

Abstract
NF-κB is a critical target of signaling downstream of the T cell receptor (TCR) complex, but how TCR signaling activates NF-κB is poorly understood. We have developed an expression cloning strategy that can identify catalytic and noncatalytic molecules that participate in different pathways of NF-κB activation. Screening of a mouse thymus cDNA library yielded CARD11, a membrane-associated guanylate kinase (MAGUK) family member containing CARD, PDZ, SH3 and GUK domains. Using a CARD-deleted variant of CARD11 and RNA interference (RNAi), we demonstrate that CARD11 mediates NF-κB activation by αCD3/αCD28 cross-linking and PMA/ionomycin treatment, but not by TNFα or dsRNA. CARD11 is not required for TCR-mediated induction of NFAT or AP-1. CARD11 functions upstream of the IκB-kinase (IKK) complex and cooperates with Bcl10 in a CARD domain-dependent manner. RNAi-rescue experiments suggest that the CARD, coiled-coil, SH3 and GUK domains of CARD11 are critical for its signaling function. These results implicate CARD11 in factor- specific activation of NF-κB by the TCR complex and establish a role for a MAGUK family member in antigen receptor signaling.
Keywords: CARD11/expression cloning/MAGUK/NF-κB/RNAi
Introduction
The NF-κB transcription factor is rapidly activated by diverse stimuli that alert a cell or organism to stressful or infectious conditions (Ghosh et al., 1998). These include UV and γ-irradiation, bacterial and viral products (e.g. lipopolysaccharide and dsRNA), pro-inflammatory cytokines (e.g. TNFα and IL-1), antigen recognition by the T and B cell receptor complexes, and apoptotic and necrotic stimuli. NF-κB regulates many genes involved in the development and function of the immune response, inflammation, cell growth control and anti-apoptotic responses.
Many stimuli activate NF-κB by causing the phosphorylation and destruction of IκBs, inhibitory molecules that retain NF-κB in the cytoplasm. The signal-induced phosphorylation of IκBs is accomplished by the IκB-kinase (IKK) complex, which is composed of two kinase subunits, IKKα and IKKβ, and a noncatalytic subunit, NEMO/IKKγ (Karin and Ben-Neriah, 2000). Phos phorylated IκB is ubiquitylated and degraded by the 26S proteasome, allowing NF-κB to translocate to the nucleus to activate target genes. The IKK complex is activated by many stimuli, but the precise mechanism in most cases has not been firmly established.
Mice deficient in NF-κB subunits, or in molecules that signal to NF-κB, have revealed that the proper regulation of NF-κB is critical for normal innate and adaptive immune responses (Gerondakis et al., 1999). In the adaptive immune response, NF-κB is a critical target of antigen receptor signaling in B and T cells. In T cells, engagement of the T cell receptor (TCR) complex in concert with costimulatory signals (CD28) leads to the activation of multiple signaling cascades which induce programs of gene expression through several transcription factors including NFAT, AP-1 and NF-κB. These signaling responses are required for normal T cell development, proliferation and activation (Alberola-Ila et al., 1997). Genetic studies have determined that NF-κB plays an important cell autonomous role in these processes. T cells lacking c-Rel, RelA or IKKβ develop, but fail to proliferate normally in response to TCR/CD28 costimulation (Kontgen et al., 1995; Doi et al., 1997; Senftleben et al., 2001). In addition, the properties of mice transgenic for non-degradable forms of IκB have suggested a role for NF-κB in TCR-mediated thymocyte selection and in pre-TCR survival signals (Hettmann and Leiden, 2000; Voll et al., 2000).
The mechanism by which TCR signaling activates the IKK complex is poorly understood. Several molecules are required, including the ZAP70 tyrosine kinase, the SLP-76 adapter and the Vav GTP/GDP exchange factor (Costello et al., 1999; Herndon et al., 2001). These proteins participate in TCR-proximal events, signal to several targets and are not involved exclusively in NF-κB activation. Protein kinase CΘ (PKCΘ) acts downstream of these molecules and is required in mature thymocytes for both NF-κB and AP-1 activation (Sun et al., 2000). A specific role in NF-κB induction by TCR triggering has been demonstrated for Bcl10, a caspase-recruitment domain (CARD)-containing protein first identified in the t(1;14)(p22;q32) translocation associated with B cell lymphomas of mucosa-associated lymphoid tissue (MALT; Willis et al., 1999; Zhang et al., 1999). This translocation places the bcl10 gene in the immunoglobulin heavy chain locus and results in its overexpression, which may provide an anti-apoptotic advantage via NF-κB activation. Mice deficient in Bcl10 are severely immunodeficient due to defects in antigen receptor-induced lymphocyte activation and NF-κB induction (Ruland et al., 2001). Although Bcl10 has been shown to act upstream of the IKK complex, its mechanism of action has not been elucidated.
As an approach to increasing our understanding of the regulation and biological roles of NF-κB, we have been interested in developing new methods for identifying signaling molecules in NF-κB-inducing pathways. Here we describe an expression cloning strategy based upon the observation that several known signaling molecules in NF-κB-inducing pathways will activate NF-κB when overexpressed in tissue culture cells. In this method, a cDNA expression library is subdivided into pools, each of which is assayed for the ability to activate an NF-κB-responsive reporter after transfection into cells. Positive pools are assayed in secondary screens to confirm their specificity and the clone responsible for an interesting pool’s activity is purified by sib selection.
We demonstrate that this strategy identifies molecules of different biochemical types and can be used to isolate components of multiple signaling pathways. Using a mouse thymus expression library we have isolated the murine CARD11 cDNA. We present evidence that CARD11 cooperates with Bcl10 and functions between the TCR complex and the IKK complex in the activation of NF-κB in T cells.
Results
Establishment of the expression cloning screen
For efficient and economical screening, we used a quantitative and highly sensitive reporter assay for NF-κB activation. In this assay, pool DNA cloned into an expression vector driven by the cytomegalovirus (CMV) promoter was transiently transfected into 293T cells with the Igκ2-IFN-LUC reporter, which contained two copies of the immunoglobulin κ light chain κB site (5′-GGGGACTTTCC-3′) upstream of the interferon-β minimal promoter (–55 to +19) (Fujita et al., 1987) driving luciferase expression. For normalization of transfection efficiency and extract recovery, the transfection included the pCSK-lacZ vector (Condie et al., 1990), which constitutively expresses β-galactosidase and is unaffected by NF-κB. To maximize the number of cDNAs that could be assayed, it was important to determine what complexity (number of cDNAs per pool) would allow reliable detection of a single active clone in a mixture of cDNAs. Pilot experiments using the cDNA for TRAF2, an adapter protein in the TNFα pathway (Rothe et al., 1995), suggested that a pool complexity of 100 cDNAs would allow detection of molecules possessing 3-fold lower specific activity than TRAF2 in this assay (data not shown). The sensitivity of detection of luciferase and β-galactosidase activities allowed us to scale down the size of the transfection and to minimize the amount of pool DNA required (see Materials and methods).
