

Abstract
IEX-1 is an early response and NF-κB target gene implicated in the regulation of cellular viability. We show here that IEX-1 is a substrate for ERKs and that IEX-1 and ERK regulate each other’s activities. IEX-1 was isolated by phosphorylation screening with active ERK2 and found subsequently phosphorylated in vivo upon ERK activation. IEX-1 interacts with phosphorylated ERKs but not with c-jun N-terminal kinase (JNK) or p38. Upon phosphorylation by ERKs, IEX-1 acquires the ability to inhibit cell death induced by various stimuli. In turn, IEX-1 potentiates ERK activation in response to various growth factors. By using various IEX-1 mutants in which the ERK phosphoacceptor and/or ERK docking sites were mutated, we show that the IEX-1 pro-survival effect is dependent on its phosphorylation state but not on its ability to potentiate ERK activation. Conversely, IEX-1-induced modulation of ERK activation requires ERK–IEX-1 association but is independent of IEX-1 phosphorylation. Thus, IEX-1 is a new type of ERK substrate that has a dual role in ERK signaling by acting both as an ERK downstream effector mediating survival and as a regulator of ERK activation.
Keywords: apoptosis/ERK/MAP kinase/phosphorylation/substrates
Introduction
Mammalian cells respond to a variety of extracellular stimuli via activation of specific mitogen-activated protein kinases (MAPK) that orchestrate the delivery of the signal from receptors at the cell surface to the nucleus. Three main classes of MAPK are recognized: the classic MAPK, also known as extra-regulated kinases, ERK1 and ERK2, c-jun N-terminal kinase (JNK) and p38 MAPK, which are activated by dual phosphorylation at neighboring threonine and tyrosine residues in the activation loop and dephosphorylation of either residue results in MAPK inactivation. The general scheme of ERK activation involves a cascade of phosphorylation events initiated by stimulation of the Ras proto-oncogene following activation of growth factors receptors. The cascade starts with the activation of one or more Raf family kinases, which phosphorylate and activate the MAPK kinases (MEK1/2). MEK in turn catalyzes dual phosphorylation and activation of ERKs (for reviews see Schaeffer and Weber, 1999; Chang and Karin, 2001). Once activated, ERKs accumulate in the nucleus by a yet to be defined mechanism that seems to involve the dissociation of ERK from MEK (Fukuda et al., 1997) and/or a blocade of its export from the nucleus by neo-synthesized nuclear anchors (Lenormand et al., 1998).
The ERK pathway has been implicated in diverse cellular processes including proliferation, differentiation and survival (for reviews see Marshall, 1995; Schaeffer and Weber, 1999; Ballif and Blenis, 2001). The variable responses elicited by this cascade in different cell types is presumably determined by the cell-specific combination of downstream substrates. More than 50 different ERK substrates have been identified so far. These include ubiquitous or lineage-restricted transcription factors, the kinases Rsk and Mnk and proteins involved in nucleotide biosynthesis, cytoskeleton organization, ribosomal transcription or membrane traffic (Sturgill et al., 1988; Marais et al., 1993; Treisman, 1996; Fukunaga and Hunter, 1997; Graves et al., 2000; Lewis et al., 2000; Stefanovsky et al., 2001).
The net result of ERK activation varies also in a given cell according to the stimulus (Marshall, 1995). This suggests the existence of mechanisms allowing ERK to discriminate between the various substrates available. Key parameters for setting this choice include the control of the amplitude and duration of the signal and the spatial organization of the components of the ERK cascade and its substrates. The importance of these mechanisms is illustrated by data showing that sustained, but not transient, ERK activation is associated with its nuclear translocation and leads to growth factor-induced gene regulation, cell cycle entry or differentiation (Traverse et al., 1992; Marshall, 1995; Rouyez et al., 1997; Brunet et al., 1999). Interestingly, MAPKs themselves regulate the strength and duration of their activation by phosphorylating various substrates: MAPKs phosphorylate, and thereby inhibit, receptor tyrosine kinases (Countaway et al., 1992), the Ras-exhange factor SOS (Waters et al., 1995) and MEK (Brunet et al., 1994); MAPK also increases the activity and half-life of the MAPK phosphatases (Pulido et al., 1998; Brondello et al., 1999). ERKs interact with their substrates through conserved docking sites (Gavin and Nebreda, 1999; Jacobs et al., 1999; Smith et al., 1999). ERK substrates could therefore also play a role in the subcellular localization of ERKs. Such a regulation has already been reported in other MAPK pathways. For instance, in the fission yeast, the transcription factor Atf1 regulates the nuclear localization of the stress-activated kinase spc1 (Gaits et al., 1998). Likewise, phosphorylation of mammalian MAPKAP kinase 2 by p38 facilitates the export of p38 from the nucleus (Ben-Levy et al., 1998). Thus, discovery of ERK phosphorylation targets will help to get a better read-out of the regulation of both ERK multiple actions and spatio-temporal activation.
To identify new ERK substrates involved in cell proliferation and survival, we used a solid phase phosphorylation assay (Fukunaga and Hunter, 1997) with active ERK2 to screen a phage expression library prepared from UT7, a cell line of hematopoietic origin, the growth of which is dependent on the presence of various cytokines such as interleukin-3, erythropoietin (EPO) and thrombopoietin (TPO) (Rouyez et al., 1997). One of the cDNAs isolated encoded IEX-1, a gene previously characterized as an early response and NF-κB-target gene regulated by X-irradiation, UV radiation and various growth factors, and reported to have contradictory functions in the control of cell survival (Charles et al., 1993; Kumar et al., 1998; Schafer et al., 1998; Wu et al., 1998; Arlt et al., 2001; Schilling et al., 2001). We show here that IEX-1 is a specific substrate of ERK1/2 and binds to the kinases in their active forms. The association of ERK and IEX-1 results in trans-regulation of their activities: IEX-1 acquires an anti-apoptotic function in response to various death triggers upon phosphorylation by ERKs, while in turn, IEX-1 potentiates ERK activation in response to growth factors. The cell survival effect of IEX-1 is dependent on its phosphorylation state but not its ability to enhance ERK signaling. Conversely, IEX-1-induced modulation of ERK activation is independent of IEX-1 phosphorylation but requires ERK binding. Thus, IEX-1 is a new type of ERK substrate that has a dual role in the ERK pathway by being both a signaling downstream effector mediating ERK survival activity and also a regulator of ERK activation.
Results
Identification of IEX-1 as a substrate of ERK MAPK in vitro and in vivo
The cDNA encoding IEX-1 (Kondratyev et al., 1996) was isolated from the UT7 cell line during a phosphorylation screening with active ERK2 (see Materials and methods). To verify that IEX-1 was a substrate for ERKs, the IEX-1 coding region was expressed as a GST fusion protein, purified by glutathione–Sepharose chromatography and incubated with purified active ERK2 in an in vitro kinase assay. As shown in Figure 1A (upper panel), the GST–IEX1 protein was readily phosphorylated by ERK2 in vitro, whereas no radioactivity was incorporated into the GST protein.

Fig. 1. ERKs phosphorylate IEX-1 in vitro. (A) One microgram of purified GST or GST–IEX-1 fusion proteins wild-type (Wt) and mutants (T18A; T123AS126A; T/SA3; ΔBD) were reacted with purified recombinant active ERK2 in the presence of [32P]ATP. The products were analyzed by autoradiography or western blotting (WB) with the indicated antibodies. (B) Schematic representation of IEX-1 Wt and ERK-phosphorylation and/or binding sites mutants. Hatched area indicates the putative IEX-1 transmembrane domain.
IEX-1 presents three putative ERK phosphorylation sites at positions T18, T123 and S126 (Kondratyev et al., 1996; Figure 1B). To determine whether ERK phosphorylates IEX-1 at one or several of these sites, we generated IEX-1 mutants in which either one, two or all three sites were converted to alanine. Whereas the GST– T123AS126A double mutant was phosphorylated to almost the same extent as the wild-type protein, the GST–T18A protein was only marginally phosphorylated by active ERK2 (Figure 1A), indicating that T18 of IEX-1 is the major ERK-catalyzed phosphorylation site in vitro. Mutation of the three ERK consensus phosphorylation sites (mutant T/SA3) completely abolished ERK phosphorylation (Figure 1A).
To confirm these results, we raised rabbit antibodies to a phosphopeptide corresponding to T18. In an immunoblot assay, the anti-phosphoT18 peptide antibody recognized the wild-type but not the T18A IEX-1 fusion protein phosphorylated by ERK2 (Figure 1A, bottom panel), whereas the anti-IEX-1 antibody recognized both proteins. Thus, the anti-phosphoT18 antibody specifically recognizes IEX-1 phosphorylated in vitro by ERK.
