) in 100 µl of 70% formic acid at room temperature, with 5 µg of endoproteinase V8 (Roche Biochemicals) in 50 µl of 25 mM sodium phosphate buffer pH 7.8 at 25°C, or with 0.5 µg of endoproteinase Arg-C in 50 µl of incubation mixture (Roche Biochemicals) at 37°C. After the reaction with CNBr, the sample was dried and then diluted in equilibration dye buffer and analyzed by 12.5% Tris–glycine SDS–PAGE in parallel with molecular weight markers ranging from 6.5 to 175 kDa (Figure 6A). The products of enzymatic digestions were diluted in dye buffer and analyzed by 16.5% Tris–tricine SDS–PAGE in parallel with ultra-low-range molecular weight markers ranging from 1.1 to 26.6 kDa (Sigma). The gels were dried and analyzed by autoradiography. As a control of proteolytic activity, 15 µg of eRF1 was digested by either endoproteinase V8 or Arg-C, separated as described above and revealed by Silver Staining Kit Protein (Amersham Pharmacia Biotech).
Acknowledgments
Acknowledgements
We thank R.Giégé and A.Theolbald-Dietrich for the kind gift of the plasmid used to obtain the yeast tRNAAsp gene transcript, and W.Merrick for the generous gift of rabbit ribosomes. We are grateful to A.Kononenko for his participation in the cloning and purification of mutant eRF1s, and to N.Ivanova and A.Poltaraus for the sequencing of human eRF1 mutant genes. The very kind assistance of A.-L.Haenni in improving the manuscript is acknowledged. The authors are indebted to Professor Lev Kisselev (L.K.) and Dr Luda Frolova (L.F.) for their encouragement throughout this work, which was supported by a ‘Chaire Internationale Blaise Pascal’ (L.K.), the Human Frontier Science Program (grant R-96-032), INTAS (grant 00-0041), the Russian Foundation for Basic Research (L.K. and L.F.) and the Russian Foundation for Support of Scientific Schools (L.K.). L.C. thanks MENESR and Fondation pour la Recherche Médicale for research fellowships.
References
- Bertram G., Bell,H.A., Ritchie,D.W., Fullerton,G. and Stansfield,I. (2000) Terminating eukaryote translation: domain 1 of release factor eRF1 functions in stop codon recognition. RNA, 6, 1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram G., Innes,S., Minella,O., Richardson,J. and Stansfield,I. (2001) Endless possibilities: translation termination and stop codon recognition. Microbiology, 147, 255–269. [DOI] [PubMed] [Google Scholar]
- Brown C.M. and Tate,W.P. (1994) Direct recognition of mRNA stop signals by Escherichia coli polypeptide chain release factor two. J. Biol. Chem., 269, 33164–33170. [PubMed] [Google Scholar]
- Bulygin K.N., Repkova,M.N., Ven’aminova,A.G., Graifer,D.M., Karpova,G.G., Frolova,L.Yu. and Kisselev,L.L. (2002) Placement of mRNA stop signal towards polypeptide chain release factors and ribosomal proteins in 80S ribosomes. FEBS Lett., 514, 96–101. [DOI] [PubMed] [Google Scholar]
- Chavatte L., Frolova,L., Kisselev,L. and Favre,A. (2001) The polypeptide chain release factor eRF1 specifically contacts the s4UGA stop codon located in the A site of eukaryotic ribosomes. Eur. J. Biochem., 268, 2896–2904. [DOI] [PubMed] [Google Scholar]
- Drugeon G., Jean-Jean,O., Frolova,L., Le Goff,X., Philippe,M., Kisselev,L. and Haenni,A.L. (1997) Eukaryotic release factor 1 (eRF1) abolishes readthrough and competes with suppressor tRNAs at all three termination codons in messenger RNA. Nucleic Acids Res., 25, 2254–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara K. and Nakamura,Y. (1999) C-terminal interaction of translational release factors eRF1 and eRF3 of fission yeast: G-domain uncoupled binding and the role of conserved amino acids. RNA, 5, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre A. (1990) 4-thiouridine as an intrinsic photoaffinity probe of nucleic acid structure and interactions. In Morrison,H. (ed.), Bioorganic Photochemistry: Photochemistry and the Nucleic Acids. John Wiley & Sons, New York, NY, pp. 379–425.