To test this methodology, a portion of an arrayed human placenta cDNA expression library was subdivided into 561 pools of ∼100 cDNA complexity. Plasmid DNA from each pool was assayed and pools were considered positive if they activated the reporter 3-fold or more relative to the activity observed with the empty expression plasmid pcDNA3. In addition, some pools were considered positive if they were 3-fold or more active than the average activity of a cohort of pools assayed in parallel. Positive pools were reassayed and their ability to activate the Igκ2-IFN-LUC reporter confirmed. Of the 561 pools assayed in this way, 41 were positive by these criteria, ranging in fold activation from 2.3-fold to 256-fold. An example of primary screening is shown in Figure 1A, in which 67 pools were assayed and those considered positive were pools 10 (4.5-fold), 12 (4.4-fold), 24 (16.7-fold), and 52 (8.3-fold).

Fig. 1. The expression cloning strategy. (A) Primary screening of pools. Pool DNA was transfected into 293T cells with the Igκ2-IFN-LUC reporter and the pCSK-lacZ control vector and fold stimulation was determined as described in Materials and methods. (B) Secondary screening of positive pools. Positive pool DNA was assayed for the ability to stimulate the Igκ2-IFN-LUC reporter, the MUT-IFN-LUC reporter or the Igκ2-IFN-LUC reporter in the presence of IKKβ K44A or TBK1 K38A as indicated. The activities in the pools illustrated here were identified to be C/EBPδ (pool 73), TRAIL (pool 224), TNFR1 (pool 473), TRAF2 (pool 24), MyD88 (pool 72) and IKK-i/ε (pool 178). (C) Clone purification. The purification of the rhoB cDNA from pool 443 is illustrated. The activities of the original pool (complexity ∼100 cDNAs) and that of the derived positive subpool (complexity of 24 cDNAs) are shown, as well as the activities observed in the clone identification matrix and for the isolated clone. In this matrix, the coordinates of the positive well were D, 2, X. A schematic of the conceptual clone identification matrix is illustrated. Actual values reading from left to right are 3.6, 39.6, 0.9, 0.7, 0.9, 39.3, 1.4, 63.4, 0.6, 61.2, 0.7 and 114.
We applied three secondary screens. First, we tested the NF-κB dependence of the activity of a pool by comparing its fold induction on the Igκ2-IFN-LUC reporter to that on the MUT-IFN-LUC reporter, which contains mutations in the Igκ κB motifs (5′-ATCCACTTTCC-3′). Secondly, we tested which activities might function upstream of the IKK complex by assessing their activity in the presence of the IKKβ K44A kinase-dead dominant negative. Thirdly, as a control, we tested each κB-specific positive pool in the presence of kinase-dead TANK-binding kinase 1 (TBK1 K38A), an IKK-related kinase (Pomerantz and Baltimore, 1999) which should not block most pathways leading to NF-κB. Examples of these secondary screens are shown in Figure 1B. Of the 41 positive pools, 34 were dependent on the κB sites for activity. Each of these specific pools was inhibited by co-expression with IKKβ K44A, and one pool (pool 178) was also inhibited by co-expression with TBK1 K38A (Figure 1B).
To identify the cDNA responsible for a pool’s activity, colonies derived from its glycerol stock were sib selected (see Materials and methods). For most pools, activity increased as the clone was purified, although the activity of some reached saturation in the assay before complete purification. An example of clone purification is shown in Figure 1C.
The identities of 25 specific clones are presented in Table I with the representative behavior of pools in primary and secondary screens, and their activities when purified. Six isolated molecules had been linked previously to pathways known to activate NF-κB. These included the ligand TRAIL, the TRAMP/DR3 and TNFR1 cell surface receptors, the TRAF2 and MyD88 adapter proteins and the IKK-i/ε kinase (see Table I for associated pathways). The other specific clones encoded the small GTPase rhoB, the MARCKS PKC substrate, the DLK cell surface protein and the Snk kinase. To our knowledge, NF-κB has not been linked to MARCKS, Snk or DLK previously, and rhoB has not been placed in a particular NF-κB-inducing pathway.
Table I. Summary of isolates from the human placenta library and representative pool characteristics.
| cDNA | No. of isolates | Type | Pathway | Rep. poola | Igκ2b | MUTc | IKKβ K44Ad | TBK1 K38Ae | Isolated clonef |
|---|---|---|---|---|---|---|---|---|---|
| TRAIL |
1 |
Ligand |
TRAIL |
224 |
3.1 |
0.6 |
1.1 |
3.0 |
19.0 |
| DLK |
2 |
Ligand |
??? |
228 |
6.2 |
0.6 |
1.5 |
11.0 |
66.0 |
| TNFR1 |
2 |
Receptor |
TNFα |
473 |
87.1 |
1.8 |
0.7 |
67.7 |
108.0 |
| TRAMP |
4 |
Receptor |
Apo3L |
110 |
74.5 |
1.0 |
0.8 |
75.4 |
109.0 |
| TRAF2 |
2 |
Adapter |
TNFα |
24 |
25.2 |
2.9 |
2.0 |
25.4 |
46.8 |
| MyD88 |
1 |
Adapter |
IL-1/Toll |
72 |
30.2 |
3.8 |
3.6 |
19.7 |
999.0 |
| IKK-i/ε |
1 |
Kinase |
PMA/TCR |
178 |
7.6 |
1.3 |
0.9 |
1.4 |
53.5 |
| rhoB |
9 |
Small GTPase |
??? |
501 |
3.2 |
0.8 |
0.3 |
2.7 |
20.3 |
| Snk |
1 |
Kinase |
??? |
270 |
6.3 |
1.1 |
1.2 |
5.2 |
24.0 |
| MARCKS | 2 | Kinase substrate | ??? | 525 | 40.9 | 4.4 | 2.9 | 47.0 | 69.0 |
See Supplementary data at The EMBO Journal Online for further discussion.
aPool identifier number.
bFold stimulation elicited by the pool on the Igκ2-IFN-LUC reporter.
cFold stimulation elicited by the pool on the MUT-IFN-LUC reporter.
dFold stimulation elicited by the pool on the Igκ2-IFN-LUC reporter in the presence of IKKβ K44A.
eFold stimulation elicited by the pool on the Igκ2-IFN-LUC reporter in the presence of TBK1 K38A.
fFold stimulation elicited by the isolated cDNA on the Igκ2-IFN-LUC reporter.