This antibody was therefore used to determine whether ERKs could phosphorylate IEX-1 at T18 in vivo. In the first set of experiments, phosphorylation was assessed in UT7 cells stably expressing an HA epitope-tagged version of IEX-1 in which ERK activation was induced by TPO stimulation. As shown in Figure 2A, the anti-phosphoT18 antibody recognized HA-IEX-1 species only in TPO-stimulated cells. The signal detected disappeared when TPO was added in the presence of the MEK inhibitor PD98059, indicating that the MEK–ERK pathway is responsible for the phosphorylation of IEX-1 at T18 in vivo upon TPO stimulation.

Fig. 2. Expression, phosphorylation and subcellular localization of IEX-1 in UT7 cells. (A and B) UT7 cells stably expressing HA-tagged IEX-1 (A) or control (B) were deprived of cytokine and stimulated with TPO for various times in the absence or presence of 20 µM PD98059, as indicated. ERK activation and the presence of phosphorylated and non-phosphorylated forms of IEX-1 were monitored by immunoblotting of total cell lysates. (C and D) UT7 cells expressing an empty vector (C), or HA-tagged Wt-IEX-1 or T/SA3-IEX-1 (D) were stimulated for 3 h with TPO, lysed and subjected to differential centrifugation. Proteins (100 µg) from each cellular fraction were loaded: heavy membrane (HM), light membrane (LM), cytoplasm (C) and nucleus (N). Antibodies specific for cytochrome c (cyt c, mitochondrial marker), caspase-3 (cytosolic marker) and PARP (nuclear marker) were used to characterize the fractions.
IEX-1 has been described as an early gene regulated by some growth factors (Charles et al., 1993). In agreement with these reports, TPO induced a rapid expression of IEX-1 in UT7 cells (Figure 2B). Western blot with anti-phosphoT18 antibodies showed that the appearance of phosphorylated IEX-1 species correlated with that of IEX-1 protein and with ERK activation. Thus, like overexpressed IEX-1, endogenous IEX-1 protein is phosphorylated at T18 upon ERK activation. Subcellular fractionation studies of UT7 cells showed that the endogenous IEX-1 protein induced by TPO stimulation was mainly located in the heavy membrane fraction that contains mitochondrial membranes, with only low levels of the protein detected in the light membrane fraction and the nucleus (Figure 2C). Since induction of IEX-1 expression by TPO required ERK activation (data not shown), the effect of IEX-1 phosphorylation on its localization was studied using stable clones of UT7 cells expressing either wild type or the non-phosphorylable T/A3-IEX-1 mutant. Like the endogenous protein, most of the HA-IEX-1 species, either wild type or T/SA3, migrated with the heavy membrane fraction whether the cells were stimulated (Figure 2D) or not (data not shown) with TPO to induce ERK activation. Thus, ERK activation does not affect IEX-1 subcellular localization.
IEX-1 binds specifically to the active forms of ERK1/2 but not to p38 and JNK MAPK
The capacity of MAPKs to phosphorylate their substrates is controlled by their docking to specific amino acid sequences on the substrates (Gavin and Nebreda, 1999; Jacobs et al., 1999; Smith et al., 1999). Therefore, we examined whether IEX-1 could interact with ERKs by reacting Sepharose-bound GST–IEX-1 fusion proteins with lysates prepared from UT7 cells that had been stimulated with TPO to activate ERKs. GST–IEX-1 precipitated ERK1 and ERK2, but only from lysates of activated cells (Figure 3A), indicating that IEX-1 associates with the active but not with the resting forms of ERK1 and ERK2. This binding to ERK was specific since the GST–IEX-1 Sepharose was unable to precipitate p38 or JNK MAPKs in their resting or anisomycin-induced phosphorylated forms (Figure 3B).
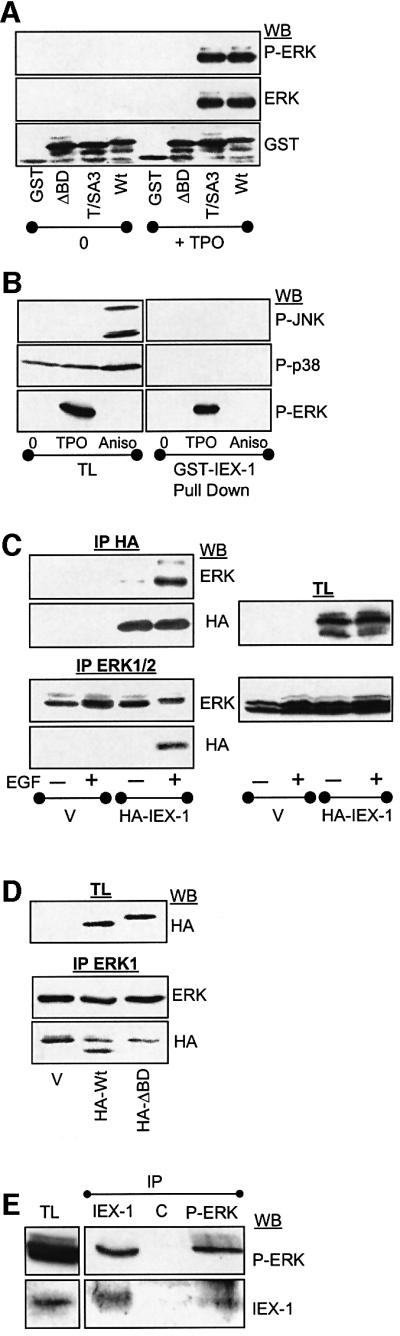
Fig. 3. IEX-1 binds specifically to the active forms of ERK1/2. (A and B) Sepharose-bound GST or GST–IEX-1 wild type and mutants were incubated with lysates from UT7 cells either untreated (0) or stimulated with 10 nM TPO or 100 ng/ml anisomycin (Aniso) for 30 min, as indicated. MAPKs were detected in GST precipitates or in samples of total lysates (TL) by immunoblotting with antibodies directed against the active forms of ERK, JNK or p38. (C and D) Cos7 cells were transfected with 2 µg of pcDNA-HA-IEX-1 (Wt or ΔBD mutant) or empty vector (V), starved overnight and stimulated (+) or not (–) with 100 ng/ml EGF for 10 min. Lysates were immunoprecipitated (IP) with anti-HA or anti-ERK1 antibodies and analyzed by western blotting. Expressions of ERK and HA-IEX-1 are shown in total lysates (TL). (E) UT7 cells (50 × 107) were treated with TPO for 3 h to induce endogenous IEX-1 protein expression. The presence of IEX-1 and phosphorylated ERK was monitored in anti-IEX-1, anti-phosphoERK or control (C) immunoprecipitates (IP), as indicated. As a control, phosphoERK and IEX-1 expressions are shown in total lysates (TL) from 1 × 106 cells.
To examine whether the interaction between IEX-1 and phosphorylated ERKs could occur within the cells, an HA-tagged IEX-1 construct was transiently transfected into Cos7 cells and the presence of ERK in anti-HA immunoprecipitates was analyzed by immunoblotting. As shown in Figure 3C, anti-HA antibodies were found to precipitate endogenous ERK1/2 only from the cells stimulated with EGF prior to lysis. Conversely, HA-IEX-1 was detected in anti-ERK1 immunoprecipitates from EGF-treated cells but not from resting cells. In TPO-stimulated UT7 cells, both endogenous IEX-1 and phosphorylated ERKs were found in the heavy membrane fraction (Figure 2C and D) and, more importantly, the two endogenous proteins could be co-immunoprecipitated from these lysates (Figure 3E). Thus, IEX-1 binds specifically to the active form of ERK1 and ERK2 MAPK in vivo.
Two different MAPK docking site consensus sequences have been identified so far in MAPK substrates: the D-domain and the FXF (or DEF) motif (Jacobs et al., 1999). Analysis of IEX-1 amino acid sequence revealed the presence of the FTF sequence just downstream of the major phosphorylation site T18. To determine whether this sequence could be responsible for the capacity of IEX-1 to bind ERK, we generated a IEX-1 mutant in which the three residues FTF were converted to alanine (Δ binding domain or ΔBD mutant; Figure 1B). In constrast to the wild-type (Wt)-IEX-1, the ΔBD-IEX-1 mutant could not precipitate ERK from stimulated cell lysates in either pull-down (Figure 3A) or co-precipitation assays (Figure 3D). This demonstrates that the FXF site is necessary for the docking of active ERK on IEX-1. Disruption of this site led to a complete loss of phosphorylation of the GST–ΔBD-IEX-1 protein by active ERK2 in vitro (Figure 1A). Thus, phosphorylation of IEX-1 is dependent on its capacity to bind ERK. In contrast, IEX-1 binding to ERK occurs independently of its phosphorylation, as shown by the capacity of Sepharose-bound GST–T/SA3-IEX-1 to pull down ERK as efficiently as Wt-IEX-1 (Figure 3A).