- Favre A., Saintome,C., Fourrey,J.L., Clivio,P. and Laugaa,P. (1998) Thionucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid–protein interactions. J. Photochem. Photobiol. B, 42, 109–124. [DOI] [PubMed] [Google Scholar]
- Freistroffer D.V., Kwiatkowski,M., Buckingham,R.H. and Ehrenberg,M. (2000) The accuracy of codon recognition by polypeptide release fctors. Proc. Natl Acad. Sci. USA, 97, 2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova L. et al. (1994) A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature, 372, 701–703. [DOI] [PubMed] [Google Scholar]
- Frolova L.Y., Simonsen,J.L., Merkulova,T.I., Litvinov,D.Y., Martensen,P.M., Rechinsky,V.O., Camonis,J.H., Kisselev,L.L. and Justesen,J. (1998) Functional expression of eukaryotic polypeptide chain release factors 1 and 3 by means of baculovirus/insect cells and complex formation between the factors. Eur. J. Biochem., 256, 36–44. [DOI] [PubMed] [Google Scholar]
- Frolova L.Y., Tsivkovskii,R.Y., Sivolobova,G.F., Oparina,N.Y., Serpinsky,O.I., Blinov,V.M., Tatkov,S.I. and Kisselev,L.L. (1999) Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA, 5, 1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova L.Y., Merkulova,T.I. and Kisselev,L.L. (2000) Translation termination in eukaryotes: polypeptide release factor eRF1 is composed of functionally and structurally distinct domains. RNA, 6, 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova L., Seit-Nebi,A. and Kisselev,L. (2002) Highly conserved NIKS tetrapeptide is functionally essential in eukaryotic translation termination factor eRF1. RNA, 8, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier M., Florentz,C., Schimmel,P. and Giege,R. (1993) Triple aminoacylation specificity of a chimerized transfer RNA. Biochemistry, 32, 14053–14061. [DOI] [PubMed] [Google Scholar]
- Fujiwara T., Ito,K. and Nakamura,Y. (2001) Functional mapping of ribosome-contact sites in the ribosome recycling factor: a structural view from a tRNA mimic. RNA, 7, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graifer D.M., Nekhai,S.Yu., Mundus,D.A., Fedorova,O.S. and Karpova,G.G. (1992) Interaction of human and Escherichia coli tRNA(Phe) with human 80S ribosomes in the presence of oligo- and polyuridylate templates. Biochim. Biophys. Acta, 1171, 56–64. [DOI] [PubMed] [Google Scholar]
- Inagaki Y. and Doolittle,W.F. (2001) Class I release factors in ciliates with variant genetic codes. Nucleic Acids Res., 29, 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki Y., Blouin,C., Doolittle,W.F. and Roger,A.J. (2002) Convergence and constraint in eukaryotic release factor 1 (eRF1) domain 1: evolution of stop codon specificity. Nucleic Acids Res., 30, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Uno,M. and Nakamura,Y. (2000) A tripeptide ‘anticodon’ deciphers stop codons in messenger RNA. Nature, 403, 680–684. [DOI] [PubMed] [Google Scholar]
- Ito K., Frolova,L., Seit-Nebi,A., Karamyshev,A., Kisselev,L. and Nakamura,Y. (2002) Omnipotent decoding potential resides in eukaryotic translation termination factor eRF1 of variant-code organisms and is modulated by the interactions of amino acid sequences within domain 1. Proc. Natl Acad. Sci. USA, 99, 8494–8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervestin S., Frolova,L., Kisselev,L. and Jean-Jean,O. (2001) Stop codon recognition in ciliates: Euplotes release factor does not respond to reassigned UGA codon. EMBO rep., 2, 680–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.K., Min,K. and Suh,S.W. (2000) Crystal structure of the ribosome recycling factor from Escherichia coli. EMBO J., 19, 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev L.L. and Buckingham,R.H. (2000) Translational termination comes of age. Trends Biochem. Sci., 25, 561–566. [DOI] [PubMed] [Google Scholar]
- Le Goff X., Philippe,M. and Jean-Jean,O. (1997) Overexpression of human release factor 1 alone has an antisuppressor effect in human cells. Mol. Cell. Biol., 17, 3164–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman N. (2001) Molecular evolution: Please release me, genetic code. Curr. Biol., 11, R63–R66. [DOI] [PubMed] [Google Scholar]
- Liang A., Brunen-Nieweler,C., Muramatsu,T., Kuchino,Y., Beier,H. and Heckmann,K. (2001) The ciliate Euplotes octocarinatus expresses two polypeptide release factors of the type eRF1. Gene, 262, 161–168. [DOI] [PubMed] [Google Scholar]