???, pathway not known.
Isolation of murine CARD11 from a mouse thymus expression library
The results from the human placenta library screen indicated that this methodology yields molecules of different biochemical types, both catalytic and non-catalytic, which mediate signaling in different pathways leading to NF-κB. With the aim of identifying molecules involved in lymphocyte signaling, we applied this screen using a mouse thymus cDNA expression library. Known molecules isolated from this library included: the TNFR1, LTβR, CD40 and TRAMP/DR3 receptors; the TRAF2, MyD88, TAB2, RIP3 and NOD1 adapters; the NIK kinase and the RelA subunit of NF-κB. In addition, we independently isolated four clones of a membrane-associated guanylate kinase (MAGUK) family member, the murine homolog of a human protein described while this work was in progress as CARD11 (Bertin et al., 2001) or CARMA1 (Gaide et al., 2001). The murine cDNA encoded 1159 amino acids (Figure 2B) and contained predicted CARD, PDZ, SH3 and GUK domains, in addition to a region predicted to form coiled coils (Figure 2A). The murine and human proteins are 88% identical.

Fig. 2. Murine CARD11. (A) Schematic of murine CARD11 showing domains predicted by sequence homology. (B) Amino acid sequence of murine CARD11 and comparison to the human homolog. Domains in (A) and (B) are color matched.
The presence of PDZ, SH3 and GUK domains is characteristic of the MAGUK family of proteins, which function to cluster and localize multiprotein signaling complexes in several systems (Dimitratos et al., 1999). The CARD is found in caspases, regulators of apoptosis and in proteins that signal to NF-κB (Reed, 2000). The isolation of murine CARD11 in our screen suggested that it might function as an adapter to coordinate signaling in an NF-κB-inducing pathway. In 293T cells, overexpression of the CARD11 cDNA activated the Igκ2-IFN-LUC reporter, but not the MUT-IFN-LUC reporter (Figure 3A). Deletion of the first 115 amino acids containing the CARD abrogated this activity (Figure 3B), confirming the import ance of this domain for NF-κB induction. Activation of NF-κB by CARD11 overexpression was inhibited by IKKβ K44A co-expression, but not by TBK1 K38A co-expression (Figure 3C), suggesting that overexpressed CARD11 activates NF-κB in 293T cells via IKK activation. Previous reports (Bertin et al., 2001; Gaide et al., 2001) also showed that overexpressed CARD11 can activate NF-κB but they did not place CARD11 in a physiological pathway.

Fig. 3. CARD11 activates the Igκ2-IFN-LUC reporter when overexpressed in 293T cells, in a CARD-dependent manner, upstream of the IKK complex. (A) Titration of the murine CARD11 expression vector (ng) in the presence of 2 ng pCSK-LacZ and 20 ng Igκ2-IFN-LUC or MUT-IFN-LUC reporters. (B) Titration of CARD11 or ΔCARD11 expression vectors in the presence of 20 ng Igκ2-IFN-LUC and 2 ng pCSK-LacZ. Western blotting confirmed comparable expression of wild-type and ΔCARD proteins. (C) Two hundred nanograms of CARD11 expression vector were transfected with 20 ng Igκ2-IFN-LUC and 2 ng pCSK-LacZ in the absence or presence of 100 ng of constructs expressing IKKβ K44A or TBK1 K38A.
Involvement of CARD11 in TCR signaling to NF-κB
The presence of CARD11 in the mouse thymus cDNA library suggested that it might function to signal to NF-κB in T cells. Since the CARD-deleted mutant of CARD11 (ΔCARD) could not activate NF-κB in 293T cells, but retained other domains of the protein, we tested its potential inhibitory effect in Jurkat T cells on various stimuli. As shown in Figure 4, ΔCARD inhibited the activation of the Igκ2-IFN-LUC reporter by αCD3/αCD28 cross-linking, while the wild-type protein enhanced the stimulation achieved with no effect on basal reporter activity (Figure 4A and B). The inhibitory effect of ΔCARD was specific; it had no effect on NF-κB activation by TNFα (Figure 4C) or dsRNA (Figure 4D), and did not inhibit the activation of an NFAT-responsive reporter, NFAT-LUC, by αCD3/αCD28 cross-linking (Figure 4E). Finally, ΔCARD also inhibited the activation of the Igκ2-IFN-LUC reporter by PMA/ionomycin treatment but not that of the NFAT-LUC reporter (Figure 4F and G). These specific effects of the ΔCARD variant suggested that CARD11 mediates pathway-specific, factor-specific activation of NF-κB by TCR/CD28 cross-linking, and that the CARD was necessary for this signaling.

Fig. 4. ΔCARD specifically blocks NF-κB activation by αCD3/αCD28 cross-linking in Jurkat T cells. The indicated amounts (ng) of expression vectors for CARD11 or ΔCARD were co-transfected with 200 ng pCSK-LacZ and 1500 ng of either Igκ2-IFN-LUC or NFAT-LUC into Jurkat T cells. Cells were stimulated with αCD3/αCD28 cross-linking, TNFα, dsRNA or PMA/ionomycin co-treatment as indicated.
CARD11 functions upstream of the IKK complex
In order to investigate how ΔCARD inhibited NF-κB reporter activation by αCD3/αCD28 cross-linking, we generated pools of Jurkat cells stably expressing myc-tagged wild-type and mutant CARD11 proteins by infection with Moloney murine leukemia virus-based retroviruses expressing these cDNAs linked to puromycin resistance (Figure 5A). A control, puro-resistant pool of Jurkat cells (puroR pool) was generated by infection with a parental virus containing no cDNA insert. Treatment of the ΔCARD Jurkat pool with αCD3/αCD28 cross-linking activated the NFAT-responsive reporter, but failed to activate the Igκ2-IFN-LUC reporter, while the wild-type CARD11 pool and control puroR pool activated both reporters (Figure 5B and D). Similar results were obtained with PMA/ionomycin treatment (Figure 5C and E). Thus, the stable pools recapitulated the phenotypes observed in transient transfections. Importantly, the levels of stably expressed myc-tagged proteins in these pools did not affect basal NF-κB activity in the absence of signaling (Figures 5 and 6).