IEX-1 increases ERK activation
While performing ERK/IEX-1 co-precipitation experiments, we noticed that ERK phosphorylation was regularly increased in cells transfected with IEX-1, suggesting that IEX-1 may modulate ERK activity. To analyze this possibility further, CHO cells were transiently transfected with vectors encoding His-tagged IEX-1 and HA-ERK1, and ERK activation was examined in anti-HA immunoprecipitates. Figure 4A shows that HA-ERK1 phosphorylation and kinase activity were significantly increased in cells transfected with Wt-IEX-1 as compared with control cells. In seven independent experiments, IEX-1 increased ERK activity by 20 ± 14-fold (mean ± SE). Transient transfection of IEX-1 into UT7 cells also stimulates endogenous ERK activity, as detected by an increase in Elk1-dependent transcription (data not shown). Likewise, endogenous ERK activity was also increased upon TPO stimulation in two stable clones of UT7 cells expressing different levels of HA-IEX-1 as compared with control cells (Neo) expressing the empty vector (Figure 4B).

Fig. 4. IEX-1 potentiates ERK activity. (A) CHO cells were co-transfected with 1 µg of pcDNA-HA-ERK1 and 1 µg of either empty vector (V) or plasmid encoding His-Wt-IEX, as indicated. Twenty-four hours later, ERK activity was measured in anti-HA immunoprecipitates (IP) by western blotting or kinase assay using MBP as a substrate. (B) Stable clones of UT7 cells expressing different levels of HA-IEX-1 or control cells (Neo) were stimulated for 30 min with TPO and ERK activation was measured by western blotting with anti-phosphoERK antibodies.
To determine whether IEX-1 acts as a constitutive activator of the ERK pathway or can only enhance the signal induced by growth factors, CHO cells expressing the EPO receptor (CHO-ER) were co-transfected with IEX-1 and ERK1 expressing vectors and analyzed after overnight starvation in a serum-free medium and EPO stimulation. No phosphorylation of ERK could be detected in non-stimulated starved cells expressing IEX-1 or the empty vector (Figure 5A). However, IEX-1 expression resulted in a great increase in EPO-induced ERK phosphorylation. Similar results were obtained in Cos7 cells stimulated with EGF (data not shown). The effect of IEX-1 was more pronounced in response to low doses of growth factors, i.e. when ERK activation is not at its maximum (Figure 5A). Kinetic studies showed that IEX-1 expression resulted in a prolonged EPO-induced ERK activation in CHO-ER cells, although the effect of IEX-1 was stronger at short time points of stimulation (Figure 5B).

Fig. 5. Effect of IEX-1 on the kinetics and dose–response of growth factor-mediated ERK activation. CHO-ER cells were transfected with HA-ERK along with either empty pcDNA (V) or pcDNA-HA-IEX-1. Twenty-four hours post-transfection, cells were either harvested directly (non-starved) or starved of serum overnight prior to stimulation with 1–10 U/ml EPO for 10 min (A), or 4 U/ml EPO for various times (B). The activity of HA-tagged ERK was analyzed in anti-HA-immuno precipitates.
The above results, together with the fact that IEX-1 is a growth factor–early gene product (Figure 2B; Charles et al., 1993) suggest that the ERK signaling might be prolonged in cells where endogenous IEX-1 is expressed after growth factor treatment. To test this hypothesis, the effect of siRNA duplexes targeting IEX-1 were examined on ERK phosphorylation at times of TPO stimulation when endogenous IEX-1 protein is not expressed (15 min) or is expressed (3, 10 and 14 h). Owing to the low transfection efficiency of UT7 cells, the effect of IEX-1 siRNA on ERK activation was assessed in cells co-transfected with HA-ERK1 and the siRNA duplex. A duplex of GFP was used as a control. As shown in Figure 6, phosphorylation of HA-ERK at late but not at early time points of TPO stimulation was greatly affected by transfection of the IEX-1 siRNA. The ability of the IEX-1 duplex to efficiently silence IEX-1 expression is indicated by the complete loss of HA-IEX-1 expression in cells co-transfected with HA-IEX-1 and IEX-1 siRNA. These results show that the induction of IEX-1 during TPO stimulation plays an important role in the the capacity of this cytokine to sustain ERK activation in UT7 cells.
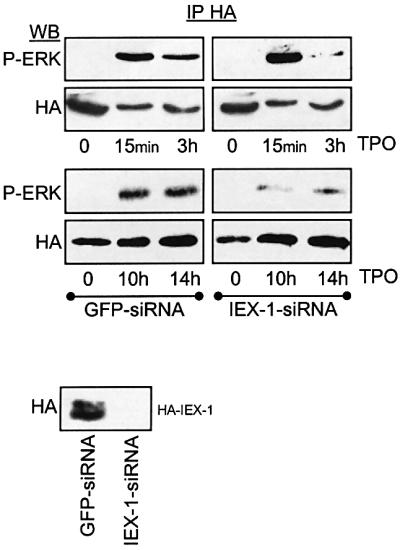
Fig. 6. IEX-1 expression is involved in the long-lasting ERK activation induced by TPO in UT7 cells. UT7 cells were electroporated with plasmids encoding HA-ERK1 (upper panel) or HA-IEX-1 (bottom panel) together with either IEX-1 or GFP siRNA duplexes, as indicated. The activity of HA-ERK was assessed in anti-HA-immunoprecipitates following various times of TPO stimulation.
In contrast to its ability to potentiate ERK activation, IEX-1 did not activate a co-transfected HA-JNK1, nor did it increase its activation by anisomycin (Figure 7A). Since this specific increase in ERK versus JNK activation recalls the ability of IEX-1 to bind ERK specifically, we examined whether ERK-stimulating activity of IEX-1 requires association with ERK. As shown in Figure 7B, the ΔBD-IEX-1 mutant was unable to enhance ERK phosphorylation. In contrast, the T/SA3-IEX-1 mutant (which still binds to ERK) was as efficient as the wild-type protein in stimulating ERK activation. Thus, IEX-1-induced stimulation of ERK activity correlates with its capacity to dock ERK kinases at the DEF motif. This stimulatory effect was specific for IEX-1 rather than a general function of ERK substrates docking ERK through a DEF motif, as shown by the inability of Elk1 (Jacobs et al., 1999) to affect ERK activation when expressed in CHO cells (Figure 7C).
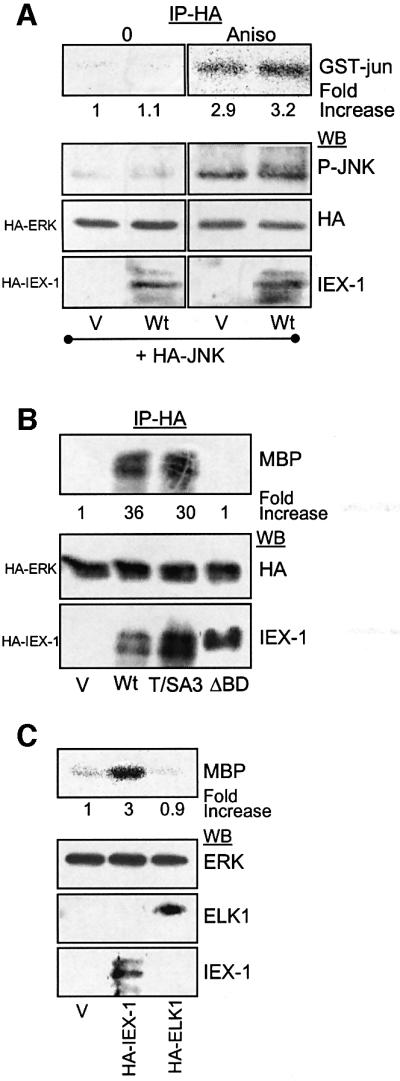
Fig. 7. The ability of IEX-1 to stimulate ERK activity is specific and requires its DEF motif. (A) CHO cells were transfected with HA-IEX-1 together with HA-JNK1 or empty vector (V) and treated or not for 10 min with 100 ng/ml anisomycin. Activated JNK was detected in anti-HA immunoprecipitates by immunoblotting with anti-phospho-JNK antibodies and by in vitro kinase assay using GST–c-jun as substrate. (B and C) CHO cells were transfected with HA-ERK, along with either empty vector (V), plasmids encoding the indicated HA-IEX-1 species or HA-Elk1. ERK activity was determined in anti-HA immunoprecipitates by in vitro kinase assay.