- Lozupone C.A., Knight,R.D. and Landweber,L.F. (2001) The molecular basis of nuclear genetic code change in ciliates. Curr. Biol., 11, 65–74. [DOI] [PubMed] [Google Scholar]
- McCaughan K.K., Poole,E.S., Pel,H.J., Mansell,J.B., Mannering,S.A. and Tate,W.P. (1998) Efficient in vitro translational termination in Escherichia coli is constrained by the orientations of the release factor, stop signal and peptidyl-tRNA within the termination complex. Biol. Chem., 379, 857–866. [DOI] [PubMed] [Google Scholar]
- Merkulova T.I., Frolova,L.Y., Lazar,M., Camonis,J. and Kisselev,L.L. (1999) C-terminal domains of human translation termination factors eRF1 and eRF3 mediate their in vivo interaction. FEBS Lett., 443, 41–47. [DOI] [PubMed] [Google Scholar]
- Moffat J.G. and Tate,W.P. (1994) A single proteolytic cleavage in release factor 2 stabilizes ribosome binding and abolishes peptidyl-tRNA hydrolysis activity. J. Biol. Chem., 269, 18899–18903. [PubMed] [Google Scholar]
- Muramatsu T., Heckmann,K., Kitanaka,C. and Kuchino,Y. (2001) Molecular mechanism of stop codon recognition by eRF1: a wobble hypothesis for peptide anticodons. FEBS Lett., 488, 105–109. [DOI] [PubMed] [Google Scholar]
- Nakamura Y. and Ito,K. (1998) How protein reads the stop codon and terminates translation. Genes Cells, 3, 265–278. [DOI] [PubMed] [Google Scholar]
- Nakamura Y., Ito,K. and Ehrenberg,M. (2000) Mimicry grasps reality in translation termination. Cell, 101, 349–352. [DOI] [PubMed] [Google Scholar]
- Poole E. and Tate,W. (2000) Release factors and their role as decoding proteins: specificity and fidelity for termination of protein synthesis. Biochim. Biophys. Acta, 1493, 1–11. [DOI] [PubMed] [Google Scholar]
- Poole E.S., Brimacombe,R. and Tate,W.P. (1997) Decoding the translational termination signal: the polypeptide chain release factor in Escherichia coli crosslinks to the base following the stop codon. RNA, 3, 974–982. [PMC free article] [PubMed] [Google Scholar]
- Sarkar G. and Sommer,S.S. (1990). The ‘megaprimer’ method of site-directed mutagenesis. Biotechniques, 8, 404–407. [PubMed] [Google Scholar]
- Seit-Nebi A., Frolova,L., Justesen,J. and Kisselev,L. (2001) Class-1 translation termination factors: invariant GGQ minidomain is essential for release activity and ribosome binding but not for stop codon recognition. Nucleic Acids Res., 29, 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmer M., Al-Karadaghi,S., Hirokawa,G., Kaji,A. and Liljas,A. (1999) Crystal structure of Thermotoga maritima ribosome recycling factor: a tRNA mimic. Science, 286, 2349–2352. [DOI] [PubMed] [Google Scholar]
- Song H., Mugnier,P., Das,A.K., Webb,H.M., Evans,D.R., Tuite,M.F., Hemmings,B.A. and Barford,D. (2000) The crystal structure of human eukaryotic release factor eRF1—mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell, 100, 311–321. [DOI] [PubMed] [Google Scholar]
- Sontheimer E.J. (1994) Site-specific RNA crosslinking with 4-thiouridine. Mol. Biol. Rep., 20, 35–44. [DOI] [PubMed] [Google Scholar]
- Tate W., Greuer,B. and Brimacombe,R. (1990) Codon recognition in polypeptide chain termination: site directed crosslinking of termination codon to Escherichia coli release factor 2. Nucleic Acids Res., 18, 6537–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda T., Tin,O.F., Ito,K., Fujiwara,T., Kumasaka,T., Yamamoto,M., Garber,M.B. and Nakamura,Y. (2000) Crystal structure combined with genetic analysis of the Thermus thermophilus ribosome recycling factor shows that a flexible hinge may act as a functional switch. RNA, 6, 1432–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard B., Van,L.B., Andersen,G.R., Nyborg,J., Buckingham,R.H. and Kjeldgaard,M. (2001) Bacterial polypeptide release factor RF2 is structurally distinct from eukaryotic eRF1. Mol. Cell, 8, 1375–1382. [DOI] [PubMed] [Google Scholar]
- Weiss R.B., Murphy,J.P. and Gallant,J.A. (1984) Genetic screen for cloned release factor genes. J. Bacteriol., 158, 362–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T. et al. (2001) Solution structure of the ribosome recycling factor from Aquifex aeolicus. Biochemistry, 40, 2387–2396. [DOI] [PubMed] [Google Scholar]
- Yu YT. (1999) Construction of 4-thiouridine site-specifically substituted RNAs for cross-linking studies. Methods, 18, 13–21. [DOI] [PubMed] [Google Scholar]