Fig. 5. Generation of puro-resistant Jurkat pools expressing wild-type murine CARD11, the ΔCARD mutant or no murine CARD11 variant. (A) Schematic of viral constructs. Following integration, the CMV enhancer/chicken β-actin promoter fusion drives expression of an mRNA containing the inserted cDNA, an internal ribosomal entry site (IRES) and the puromycin resistance gene (PURO). (B–E) Jurkat pools were transfected with 200 ng pCSK-LacZ and 2800 ng of either Igκ2-IFN-LUC or NFAT-LUC and stimulated with either αCD3/αCD28 cross-linking or PMA/ionomycin co-treatment as indicated.

Fig. 6. ΔCARD blocks αCD3/αCD28 cross-linking-induced IKK activation, IκBα degradation and nuclear translocation of NF-κB. Jurkat pools were treated with αCD3/αCD28 cross-linking or TNFα for the indicated times in minutes, and nuclear and cytoplasmic extracts were prepared. (A) EMSA assays were performed with nuclear extracts using probes for NF-κB, NFAT and AP-1. The arrows indicate the specific inducible complexes as verified by their lack of binding to control probes containing binding site mutations (data not shown). Unbound probes are not shown. (B) Western blot assays were performed on cytoplasmic extracts using antibodies for IκBα, IκBβ or myc-tagged proteins. The asterisk indicates an ∼77 kDa truncation of wild-type CARD11 that contains the myc-tagged N-terminus. No truncation was observed in the ΔCARD-expressing pool. (C) Jurkat pools were treated with αCD3/αCD28 cross-linking for the indicated times in minutes, and IKK IP-kinase assays were performed. The amount of radioactivity incorporated into substrate (top panel) was quantitated and fold activations were, from left to right, 1.0, 3.9, 4.8, 1.0, 2.9, 3.1, 1.0, 1.3 and 1.2. The lower panel shows a western blot with αIKKα to indicate the relative amount of IKKα in each sample.
We treated these Jurkat pools with a time course of αCD3/αCD28 cross-linking, and made nuclear and cytoplasmic extracts to assess the induction of NF-κB, AP-1 and NFAT DNA-binding activity. As shown in Figure 6A, the puroR control and wild-type CARD11-expressing pools activated NF-κB DNA-binding activity in response to αCD3/αCD28 cross-linking, while the ΔCARD pool failed to induce NF-κB. In contrast, all three pools induced NF-κB in response to TNFα treatment. The failure of the ΔCARD pool to induce NF-κB in response to αCD3/αCD28 cross-linking was specific since AP-1 and NFAT activities were induced in a manner comparable with that observed with the control and wild-type CARD11 pools (Figure 6A, lower two panels). Western blot analysis of cytoplasmic extracts revealed degradation of IκBα in the puroR control and wild-type CARD11 pools in response to αCD3/αCD28 cross-linking and TNFα treatment, while degradation of IκBα was only observed in the ΔCARD pool in response to TNFα (Figure 6B). Degradation of IκBβ was not observed in response to these stimuli. Western blotting with anti-c-myc antibodies revealed slightly lower expression of the myc-tagged ΔCARD protein than the myc-tagged wild-type CARD11 protein in their respective pools (Figure 6B, lower panel).
IKK IP-kinase assays confirmed a failure to activate the IKK complex in the ΔCARD pool in response to αCD3/αCD28 cross-linking, as compared with that observed in the control and wild-type CARD11 pools (Figure 6C). These results indicated that the ΔCARD mutant blocks NF-κB induction upstream of IKK complex activation, IκBα degradation and nuclear localization of NF-κB.
Functional cooperation and association of CARD11 with Bcl10
Bcl10-deficient T cells display a factor-specific defect in NF-κB activation in response to TCR triggering or PMA/ionomycin treatment (Ruland et al., 2001). These cells fail to activate the IKK complex in response to TCR triggering, while other signaling events are unperturbed. To assess a functional relationship between Bcl10 and CARD11, we transfected a Bcl10 expression vector into the puroR control, CARD11- and ΔCARD11-expressing Jurkat pools and assayed Igκ2-IFN-LUC activity in the absence and presence of αCD3/αCD28 cross-linking. As shown in Figure 7A, transfection of Bcl10 into the control puroR Jurkat pool had no significant effect on reporter activity either in the absence or presence of stimulation by αCD3/αCD28 cross-linking. However, in the CARD11-expressing pool, transfection of Bcl10 resulted in a robust induction in the absence of TCR triggering, which was not significantly enhanced by αCD3/αCD28 cross-linking. In contrast, in the ΔCARD-expressing pool, transfection of Bcl10 resulted in no stimulation in the absence of αCD3/αCD28 treatment and no rescue of reporter induction by αCD3/αCD28 cross-linking. These results indicate that Bcl10 and CARD11 can cooperate in T cells to signal to NF-κB in a manner dependent on the CARD11 CARD. In addition, the block in TCR signaling to NF-κB that is imposed by the ΔCARD variant cannot be bypassed by Bcl10 overexpression. These data suggest that CARD11 and Bcl10 function at the same level of the signaling pathway between the TCR and IKK complexes.

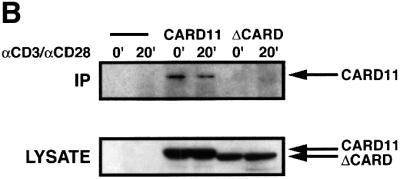
Fig. 7. CARD11 and Bcl10 functionally cooperate and associate in T cells. (A) Igκ2-IFN-LUC (1800 ng) was transfected into Jurkat pools expressing wild-type CARD11, ΔCARD or no cDNA insert in the presence of the indicated amounts (nanograms) of a Bcl10 expression vector (pc-FL-CIPER) (Koseki et al., 1999). Cells were treated with αCD3/αCD28 cross-linking as indicated. (B) Jurkat pools were treated with αCD3/αCD28 cross-linking for the indicated times in minutes, and immunoprecipitations using αBcl10 antibodies were performed. The immunoprecipitates (top panel) and lysates (6.5% of the IP input; bottom panel) were developed with α-myc primary antibody.