Taken together these data show that IEX-1 is a direct phosphorylation target of ERK that has the ability to potentiate ERK activation in response to various growth factors (serum, EPO, TPO, EGF). The binding of active ERK to the DEF binding site of IEX-1 is necessary for both events to occur.
IEX-1 is an inhibitor of apoptosis induced by various stimuli
Previous studies have described conflicting data on the role of IEX-1 in cellular viability establishing IEX-1 either as an inducer or as an inhibitor of apoptosis (Wu et al., 1998; Arlt et al., 2001; Schilling et al., 2001). The above data showing that IEX-1 is a target of ERKs that potentiates growth factor-induced ERK activation prompted us to re-explore the possible involvement of IEX-1 in the control of apoptosis.
First, we analyzed apoptosis in UT7 cells stably transfected with HA-tagged Wt-IEX-1 or with an empty vector. Death was induced by removing the cytokine (EPO) from the culture medium while maintaining the presence of serum, and was monitored by annexin V–FITC staining (Figure 8A) and by the appearance of active caspase-3 fragments (Figure 8B). Several clones expressing IEX-1 were uniformly more resistant to apoptosis induced by cytokine deprivation than control (Neo) cells, with the extent of protection correlating with the levels of HA-IEX-1 protein.

Fig. 8. IEX-1 inhibits cell death induced by various stimuli. (A and B) Clones of UT7 cells stably transfected with empty (Neo) or HA-IEX-1 (Wt) encoding plasmids were cultured with EPO. Apoptosis was induced by removing EPO from the culture medium and measured by annexin V–FITC staining (A) or immunoblotting with anti-caspase-3 antibodies (B). Anti-HA immunoblots show expression of the transgene in the different clones (lower panels). (C) CHO or (D) HeLa cells were transiently transfected with pEGFP or pEGFP-IEX-1. Twenty-four hours post-transfection, the cells were treated with STS (C) or TNF + CHX (D), as indicated. Apoptosis of the EGFP-positive transfected cells was determined as described in Materials and methods. (D) Mean ± SE of three independent transfections.
The impact of IEX-1 overexpression on cell survival was also explored in cells transiently transfected with EGFP-IEX-1 proteins. As shown in Figure 8C, CHO cells transfected with EGFP-IEX-1 were considerably more resistant to apoptosis induced by staurosporin (STS) than cells expressing EGFP. Likewise, HeLa cells expressing EGFP-IEX-1 were partially protected against death induced by tumor necrosis factor (TNF) in the presence of cycloheximide (CHX) (Figure 8D).
Thus, IEX-1 is able to decrease cell death in response to many stimuli and in different cell types.
IEX-1 phosphorylation at T18 is required for its survival effect
To determine whether IEX-1 phosphorylation by ERK is required for its anti-apoptotic effect, we examined the ability of IEX-1 phosphorylation mutants to decrease cell death in the various apoptotic assays described above. In contrast to the limited cell death observed in cells expressing Wt-IEX-1, apoptosis induced upon cytokine deprivation was similar in UT7 clones expressing HA- T/SA3-IEX-1 and HA-ΔBD-IEX-1, and in the Neo controls (Figure 9A). This indicates that phosphorylation of IEX-1 at ERK consensus sites is required for its pro-survival activity.
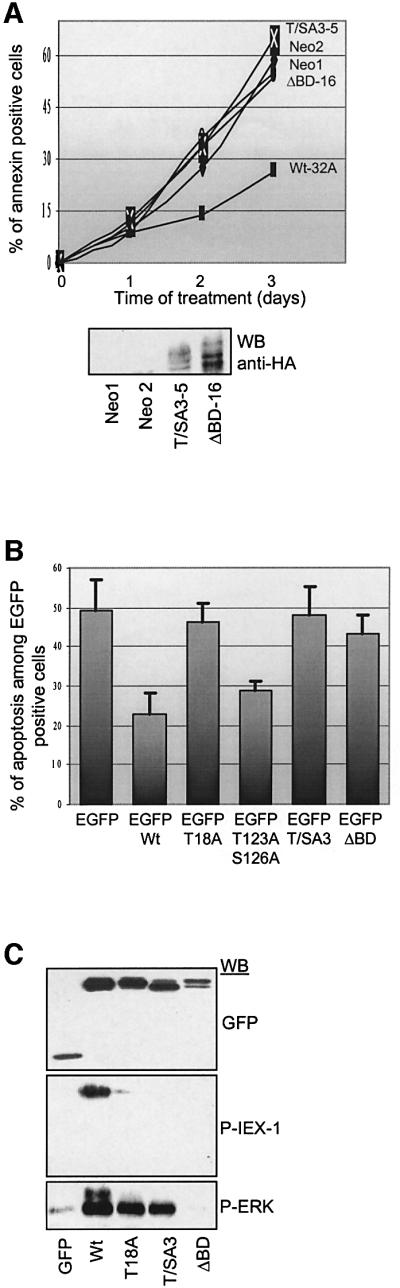
Fig. 9. IEX-1 phosphorylation is required for its survival effect. (A) UT7 clones stably expressing HA-tagged Wt-IEX-1, T/SA3-IEX-1, ΔBD-IEX-1 or an empty vector (Neo) were deprived of cytokine and stained with annexin V–FITC at the indicated times. Similar results were obtained in three independent experiments. (B) CHO cells were transiently transfected with EGFP or EGFP-IEX-1 wild-type or mutant fusion proteins. Apoptosis was induced by treatment with STS for 7 h and measured in EGFP-transfected cells as described in Materials and methods. Results are mean ± SE of values from at least four independent transfections. (C) Total lysates from CHO cells transfected and treated as in (B) were analyzed by western blotting with anti-phosphoIEX-1 and anti-GFP antibodies.
Likewise, in contrast to Wt-IEX-1, transient expression of neither the T/SA3 nor the ΔBD forms of IEX-1 provided protection of CHO cells against STS-triggered apoptosis. The capacity of IEX-1 to inhibit STS-induced death was also completely lost in the T18A single mutant, whereas mutation of T123 and S126 sites did not affect IEX-1 pro-survival activity (Figure 9B). Thus, phosphorylation of IEX-1 at the major ERK phosphorylation site T18 is required for its anti-apoptotic effect.
These results suggest that the anti-apoptotic function of IEX-1 is dependent on the basal ERK activity, as it exists in cells cultured in the presence of FCS, and that this activity is sufficient to allow IEX-1 phosphorylation. Indeed, western blotting of lysates of transfected STS-treated CHO cells revealed the presence of EGFP–Wt-IEX-1 species, which reacted with the anti-phosphoT18 IEX-1 antibodies (Figure 9C). As expected, the anti-phosphoT18 antibody did not react with GFP alone or the EGFP–IEX-1 mutants T18A, T/SA3 and ΔBD-IEX-1. In further support of these results, activated ERK could be detected in STS-treated CHO cells (Figure 10A). This is in agreement with previous studies showing that MEK and ERK are resistant to inhibition by 1 µM STS (Deng et al., 2000). To confirm that activation of the ERK MAPK pathway is controlling the pro-survival activity of IEX-1, CHO cells transfected with EGFP–Wt-IEX-1 were treated with the MEK inhibitor PD98059 prior to induction of cell death by STS. Although it had no pro-apoptotic effect by itself in these cells (data not shown), PD98059 abolished completely both IEX-1 phosphorylation at T18 (Figure 10A) and the capacity of IEX-1 to protect CHO cells against apoptosis (Figure 10B).

Fig. 10. IEX-1 inhibits apoptosis independently of its ability to potentiate ERK activation. (A, B and E) CHO cells were transfected with the indicated plasmids encoding EGFP-tagged wild-type or mutant forms of IEX-1. After a 7 h treatment with STS in the presence or absence of 30 µM PD98059, cells were either lysed for western blotting analysis (A) or fixed for determination of cell death (B and E). Results of apoptosis are expressed as mean ± SE of four independent experiments. (C and D) CHO cells were transfected with HA-IEX-1 wild type or mutants, with (C) or without (D) HA-ERK1. HA immunoprecipitates were subjected to kinase assay and western blotting, as indicated.
The capacity of IEX-1 to potentiate ERK activity is not necessary for its anti-apoptotic effect
Since IEX-1 binds to phosphorylated ERK and increases its activity, one role of IEX-1 in the control of apoptosis might be to bring active ERK close to other substrates involved in cell survival. To test whether the ability of IEX-1 to potentiate ERK activation was necessary for its anti-apoptotic function, we generated new IEX-1 mutants in which phosphorylation was mimicked by negatively charged amino acids (T/SD3 and T18D mutants; Figure 1B). T/SD3-IEX-1 species induced a strong increase in ERK1 phosphorylation (Figure 10C) and co-precipitated with endogenous ERK1 (Figure 10D) when transfected into CHO cells. Similar results were observed for the T18D mutant (data not shown). Both the T/SD3 and the T18D mutants retained the ability to inhibit STS-induced apoptosis in cells in which MAPK activation was impeded by the MEK inhibitor (Figure 10B). This further indicates that IEX-1 phosphorylation at T18 is critical for its pro-survival effect and strongly suggests that its ability to increase the growth factor-induced ERK signal is not required for this process. To confirm these data, mutation of the ERK binding site was inserted into the T/SD3-IEX-1 mutant. The resulting double mutant, T/SD3ΔBD-IEX-1 was unable to potentiate ERK1 activation or to associate with endogenous ERK when transiently expressed in CHO cells (Figure 10C and D). However, this mutant conferred resistance to STS-induced cell death to the same extent as T/SD3-IEX-1 (Figure 10E).
Thus, in different cellular contexts, the ERK MAPKs directly control IEX-1 ability to prevent cell death by catalyzing IEX-1 phosphorylation. Phosphorylation of IEX-1 at ERK target sites is necessary and sufficient to confer anti-apoptotic activity to IEX-1. In contrast, the anti-apoptotic function of IEX-1 is independent of its ability to enhance ERK activity in response to growth factor stimulation.
Discussion
The past few years have brought a considerable advancement in our knowledge of the mechanisms controlling apoptosis by growth factors, and have revealed the existence of tight and unexpected connections between death and signaling pathways. Indeed, not only does it appear that kinase cascades activated by growth factors modulate the function or the expression of proteins directly involved in the control of apoptosis, but also that these proteins can act as positive or negative regulators of signaling modules. Examples include some members of the inhibitor of apoptosis family, which, in addition to their ability to inhibit caspase activation, stimulate NF-κB activity (Hofer-Warbinek et al., 2000), and regulate the extent and duration of JNK activation in response to TNF (Tang et al., 2001) and the direct activation of Raf-1 by the Bcl2-partner Bag1 (Wang et al., 1996). Here we characterize the product of the early gene IEX-1 as a substrate for ERK MAPK and reveal a new connection between apoptosis and the regulation of MAPK signaling that operates directly at the level of this substrate. Association of ERK and IEX-1 results in trans-regulation of their biological activities: ERK-catalyzed phosphorylation of IEX-1 induces IEX-1’s ability to prevent cell death while, by docking active ERKs, IEX-1 provides a positive regulatory loop on the MAPK cascade leading to enhanced ERK activation in response to growth factors.
Phosphorylation of IEX-1 at ERK target sites is necessary and sufficient to confer anti-apoptotic functions to IEX-1. Indeed, mutants of IEX-1 in which phosphorylation was constitutively mimicked (T18D and T/SD3 mutants) protected the cells against STS-induced apoptosis even when the MAPK pathway was inhibited. In addition, mutation of the ERK docking site in this context led to IEX-1 species that remained fully able to confer resistance to cell death, while their capacity to stimulate ERK activity was completely abolished. Thus, the anti-apoptotic function of IEX-1 is independent of its ability to increase ERK activation and cannot be due to ERK-mediated induction and/or phosphorylation of other known ERK targets involved in apoptotic pathways, such as members of the Bcl2 family (Bonni et al., 1999; Breitschopf et al., 2000; Deng et al., 2000; Ballif and Blenis, 2001). How phosphorylation of IEX-1 by ERK may control its anti-apoptotic function is presently unknown, and awaits the elucidation of the molecular mechanism by which IEX-1 acts in the apoptotic pathways. The ability of IEX-1 to inhibit death induced by cytokine removal, STS, TNF or etoposide suggests that it affects a general step in the mitochondrial apoptotic pathway. Indeed, IEX-1 can prevent the release of cytochrome c from mitochondria (Y.Ye and F.Porteu, unpublished data). IEX-1 has no homology with other proteins and does not possess any of the known domains present in effectors or inhibitors of apoptosis. IEX-1 may associate with membranes through its transmembrane domain (Kondratyev et al., 1996). We have found by cell fractionation studies that, in UT7 cells, both endogenous IEX-1 induced by TPO and HA-tagged IEX-1 are located mainly to the membrane fraction enriched in mitochondria. In agreement with previous studies (Kumar et al., 1998; Wu et al., 1998), a small amount of IEX-1 was also detected in the light membrane fraction containing the endoplasmic reticulum and in the nucleus. However, we have been unable to confirm IEX-1 localization by immunofluorescence, probably due to the small amount of IEX-1 protein present in these cells and/or the poor ability of our antibody to recognize the native form of IEX-1. IEX-1 localization is not dependent on its phosphorylation state since it remained unchanged upon activation of the MAPK pathway and was similar for wild-type and T/SA3 forms of IEX-1. The similarities in the localizations of IEX-1 and of some members of the Bcl2 family (Krajewski et al., 1993) suggest that upon ERK-induced phosphorylation IEX-1 might associate with the direct effectors of cell survival. This possibility is currently being studied.
Like most substrates, IEX-1 associates with ERK. ERK binding to IEX-1 requires prior ERK activation, since IEX-1 was found associated with the doubly phosphorylated forms, but not with the resting forms, of ERK1/2. Crystallographic data indicate that the extended substrate-binding region of ERK2 is not regulated by phosphorylation and thus predict that its association with substrates should occur independently of the activation state (Canagarajah et al., 1997). Although this indeed seems to be the case for most ERK substrates for which interaction with ERKs has been studied so far, such a specific affinity for activated ERKs is not without precedent. Notably, it has been described for the binding of ERK2 to Elk1 (Yang et al., 1998). ERKs can be targeted to their substrates through two conserved docking sites: the D-domain, a stretch of positively charged amino acids that is commonly found in MAPK kinases, phosphatases and substrates (Pulido et al., 1998; Gavin and Nebreda, 1999; Jacobs et al., 1999; Smith et al., 1999); or the DEF motif, defined by the conserved FXF consensus sequence that is found in some ERK substrates such as Ksr and Elk1 (Jacobs et al., 1999). These sites can function independently or in combination. In contrast to the D-domain, which binds both JNK and ERK, the DEF motif is specific for ERK. Mutation of the FTF residues to alanine in IEX-1 (ΔBD mutant) led to a complete loss of ERK–IEX-1 association in both pull-down and co-precipitation assays, showing that this sequence represents the unique ERK-interaction motif of IEX-1. This is in agreement with the fact that IEX-1 does not bind JNK MAPK.
Association of IEX-1 with ERK1/2 is essential for subsequent phosphorylation of phosphoacceptor motifs of IEX-1, as shown by the complete loss of IEX-1 phosphorylation upon mutation of the FTF sequence. The concommittant loss of both ERK binding and IEX-1 phosphorylation in the ΔBD mutant is most likely not the result of a gross non-specific alteration of IEX-1 protein, since the constitutively phosphorylated ΔBDT/SD3-IEX-1 mutant remains fully functional for protecting cells from apoptosis. Association of active ERKs with IEX-1 is also required for its ability to trans-regulate ERK activity. However, the mechanism by which this regulation occurs is not yet understood. IEX-1 alone does not stimulate ERK activity in the absence of growth factors. Thus, IEX-1 could act by increasing the rate of ERK activation by its upstream kinases. For example, IEX-1 could be a scaffold protein for the ERK signaling module by facilitating the recruitment of multiple members of the cascade. Some scaffold proteins acting on the JNK or ERK pathways, such as JSAP or Ksr, respectively, are themselves substrates of MAPK (Ito et al., 1999; Jacobs et al., 1999; Stewart et al., 1999). However, the capacity of IEX-1 to bind only to activated ERKs is not in favor of such a function. In addition, we were not able to detect any association of IEX-1 with MEK or any effect of recombinant IEX-1 on ERK activation induced in vitro by activated MEK (F.Porteu, unpublished data). Alternatively, IEX-1 could stabilize the kinase in an active state by preventing its inactivation. Amongst the mechanisms that could affect ERK activation are those involving the retro-phosphorylation and inhibition of SOS and MEK (Brunet et al., 1994; Waters et al., 1995). These mechanisms, however, most probably do not contribute to the IEX-1-induced increase in ERK phosphorylation, since IEX-1 was found not to affect MEK activation (F.Porteu, unpublished data). ERK inactivation can also occur upon dephosphorylation of either one or both of its phosphorylated residues by phosphatases. Just as for MAPK substrates and upstream kinases, the MAPK phosphatases bind to ERKs, and this interaction is required for the catalytic activation of the phosphatase and ERK inactivation (Camps et al., 1998; Pulido et al., 1998; Zuniga et al., 1999; Tanoue et al., 2000; Slack et al., 2001). Thus, IEX-1 could compete with MAPK phosphatases for the binding to the kinase. However, the inability of IEX-1 to protect ERK from inactivation induced by overexpression of MKP1 or MKP3 in CHO cells renders this possibility unlikely. Finally, no change in the subcellular localization of phosphorylated ERK was observed upon expression of wild-type IEX-1 or ΔBD-IEX-1 (P.Lenormand, personal communication), suggesting that IEX-1 does not influence the exent of ERK activation by sequestering ERKs in a cellular compartment that is not accessible to inactivators. Whatever the mechanism involved, preventing IEX-1 expression during TPO stimulation through siRNA greatly affected the capacity of TPO to sustain ERK signaling in UT7 cells. Since IEX-1 is also induced by other growth factors (Charles et al., 1993), potentiation of ERK activation following IEX-1 expression may represent an important means by which various growth factors can regulate the amplitude and the duration of ERK signaling.