To examine whether CARD11 and Bcl10 associate during TCR signaling, we assayed for myc-tagged proteins precipitating with anti-Bcl10 antibodies in lysates from puroR control, myc-tagged CARD11- and ΔCARD-expressing Jurkat pools. As shown in Figure 7B, we detected wild-type CARD11 co-precipitating with endogenous Bcl10 to an extent that was not reproducibly enhanced by αCD3/αCD28 cross-linking. In contrast, we failed to detect co-precipitation of the ΔCARD variant with endogenous Bcl10. These data suggest that CARD11 and Bcl10 associate in a manner that is dependent on the CARD of CARD11 and independent of TCR signaling.
RNA interference of CARD11 inhibits TCR signaling to NF-κB
To independently assess a role for CARD11 in TCR signaling to NF-κB, we used RNA interference (RNAi). Short hairpin RNAs can be expressed from RNA pol III promoters in vivo and processed to small interfering RNAs that silence genes in a sequence-specific manner (Brummelkamp et al., 2002). We designed two short hairpin RNAs to silence the endogenous human CARD11 mRNA (Figure 8A) in Jurkat T cells and expressed them downstream of the human H1 RNA promoter. The sihCARD11-1 hairpin targets a region 260 nucleotides 5′ to the termination codon while sihCARD11-2 targets a region 1400 nucleotides 3′ to the initiation codon. As shown in Figure 8B, transient transfection of Jurkat T cells with either sihCARD11-1 or sihCARD11-2 inhibited induction of the Igκ2-IFN-LUC reporter by TCR cross-linking by 70–75%. A control hairpin targeted to the luciferase mRNA of Renilla reniformis (siRenLuc) did not significantly inhibit. Co-transfection of sihCARD11-1 and sihCARD11-2 did not inhibit more than either transfected alone.

Fig. 8. siRNAs targeted to CARD11 inhibit NF-κB activation by αCD3/αCD28 cross-linking. (A) The sihCARD11-1 and sihCARD11-2 shRNAs are depicted with the murine sequence corresponding to the human target sequence of sihCARD11-1 (mismatches indicated in green). (B) The indicated amounts (nanograms) of Pol III expression constructs for shRNAs were co-transfected with 200 ng pCSK-LacZ and 2700 ng Igκ2-IFN-LUC into Jurkat T cells. Cells were stimulated with αCD3/αCD28 as indicated. (C) Jurkat T cells were transfected with 200 ng pCSK-LacZ and 2500 ng Igκ2-IFN-LUC in the absence or presence of 100 ng of the sihCARD11-1 expression construct and 200 ng of the indicated CARD11 variant expression constructs. Cells were stimulated with αCD3/αCD28 as indicated. Western blot analysis indicated that expression constructs for CARD11 deletion variants express comparable protein levels within a ∼2-fold range. (D) Jurkat T cells were transfected with 200 ng pCSK-LacZ and 2700 ng NFAT-LUC in the absence or presence of 100 ng of the sihCARD11-1 expression construct and stimulated with αCD3/αCD28 as indicated. (E) Jurkat T cells were transfected with 200 ng pCSK-LacZ and 2700 ng Igκ2-IFN-LUC in the absence or presence of 100 ng of the sihCARD11-1 expression construct and stimulated with TNFα as indicated.
The sihCARD11-1 hairpin specifically targets the human CARD11 mRNA and not the murine CARD11 mRNA because it targets a 23 bp region in which the two homologues differ at five positions (Figure 8A). This allowed us to confirm the specificity of the inhibition mediated by sihCARD11-1 by rescuing its inhibitory effect with co-transfection of the murine CARD11 cDNA (Figure 8C). We compared several deletion variants for their ability to rescue sihCARD11-1-mediated inhibition of TCR signaling. As expected, the ΔCARD variant failed to rescue. Furthermore, deletion of the coiled-coil domain (ΔCC), the SH3 domain (ΔSH3) or the GUK domain (ΔGUK) also abrogated the rescuing activity of the CARD11 cDNA, while deletion of the PDZ domain (ΔPDZ) had no effect. These results suggest that the CARD, coiled-coil, SH3 and GUK domains are all important for the signaling function of CARD11. Finally, the sihCARD11-1 hairpin did not significantly inhibit the induction of the Igκ2-IFN-LUC reporter by TNFα, or the induction of the NFAT-LUC reporter by αCD3/αCD28 cross-linking, consistent with a pathway-specific, factor-specific role of CARD11 in NF-κB activation (Figure 8D and E). We have used lentiviruses to establish Jurkat pools that stably express short hairpin RNAs directed against CARD11. These pools display an 80% inhibition of Igκ2-IFN-LUC reporter induction by TCR cross-linking, and an ∼80% decrease in endogenous CARD11 protein levels (data not shown).
Discussion
Many pathways activate NF-κB by transducing signals to a core activation module consisting of the IKK complex, IκB, ubiquitylation machinery, proteasome, rel homo- or heterodimer and nuclear import factors. Pathway-specific molecules function in the link between stimulus recognition and IKK complex activation in an interplay that must elicit an appropriate response to the stimulus, in part through the coordinate regulation of other transcription factor systems. In an effort to better understand the disparate mechanisms for NF-κB activation, we have explored an expression cloning approach with the hope of finding new players, new mechanistic insights and, potentially, new pathways. To probe components used for lymphocyte signaling to NF-κB, we screened a mouse thymus expression library and cloned the murine CARD11 cDNA.
Our data indicate that CARD11 functions in the activation of NF-κB by TCR ligation and CD28 co-stimulation. Expression of a CARD-deleted CARD11 in Jurkat T cells specifically inhibits IKK activation, IκBα degradation, nuclear translocation of NF-κB and induction of a κB-dependent reporter by TCR/CD28 costimulation. The ΔCARD protein does not inhibit NF-κB activation by TNFα or dsRNA, nor does it block NFAT or AP-1 induction by TCR/CD28 costimulation. Two different short hairpin RNAs targeted to suppress the human CARD11 mRNA by RNAi also specifically inhibit κB reporter induction by TCR cross-linking, and this effect can be rescued with murine CARD11. These data assign CARD11 a pathway-specific, factor-specific role in the induction of NF-κB by TCR/CD28 signaling.