Conflicting data have been reported concerning the role of IEX-1 in both apoptosis and cell proliferation. Indeed, IEX-1 was found to increase apoptosis induced by UV radiation, TNF, or upon serum deprivation (Segev et al., 2000; Arlt et al., 2001; Schilling et al., 2001), but it also plays a key role in cellular resistance to TNF and Fas-induced apoptosis (Wu et al., 1998; Domachowske et al., 2000). In agreement with the latter studies, we observed that IEX-1 protects different cell types against various apoptotic triggers. Also supporting this anti-apoptotic role is a recent study showing that transgenic mice expressing IEX-1 in lymphocytes develop an auto-immune disease due to a striking deficiency in apoptosis of activated T cells (Zhang et al., 2002). Our finding that the IEX-1 anti-apoptotic function is activated by the MAPK pathway and is dependent on the presence of at least some active ERK in the cells may reconcile, at least in part, these contradictory data. Supporting this conclusion, IEX-1-induced increase in susceptibility to death has been observed in serum-deprived cells or upon cellular stress, conditions that probably prevent any activation of the ERK pathway, and is abolished in the presence of serum (Arlt et al., 2001; Schilling et al., 2001). On the other hand, IEX-1 has been shown to contribute to proliferation in vivo and to accelerate cell cycle progression upon serum stimulation in some cells in vitro (Schafer et al., 1996; Kumar et al., 1998; Arlt et al., 2001). Given the well known role of the ERK pathway in controlling proliferation, the ability of IEX-1 to potentiate ERK activity demonstrated here provides a potential molecular mechanism for IEX-1 growth promoting action.
IEX-1 expression is transcriptionally regulated by NF-κB and is involved in the NF-κB-dependent resistance to apoptosis (Wu et al., 1998; Zhang et al., 2002). Our results show that IEX-1 is also a target of regulation by ERK, suggesting that these two pathways could synergize to induce full protection against apoptosis in some cells. By stimulating ERK activation, IEX-1 could also contribute to the control of cell growth by NF-κB (Guttridge et al., 1999). Increased expression or subvertion of IEX-1 activities in tumor cells may result in deficient apoptosis and/or enhanced proliferation and contribute to malignant transformation.
Materials and methods
Antibodies
Antibodies against IEX-1 were generated by immunizing rabbits with recombinant full-length GST–IEX-1 fusion protein purified on glutathione–Sepharose (Amersham Pharmacia Biotech). Antibodies recognizing specifically IEX-1 phosphorylated at T18 were produced by injecting rabbits with the phosphopeptide P-T18 (CMTILQAPp TPAPSTI) coupled to KLH (Genosphere Biotechnologies). Other antibodies used were from commercial sources: anti-phospho antibodies to p38, JNK and ERK (Cell Signaling Technology); anti-p38, anti-ERK1 and anti-JNK (Santa Cruz Biotechnology, Inc.); anti-caspase-3, anti-cytochrome c and anti-PARP (PharMingen International); anti-HA (12CA5 and 3F10) (Boehringer Mannheim).
Production and purification of active recombinant ERK2
The biscistronic vector pETHis6/MEK1-R4F (Khokhlatchev et al., 1997), allowing expression of His-tagged-rat ERK2 in bacteria, is a generous gift from Dr M.Cobb (University of Texas Southwestern Medical Center, Dallas, TX). Recombinant doubly phosphorylated His6-ERK2 was purified as described previously (Khokhlatchev et al., 1997).
Cell culture
The cytokine-dependent human megakaryoblastic cell line expressing the TPO receptor (UT7) in which TPO can promote both proliferation and differentiation signals has been described previously (Rouyez et al., 1997). It was routinely cultured in α-minimum essential medium supplemented with 10% FCS and 1 U/ml EPO (Boehringer Mannheim). For TPO stimulation, the cells were washed three times and switched to complete medium containing 10 nM TPO-mimetic peptide (Garcia et al., 2001). CHO cells were cultured in Dulbecco’s MEM nut Mix F-12 (HAM) medium containing 7% FCS. Cos7 and HeLa cells were maintained in Dulbecco’s MEM supplemented with 10% FCS. Stable clones of CHO-ER cells were a gift from C.Guillard (Institut Cochin, Paris, France).
Tranfections and transfection-based assays
Stable clones of UT7 cells expressing HA-tagged wild-type or mutant forms of IEX-1 were generated by electroporation and selected in G418 as described previously (Rouyez et al., 1997). To test the effect of IEX-1 siRNA on TPO-induced ERK activation, UT7 cells were electroporated with 1 µg of IEX-1 or GFP siRNA duplexes (Xeragon, Inc.), along with 5 µg of HA-ERK. Eight to 12 h post-transfection, the cells were stimulated for various times with TPO and ERK activity was analyzed in anti-HA immunoprecipitates. Transient transfections into CHO, Cos7 and HeLa cells were performed with LipofectAMINE plus reagent (Life Technologies) according to the manufacturer’s instructions. In some experiments, 24 h post-transfection, cells were deprived of serum overnight, and stimulated with 10 ng/ml EGF (PeproTech, Inc.) or 1–10 U/ml EPO for various times before lysis and immunoprecipitation.
Construction of λGEX5 UT7 cDNA library and screening with active ERK2
The phage expression vector λGEX5, a λgt11-derived vector modified by insertion of pGEX-PUC-3T plasmid nucleotide sequence (Fukunaga and Hunter, 1997), was a generous gift from T.Hunter (The Salk Institute, San Diego, CA). A cDNA library was constructed from UT7 cells cultured for 12 h in the presence of TPO. Poly(A)+ RNA was obtained using a mRNA separator kit (Clontech) and double-stranded cDNA was synthesized with oligo(dT) primers using the Superscript choice system kit (Gibco-BRL). cDNA was sized-fractionated on sucrose gradient and cDNA over 1 kb was ligated to λGEX5 arms and packaged into bacteriophage λ particles. The cDNA library was amplified by growth in Escherichia coli BB4 strain and plated at a density of 1.5 × 104 plaques per 150 mm agar plates. After 3.5 h at 37°C, the plates were overlaid with nitrocellulose impregnated with 10 mM IPTG and returned to 37°C overnight. Filters were saturated and reacted with 1 µg/ml purified recombinant active ERK2 and radiolabeled ATP as described previously (Fukunaga and Hunter, 1997). After secondary screening, phage DNA was prepared and the cDNA-containing plasmid was recovered and transformed into bacteria. The insert DNA sequence was determined and analyzed with the BLAST program. Of the positive clones isolated, one contained the entire coding sequence of IEX-1 (Kondratyev et al., 1996).
Constructs
pcDNA3 expression plasmids encoding HA-tagged ERK1, JNK1 and Elk1 were gifts from Drs J.Pouyssegur, B.Derijard (UMR CNRS 6543 and 6548; Nice, France) and R.Hipskind (IGMM, Montpellier, France), respectively. Forward and reverse primers containing BamHI and XbaI restriction sites, were used to amplify the pGEX-PUC-3T-IEX-1 cDNA by PCR. This fragment was subcloned into pcDNA3-HA, pcDNA3-His (Invitrogen) and pEGFP-C1 (Clontech) to generate IEX-1 tagged at its N-terminus with HA, His and EGFP, respectively. Mutants of each IEX-1 phosphorylation site and ERK binding site were generated by PCR and subcloned in pGEX4T3, pCDNA3-HA and pEGFP-C1.