Importantly, ΔCARD also blocks NF-κB activation by phorbol ester/ionomycin treatment, which can bypass the requirements for other molecules, such as ZAP70, SLP-76 and Vav (Costello et al., 1999; Herndon et al., 2001), that function in TCR-proximal events. Even the block imposed by PKCΘ deficiency can be bypassed by phorbol ester/ionomycin co-treatment, presumably through the recruitment of other PKC isoforms (Sun et al., 2000). The effect of ΔCARD was reminiscent of that observed in Bcl10-deficient T cells, in which IKK and NF-κB activation by TCR cross-linking and PMA/ionomycin are blocked (Ruland et al., 2001). CARD11 and Bcl10 probably function together in this pathway, downstream of PKCΘ. Overexpression of Bcl10 with CARD11 in Jurkat T cells synergistically activated NF-κB in a manner that was dependent on the CARD of CARD11, and the block in TCR/CD28 signaling imposed by ΔCARD expression could not be bypassed by Bcl10 overexpression. In addition, CARD11 could be immunoprecipitated with endogenous Bcl10 but ΔCARD could not. We conclude that the role of the CARD of CARD11 is to function with Bcl10 to transduce a signal from the TCR complex to the IKK complex. ΔCARD blocks specific signaling to NF-κB probably by decoupling the association of Bcl10 with other portions of the CARD11 protein and other CARD11-associated factors.
The precise mechanism by which Bcl10 and CARD11 function together in TCR signaling is presently unclear, but it may involve MALT1, a protein containing caspase-like, Ig-like and death domains (Akagi et al., 1999; Morgan et al., 1999). MALT1 and Bcl10 have been shown to associate and synergistically activate NF-κB when overexpressed in 293T cells (Lucas et al., 2001). The t(11;18)(q21;q21) translocation, also associated with MALT lymphoma, results in the fusion of the C-terminus of the MALT1 protein to the N-terminus of the API2 protein. The API2–MALT1 fusion can also activate NF-κB when overexpressed (Lucas et al., 2001).
It is intriguing to establish a role for a MAGUK family member in antigen–receptor signaling. MAGUK proteins function in other systems as molecular scaffolds for the assembly and clustering of signaling molecules into localized complexes (Dimitratos et al., 1999). MAGUK proteins can interact with other proteins through each of their characteristic PDZ, SH3 and GUK domains as well as through specialized domains like the CaM kinase and calmodulin-binding domains found in the Lin-2 MAGUK subfamily. In Caenorhabditis elegans, Lin-2 is required for vulval induction and functions in a complex with Lin-7 and Lin-10 which is necessary for proper localization and aggregation of the Let-23 receptor tyrosine kinase at the basolateral membrane of vulval precursor cells (Kaech et al., 1998). Mutations that affect this complex reduce Let-23 signaling and result in a vulvaless phenotype. Similarly, the mammalian PSD-95 MAGUK protein and related proteins bind to and cluster NMDA receptors and potassium channels at appropriate membrane locations in neurons (Sheng, 1996). In T cells, CARD11 may serve a similar role to coordinate complex assembly or appropriate membrane association of signaling molecules at the immunological synapse. Our RNAi-rescue experiments suggest that in addition to the CARD, the coiled-coil, SH3 and GUK domains each contribute to CARD11 function in this pathway. The investigation of their interaction partners will be required for an understanding of the mechanism of CARD11 signaling.
It will be important to assess CARD11 function in the developing mouse and in primary T cells. The closest homologs to CARD11 include CARD10 (Bimp1; McAllister-Lucas et al., 2001; Wang et al., 2001) and CARD14 (Bimp2; Bertin et al., 2001; McAllister-Lucas et al., 2001), each of which contains coiled-coil, PDZ, SH3 and GUK domains and a CARD that can interact with Bcl10. While CARD11 appears to be the predominant family member expressed in lymphoid tissues, CARD14 and CARD10 may play similar roles to transmit signals from PKC-dependent pathways to NF-κB in tissues in which they are expressed (McAllister-Lucas et al., 2001). Targeted mutagenesis of these genes in mice will be required to establish their nonredundant and overlapping functions.
The expression cloning strategy we employed should be versatile. The chief advantage of the screen is that it quantitatively screens for function—the ability to activate a κB-dependent reporter—and it applies no constraints on how the assayed pool achieves that function. As such, it has the ability to detect molecules that have distinct biochemical mechanisms and that serve in disparate signaling pathways. Importantly, the approach provides for facile isolation of the cDNA of interest through sib selection. It is worth noting that >90% of our isolated κB-specific clones contained full-length cDNAs. An attractive feature of the screen is the straightforward manner in which secondary screens can be performed at the level of the pool. Because the assay is quantitative, it is easy to assess dependence on κB sites for activity and also to measure and compare the effects of co-transfected molecules such as dominant negatives. In principle, any dominant-negative molecule could be used to target the discovery of cDNAs encoding components of a particular pathway, or components that function at a particular epistatic level of a pathway. Other secondary screens may give additional insight. For example, pools can be screened for the ability to synergistically activate the reporter with a particular co-transfected molecule or with an applied stimulus.
The screen employs modular components (cell line, reporter, cDNA library) and may be adaptable for the isolation of molecules that activate other transcription factors. Important criteria for establishing an analogous system include the choice of assay cell type, the cDNA library source tissue, and the sensitivity of the assay as determined by the background and induced activities of the transcription factor of interest. The latter will determine the optimal pool complexity for efficient screening and will be influenced by the number of transcription factor binding sites in the reporter.
Materials and methods
Preparation of the expression library for screening
Escherichia coli (DH10B) was transformed with DNA from 16 wells of an arrayed human placenta cDNA library (OriGene Technologies, Inc.) and plated on LB-agar plus ampicillin (100 µg/ml) to obtain ∼100 colonies per plate. Colonies were scraped and a fraction of the pooled bacteria was stored as a 50% (w/v) glycerol stock at –80°C. Plasmid DNA was prepared from the remainder of the bacterial prep by the Qiagen QIAprep 8 Miniprep kit according to the manufacturer’s protocol. The cDNAs in this expression library have been oligo(dT) primed, size fractionated and directionally cloned into pCMV6-XL3, which transcribes the cDNA under the control of the CMV promoter and contains an SV40 origin of replication. Sixteen wells represents 17% of the total library.
Primary screen
293T cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (FCS), 100 U/ml each of penicillin and streptomycin, and 2 mM glutamine in humidified 5% CO2 at 37°C. Cells were plated at 9 × 104 per well in 24-well dishes, 24 h before transfection by the calcium phosphate method. A total of 372 ng of DNA was transfected, including 2 ng of pCSK-LacZ, 20 ng of Igκ2-IFN-LUC and 350 ng of pool DNA. The reference control transfection contained 350 ng pcDNA3 instead of pool DNA. The medium was changed 20–24 h following transfection, and 40–48 h following transfection, cells were lysed in 100 µl of reporter lysis buffer (Promega) at room temperature. Cells were scraped and spun at 13 000 r.p.m. at room temperature for 5 min to pellet debris. Twenty microliters of extract were used to measure luciferase activity using the luciferase assay system (Promega) and a luminometer (Optocomp I, MGM instruments) integrating for 10 s after a 3 s delay according to the manufacturers’ instructions. β-galactosidase activity was determined using 30 µl of extract and the chemiluminescent β-gal reporter gene assay (Roche) according to the manufacturer’s instructions. Fold stimulation was calculated for each sample by dividing the luciferase activity, normalized to β-gal activity, to that observed in the pcDNA3 control sample.
Secondary screens
To determine specificity, pool DNA was assayed as described above with the MUT-IFN-LUC reporter. Positive pools were considered specific if their activity on the MUT-IFN-LUC reporter was ≤30% of that observed on the Igκ2-IFN-LUC reporter or if they stimulated the MUT-IFN-LUC reporter <1.5-fold. To determine the effect of IKKβ K44A or TBK1 K38A, transfections included 75 ng of either pRK-IKKβK44A (Woronicz et al., 1997) or pc-TBK1 K38A (Pomerantz and Baltimore, 1999). Pools were considered inhibited by either dominant negative if in its presence the pool’s activity on the Igκ2-IFN-LUC reporter was reduced by ≥70% or if in its presence the pool stimulated the Igκ2-IFN-LUC reporter <1.5-fold.
Clone purification
A portion of the glycerol stock of a positive pool was plated on LB-agar ampicillin and individual colonies were grown in 1 ml LB-amp cultures in 24-well plates. Each 24-well plate was treated as a subpool; aliquots of each well culture were pooled and assayed in a single transfection. Once a 24-well plate subpool was identified as positive, new aliquots were pooled from the individual wells as rows, columns and levels in a conceptual 4 × 3 × 2 matrix, for a total of nine axis pools. DNA from each axis pool was assayed, yielding the coordinates of the culture well containing the cDNA clone responsible for the activity of the subpool. DNA from the appropriate well culture was prepared, assayed and sequenced with an automated sequencer (Applied Biosystems) to obtain sequence at the 5′ and 3′ ends of the cDNA.
Isolation of the murine CARD11 cDNA
An arrayed mouse thymus cDNA library (OriGene Technologies, Inc.) was screened essentially as described above. A single murine CARD11 cDNA clone was completely sequenced. It encoded an entire open reading frame with an in-frame stop codon 20 codons upstream of the first ATG. Sequence data have been submitted to DDBJ/EMBL/GenBank under accession No. AY135367. Residues 2–1159 were cloned into pIRESPURO (Clontech) fused in-frame to an N-terminal c-myc epitope (EQKLISEEDL) to generate pmCARD11. The ΔCARD expression construct (pΔCARD11) deletes residues 2–115 and retains the N-terminal myc epitope. The ΔCC expression construct (pΔCC) deletes residues 152–447, the ΔPDZ expression construct (pΔPDZ) deletes residues 669–752, the ΔSH3 expression construct (pΔSH3) deletes residues 779–844 and the ΔGUK expression construct (pΔGUK) deletes residues 956–1159.
Transient transfections of Jurkat T cells
Jurkat T cells were grown in RPMI supplemented with 10% FCS, 100 U/ml each of penicillin and streptomycin, 2 mM glutamine and 50 µM β-mercaptoethanol in humidified 5% CO2 at 37°C. On the day of transfection, 5 × 105 cells were plated in 2 ml in each well of a 6-well plate prior to incubation with DNA–Fugene 6 (Roche) complexes according to the manufacturer’s instructions using a total of 3 µg of DNA and 9 µl of Fugene 6. Forty to forty-eight hours after transfection, cells were resuspended in 1 ml media with or without 10 ng/ml TNFα (R&D Systems, Inc.), 50 ng/ml PMA (Sigma) plus 1 µM ionomycin (Calbiochem), or 1 µg/ml each of αhuman CD3 (BD PharMingen 555329), αhuman CD28 (BD PharMingen 555725) and αmouse IgG1 (BD PharMingen 02231D), and incubated for 4–6 h. For dsRNA treatment, 35 h after transfection, double-stranded poly(I)–poly(C) (Amersham Pharmacia Biotech) was added to the cells to a final concentration of 100 µg/ml and incubated for 15 h. Following stimulation, cells were lysed in 150 µl of reporter lysis buffer (Promega) for 10 min at room temperature. Debris was removed by centrifugation at 13 000 r.p.m. for 5 min at room temperature. Luciferase and β-gal activity were measured as described above using 20 and 50 µl of lysate, respectively. In each experiment, each sample was supplemented with the appropriate empty parental expression vector (pIRESPURO, pcDNA3 or pBSK) to keep the total amount of expression vector constant. Fold stimulation was calculated for each sample by dividing the luciferase activity, normalized to β-gal activity, by that observed in the unstimulated sample containing only empty expression vector.
Generation of Jurkat pools stably expressing murine CARD11 and ΔCARD proteins
N-terminally myc-tagged wild-type and ΔCARD cDNAs were cloned into pCL-LZRS-Lox-IRES-PURO (provided by C.Lois) as depicted in Figure 5A. To package retroviruses, 293T cells were plated at 2 × 106 per 10 cm plate 24 h prior to calcium phosphate-mediated transfection with 3 µg pCL-Ampho (Naviaux et al., 1996) and 8 µg of either parental (no cDNA insert), murine CARD11 or ΔCARD retroviral construct. The medium was changed 24 h after transfection, and at 48 h after transfection viral supernatant was supplemented with polybrene (8 µg/ml) and added to Jurkat cultures. Forty-eight hours after addition of viral supernatant, Jurkat cells were resuspended in fresh medium containing puromycin at 0.5 µg/ml and selected for two weeks. Puro-resistant Jurkat pools were maintained in media containing 0.5 µg/ml puromycin.