Cell death assays
In the UT7 stable clones expressing wild-type or mutant forms of IEX-1, apoptosis was induced by withdrawing the cytokine from the medium: cells were washed three times in serum-free αMEM and resuspended at 1 × 105 cells/ml in medium supplemented with 10% FCS in the absence of EPO or TPO. The cells were harvested at various time intervals, stained with annexin V–FITC (Immunotech, France) and analyzed by flow cytometry. Cleavage of pro-caspase-3 was assessed in the same samples by visualizing the appearance active fragments upon western blotting of total cell lysates with anti-caspase-3 antibodies. CHO cells were seeded on 4 cm2 Labteck slides and transfected with 0.7 µg of EGFP-C1 or EGFP-IEX-1 plasmids. Alternatively, cells were co-transfected with pcDNA plasmids encoding IEX-1 (0.5 µg) and empty EGFP vector (0.2 µg). One day following transfection, cells were treated with staurosporin (1 µM; Sigma) for 4–7 h. After three washes in PBS, adherent cells were fixed with 4% paraformaldehyde and stained with DAPI to assess nuclear morphology. The percentage of GFP-positive cells with fragmented or condensed nuclei was determined by blind counting of at least 500 GFP-positive cells under a fluorescence microscope. To measure apoptosis in HeLa cells, cells were seeded into 6-well tissue culture dishes, transfected with EGFP or EGFP-IEX-1 encoding plasmids. One day later cells were subjected to TNF (20 ng/ml; PeproTech, Inc.) alone or together with CHX (30 µg/ml) for 8 h. After three washes with PBS to remove floating apoptotic cells, the remaining adherent (viable) cells were fixed and stained with DAPI. GFP-positive cells were counted from a minimum of 20 fields chosen at random.
Immunoprecipitation and western blotting
Transfected CHO and Cos cells were harvested 24 h after transfection and lysed in 50 mM Tris pH 7.5 buffer containing 137 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, 1 mM orthovanadate, 25 mM β-glycerophosphate and a protease inhibitor cocktail (Roche). For co-immunoprecitation of endogenous ERKs and IEX-1 proteins, 5 × 107 UT7 cells were treated with TPO for 3 h to induce expression of endogenous IEX-1 and ERK activation prior to lysis. In some experiments, the proteasome inhibitors Mg132 or LLnL were added during TPO stimulation to prevent IEX-1 degradation. Clear lysates were obtained by centrifugation and immunoprecipitation, gel electrophoresis and immunoblotting were performed as described previously (Garcia et al., 2001).
Subcellular fractionation
Parental UT7 cells or clones expressing HA-IEX-1 wild type or mutant were treated for 3 h with TPO to induce endogenous IEX-1 expression. Homogenization was performed by passing the cells through a ball-bearing homogenizer (cell cracker; EMBL) as described previously (Meunier et al., 2002).
GST phosphorylation and pull-down assays
One to three micrograms of purified GST fusion proteins for wild-type or mutant forms of IEX-1 were incubated with 0.5 µg of active ERK in 50 µl of kinase buffer (HEPES 10 mM, pH 7.5, 10 mM MgCl2 and 1 mM dithiothreitol) in the presence of 50 µM ATP and 3 µCi of [γ-32P]ATP for 20 min at 30°C. The products were resolved on polyacrylamide gels and transferred to nitrocellulose membranes. Radioactive bands were visualized by autoradiography and quantitated by phosphoimaging. For pull-down assays, GST–IEX-1 fusion proteins bound to glutathione– Sepharose (20 µg) were incubated for 2 h at 4°C with lysates from UT7 cells (107) that had been stimulated or not with various agents known to activate ERK, p38 or JNK MAPK. The beads were then washed four times in lysis buffer and sample analyzed by western blotting with antibodies directed against the active forms of these kinases.
Aknowledgements
We are grateful to Drs T.Hunter, M.Cobb, N.Ahn, J.Pouyssegur, B.Dérijard and R.Hipskind for the gifts of expression plasmids, and C.Guillard for CHO-ER cells. We also wish to thank Dr P.Lenormand (CRNS 6543, Nice, France) for personal communication of data, and Drs V.Duprez and S.Gisselbrecht for careful reading of the manuscript. This work was supported by the Institut National de la Santé et de la Recherche Médicale and by a grant from the Ligue Nationale Contre le Cancer (associated laboratory). J.G. is supported by a fellowship from the Ligue Nationale Contre le Cancer.
References
- Arlt A., Grobe,O., Sieke,A., Kruse,M.L., Folsch,U.R., Schmidt,W.E. and Schafer,H. (2001) Expression of the NF-κ B target gene IEX-1 (p22/PRG1) does not prevent cell death but instead triggers apoptosis in HeLa cells. Oncogene, 20, 69–76. [DOI] [PubMed] [Google Scholar]
- Ballif B.A. and Blenis,J. (2001) Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ., 12, 397–408. [PubMed] [Google Scholar]
- Ben-Levy R., Hooper,S., Wilson,R., Paterson,H.F. and Marshall,C.J. (1998) Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol., 8, 1049–1057. [DOI] [PubMed] [Google Scholar]
- Bonni A., Brunet,A., West,A.E., Datta,S.R., Takasu,M.A. and Greenberg,M.E. (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science, 286, 1358–1362. [DOI] [PubMed] [Google Scholar]
- Breitschopf K., Haendeler,J., Malchow,P., Zeiher,A.M. and Dimmeler,S. (2000) Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway. Mol. Cell. Biol., 20, 1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brondello J.M., Pouyssegur,J. and McKenzie,F.R. (1999) Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science, 286, 2514–2517. [DOI] [PubMed] [Google Scholar]
- Brunet A., Pages,G. and Pouyssegur,J. (1994) Growth factor-stimulated MAP kinase induces rapid retrophosphorylation and inhibition of MAP kinase kinase (MEK1). FEBS Lett., 346, 299–303. [DOI] [PubMed] [Google Scholar]
- Brunet A., Roux,D., Lenormand,P., Dowd,S., Keyse,S. and Pouyssegur,J. (1999) Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J., 18, 664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M., Nichols,A., Gillieron,C., Antonsson,B., Muda,M., Chabert,C., Boschert,U. and Arkinstall,S. (1998) Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science, 280, 1262–1265. [DOI] [PubMed] [Google Scholar]
- Canagarajah B.J., Khokhlatchev,A., Cobb,M.H. and Goldsmith,E.J. (1997) Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell, 90, 859–869. [DOI] [PubMed] [Google Scholar]
- Chang L. and Karin,M. (2001) Mammalian MAP kinase signalling cascades. Nature, 410, 37–40. [DOI] [PubMed] [Google Scholar]
- Charles C.H., Yoon,J.K., Simske,J.S. and Lau,L.F. (1993) Genomic structure, cDNA sequence and expression of gly96, a growth factor-inducible immediate-early gene encoding a short-lived glycosylated protein. Oncogene, 8, 797–801. [PubMed] [Google Scholar]
- Countaway J.L., Nairn,A.C. and Davis,R.J. (1992) Mechanism of desensitization of the epidermal growth factor receptor protein-tyrosine kinase. J. Biol. Chem., 267, 1129–1140. [PubMed] [Google Scholar]
- Deng X., Ruvolo,P., Carr,B. and May,W.S.,Jr (2000) Survival function of ERK1/2 as IL-3-activated, staurosporine-resistant Bcl2 kinases. Proc. Natl Acad. Sci. USA, 97, 1578–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domachowske J.B., Bonville,C.A., Mortelliti,A.J., Colella,C.B., Kim,U. and Rosenberg,H.F. (2000) Respiratory syncytial virus infection induces expression of the anti-apoptosis gene IEX-1L in human respiratory epithelial cells. J. Infect. Dis., 181, 824–830. [DOI] [PubMed] [Google Scholar]
- Fukuda M., Gotoh,Y. and Nishida,E. (1997) Interaction of MAP kinase with MAP kinase kinase: its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J., 16, 1901–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga R. and Hunter,T. (1997) MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J., 16, 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaits F., Degols,G., Shiozaki,K. and Russell,P. (1998) Phosphorylation and association with the transcription factor Atf1 regulate localization of Spc1/Sty1 stress-activated kinase in fission yeast. Genes Dev., 12, 1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J., de Gunzburg,J., Eychene,A., Gisselbrecht,S. and Porteu,F. (2001) Thrombopoietin-mediated sustained activation of extracellular signal-regulated kinase in UT7-Mpl cells requires both Ras-Raf-1- and Rap1-B-Raf-dependent pathways. Mol. Cell. Biol., 21, 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A.C. and Nebreda,A.R. (1999) A MAP kinase docking site is required for phosphorylation and activation of p90rsk/MAPKAP kinase-1. Curr. Biol., 9, 281–284. [DOI] [PubMed] [Google Scholar]
- Graves L.M. et al. (2000) Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature, 403, 328–332. [DOI] [PubMed] [Google Scholar]
- Guttridge D.C., Albanese,C., Reuther,J.Y., Pestell,R.G. and Baldwin,A.S.,Jr (1999) NF-κB controls cell growth and differ entiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol., 19, 5785–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer-Warbinek R., Schmid,J.A., Stehlik,C., Binder,B.R., Lipp,J. and de Martin,R. (2000) Activation of NF-κB by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J. Biol. Chem., 275, 22064–22068. [DOI] [PubMed] [Google Scholar]
- Ito M. et al. (1999) JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol. Cell. Biol., 19, 7539–7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D., Glossip,D., Xing,H., Muslin,A.J. and Kornfeld,K. (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev., 13, 163–175. [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev A., Xu,S., English,J., Wu,P., Schaefer,E. and Cobb,M.H. (1997) Reconstitution of mitogen-activated protein kinase phosphorylation cascades in bacteria. Efficient synthesis of active protein kinases. J. Biol. Chem., 272, 11057–11062. [DOI] [PubMed] [Google Scholar]
- Kondratyev A.D., Chung,K.N. and Jung,M.O. (1996) Identification and characterization of a radiation-inducible glycosylated human early-response gene. Cancer Res., 56, 1498–1502. [PubMed] [Google Scholar]
- Krajewski S., Tanaka,S., Takayama,S., Schibler,M.J., Fenton,W. and Reed,J.C. (1993) Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum and outer mitochondrial membranes. Cancer Res., 53, 4701–4714. [PubMed] [Google Scholar]
- Kumar R., Kobayashi,T., Warner,G.M., Wu,Y., Salisbury,J.L., Lingle,W. and Pittelkow,M.R. (1998) A novel immediate early response gene, IEX-1, is induced by ultraviolet radiation in human keratinocytes. Biochem. Biophys. Res. Commun., 253, 336–341. [DOI] [PubMed] [Google Scholar]
- Lenormand P., Brondello,J.M., Brunet,A. and Pouyssegur,J. (1998) Growth factor-induced p42/p44 MAPK nuclear translocation and retention requires both MAPK activation and neosynthesis of nuclear anchoring proteins. J. Cell Biol., 142, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis T.S., Hunt,J.B., Aveline,L.D., Jonscher,K.R., Louie,D.F., Yeh,J.M., Nahreini,T.S., Resing,K.A. and Ahn,N.G. (2000) Identification of novel MAP kinase pathway signaling targets by functional proteomics and mass spectrometry. Mol. Cell, 6, 1343–1354. [DOI] [PubMed] [Google Scholar]
- Marais R., Wynne,J. and Treisman,R. (1993) The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell, 73, 381–393. [DOI] [PubMed] [Google Scholar]
- Marshall C.J. (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell, 80, 179–185. [DOI] [PubMed] [Google Scholar]
- Meunier C., Bordereaux,D., Porteu,F., Gisselbrecht,S., Chretien,S. and Courtois,G. (2002) Cloning and characterization of a family of proteins associated with Mpl. J. Biol. Chem., 277, 9139–9147. [DOI] [PubMed] [Google Scholar]
- Pulido R., Zuniga,A. and Ullrich,A. (1998) PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J., 17, 7337–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouyez M.C., Boucheron,C., Gisselbrecht,S., Dusanter-Fourt,I. and Porteu,F. (1997) Control of thrombopoietin-induced megakaryocytic differentiation by the mitogen-activated protein kinase pathway. Mol. Cell. Biol., 17, 4991–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer H.J. and Weber,M.J. (1999) Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol., 19, 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer H., Trauzold,A., Siegel,E.G., Folsch,U.R. and Schmidt,W.E. (1996) PRG1: a novel early-response gene transcriptionally induced by pituitary adenylate cyclase activating polypeptide in a pancreatic carcinoma cell line. Cancer Res., 56, 2641–2648. [PubMed] [Google Scholar]
- Schafer H., Trauzold,A., Sebens,T., Deppert,W., Folsch,U.R. and Schmidt,W.E. (1998) The proliferation-associated early response gene p22/PRG1 is a novel p53 target gene. Oncogene, 16, 2479–2487. [DOI] [PubMed] [Google Scholar]
- Schilling D., Pittelkow,M.R. and Kumar,R. (2001) IEX-1, an immediate early gene, increases the rate of apoptosis in keratinocytes. Oncogene, 20, 7992–7997. [DOI] [PubMed] [Google Scholar]
- Segev D.L., Ha,T.U., Tran,T.T., Kenneally,M., Harkin,P., Jung,M., MacLaughlin,D.T., Donahoe,P.K. and Maheswaran,S. (2000) Mullerian inhibiting substance inhibits breast cancer cell growth through an NF-κB-mediated pathway. J. Biol. Chem., 275, 28371–28379. [DOI] [PubMed] [Google Scholar]
- Slack D.N., Seternes,O.M., Gabrielsen,M. and Keyse,S.M. (2001) Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J. Biol. Chem., 276, 16491–16500. [DOI] [PubMed] [Google Scholar]
- Smith J.A., Poteet-Smith,C.E., Malarkey,K. and Sturgill,T.W. (1999) Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J. Biol. Chem., 274, 2893–2898. [DOI] [PubMed] [Google Scholar]
- Stefanovsky V.Y., Pelletier,G., Hannan,R., Gagnon-Kugler,T., Rothblum,L.I. and Moss,T. (2001) An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol. Cell, 8, 1063–1073. [DOI] [PubMed] [Google Scholar]
- Stewart S., Sundaram,M., Zhang,Y., Lee,J., Han,M. and Guan,K.L. (1999) Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol. Cell. Biol., 19, 5523–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill T.W., Ray,L.B., Erikson,E. and Maller,J.L. (1988) Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature, 334, 715–718. [DOI] [PubMed] [Google Scholar]
- Tang G., Minemoto,Y., Dibling,B., Purcell,N.H., Li,Z., Karin,M. and Lin,A. (2001) Inhibition of JNK activation through NF-κB target genes. Nature, 414, 313–317. [DOI] [PubMed] [Google Scholar]
- Tanoue T., Adachi,M., Moriguchi,T. and Nishida,E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol., 2, 110–116. [DOI] [PubMed] [Google Scholar]
- Traverse S., Gomez,N., Paterson,H., Marshall,C. and Cohen,P. (1992) Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem. J., 288, 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman R. (1996) Regulation of transcription by MAP kinase cascades. Curr. Opin. Cell Biol., 8, 205–215. [DOI] [PubMed] [Google Scholar]
- Wang H.G., Takayama,S., Rapp,U.R. and Reed,J.C. (1996) Bcl-2 interacting protein, BAG-1, binds to and activates the kinase Raf-1. Proc. Natl Acad. Sci. USA, 93, 7063–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters S.B., Holt,K.H., Ross,S.E., Syu,L.J., Guan,K.L., Saltiel,A.R., Koretzky,G.A. and Pessin,J.E. (1995) Desensitization of Ras activation by a feedback disassociation of the SOS–Grb2 complex. J. Biol. Chem., 270, 20883–20886. [DOI] [PubMed] [Google Scholar]
- Wu M.X., Ao,Z., Prasad,K.V., Wu,R. and Schlossman,S.F. (1998) IEX-1L, an apoptosis inhibitor involved in NF-κB-mediated cell survival. Science, 281, 998–1001. [DOI] [PubMed] [Google Scholar]
- Yang S.H., Yates,P.R., Whitmarsh,A.J., Davis,R.J. and Sharrocks,A.D. (1998) The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol. Cell. Biol., 18, 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Schlossman,S.F., Edwards,R.A., Ou,C.N., Gu,J. and Wu,M.X. (2002) Impaired apoptosis, extended duration of immune responses and a lupus-like autoimmune disease in IEX-1-transgenic mice. Proc. Natl Acad. Sci. USA, 99, 878–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga A., Torres,J., Ubeda,J. and Pulido,R. (1999) Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides substrate specificity and retains ERK2 in the cytoplasm. J. Biol. Chem., 274, 21900–21907. [DOI] [PubMed] [Google Scholar]