Electrophoretic mobility shift assays and western blot analysis of IκB degradation
For each timepoint, 107 Jurkat cells were treated in 1 ml as described in the figure legends, pelleted, frozen, thawed and resuspended in 400 µl of buffer A [10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT), and complete protease inhibitors (Roche)]. After 15 min on ice, NP-40 was added to a final 0.5%, the cells were vortexed and pelleted at 6000 r.p.m. for 2 min at 4°C. The cytoplasmic extract supernatant was used for western blot analysis. Nuclear pellets were resuspended in 25 µl buffer C (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 450 mM NaCl, 0.2 mM EDTA, 0.5 mM DTT, 25% glycerol and complete protease inhibitors) and incubated on ice for 20 min, vortexing at 5 min intervals. After centrifugation at 12 000 r.p.m. for 2 min at 4°C, protein concentration of the nuclear extract supernatant was determined by Bradford assay. NF-κB DNA-binding reactions contained 15 mM Tris pH 7.5, 1.5 mM EDTA, 1.5 mM DTT, 5% glycerol, 20 µg/ml bovine serum albumin (BSA), 150 µg/ml dIdC, ∼0.1 ng of labeled probe and 3 µg nuclear extract in a total volume of 20 µl. Binding reactions were incubated at 30°C for 30 min and resolved on 4% polyacrylamide gels in 0.25× TBE. NFAT DNA-binding reactions were performed similarly and contained 15 mM Tris pH 7.5, 1.5 mM EDTA, 1.5 mM DTT, 5% glycerol, 20 µg/ml BSA, 100 mM KCl, 0.5 mM MgCl2 and 200 µg/ml dIdC. AP-1 DNA-binding reactions were performed similarly and contained 15 mM Tris pH 7.5, 1.5 mM EDTA, 1.5 mM DTT, 5% glycerol, 20 µg/ml BSA, 0.5 mM MgCl2 and 200 µg/ml dIdC. Probe sequences are as follows. For NF-κB: 5′-AATTCATGCAGTTGAGGGACTTTCCCAGGCATGCAAGCT-3′; for NFAT: 5′-AATTCGGAGGAAAAACTGTTTCATACAGAAGGCGAAGCT-3′; for AP-1: 5′- AATTCGCTTGATGACTCAGCCGGAACGAAGCT-3′. Cytoplasmic extracts were resolved on 10% SDS–PAGE gels, transferred to immobilon-P membranes (Millipore) and probed with antibodies to IκBα (Santa Cruz sc-371), IκBβ (sc-945) and the c-myc epitope (sc-40).
IKK kinase assays
For each time point, 2 × 106 Jurkat cells were resuspended in 1 ml media in the absence or presence of 1 µg/ml each of αCD3, αCD28 and αIgG for the indicated times at 37°C. Cells were pelleted and lysed in 1 ml IP lysis buffer (50 mM HEPES pH 7.9, 250 mM NaCl, 1% NP40, 1 mM EDTA, 50 µM DTT, 50 µM Na3VO4 and complete protease inhibitors) for 5 min at 4°C. Following removal of debris by centrifugation at 13 000 r.p.m. for 5 min at 4°C, 1 µg of αIKKα antibodies(sc-7218) were added and incubated at 4°C for 30 min. Subsequently, 10 µl bed of protein G–Sepharose (Amersham Pharmacia) was added and samples were rotated at 4°C for 1 h. Beads were washed twice with 1 ml IP lysis buffer, followed by a 1 ml wash with reaction buffer (10 mM HEPES pH 7.9, 5 mM MgCl2, 1 mM MnCl2, 12.5 mM β-glycerophosphate, 2 mM NaF, 50 µM DTT, 50 µM Na3VO4). Kinase reactions were initiated by adding 30 µl of reaction buffer plus 10 µM ATP, 5 µCi [γ-32P]ATP and 1 µg of glutathione S-transferase–IκBα(1–62). After 30 min at 30°C, reactions were terminated with SDS–PAGE loading buffer, resolved on 12% SDS–PAGE gels, transferred to PVDF membranes and then exposed for quantitation using a Storm 860 PhosphorImager (Molecular Dynamics). Subsequently, membranes were western blotted using αIKKα antibody (sc-7606) to visualize the relative amount of IKK in each sample.
Immunoprecipitations
For each time point, 4 × 107 Jurkat cells were resuspended in 1.5 ml media in the absence or presence of 1 µg/ml each of αCD3, αCD28 and αIgG for the indicated times at 37°C. Cells were washed with 1 ml cold phosphate-buffered saline and lysed in 1 ml IP lysis buffer for 5 min at 4°C. Following removal of debris by centrifugation at 13 000 r.p.m. for 5 min at 4°C, 1 µg of αBcl10 antibodies (sc-5611) were added to 400 µl supernatant and incubated at 4°C for 1 h. Subsequently, 10 µl bed of protein G–Sepharose was added and samples were rotated at 4°C for 2 h. Beads were washed three times with 1 ml IP lysis buffer, boiled in SDS–PAGE loading buffer, resolved on 10% SDS–PAGE gels and transferred to PVDF membranes for western blotting with α-myc antibodies.
RNA interference
Oligonucleotides were cloned into pSKKD1 (provided by X.F.Qin) to express the sihCARD11-1 and sihCARD11-2 hairpins downstream of the human H1 RNA promoter essentially as described (Brummelkamp et al., 2002). The siRenLuc short hairpin RNA (provided by X.F.Qin) targets the sequence 5′-AAACATGCAGAAAATGCTG-3′. These constructs were cotransfected into Jurkat T cells with the appropriate reporters as described above.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank members of the Baltimore laboratory for helpful discussions and support, G.Nuñez for pc-FL-CIPER and J.Alberola-Ila, M.Boldin, L.Gammill, J.Kim, M.Laurent, M.Meffert, P.Sternberg and T.-M.Yi for critical reading of the manuscript. During the course of this work, J.L.P. was supported by a postdoctoral fellowship from the Helen Hay Whitney Foundation and is currently a Special Fellow of the Leukemia and Lymphoma Society. This work was supported by NIH grant AI42549-04.
References
- Akagi T. et al. (1999) A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene, 18, 5785–5794. [DOI] [PubMed] [Google Scholar]
- Alberola-Ila J., Takaki,S., Kerner,J.D. and Perlmutter,R.M. (1997) Differential signaling by lymphocyte antigen receptors. Annu. Rev. Immunol., 15, 125–154. [DOI] [PubMed] [Google Scholar]
- Bertin J., Wang,L., Guo,Y., Jacobson,M.D., Poyet,J.L., Srinivasula,S.M., Merriam,S., DiStefano,P.S. and Alnemri,E.S. (2001) CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-κB. J. Biol. Chem., 276, 11877–11882. [DOI] [PubMed] [Google Scholar]
- Brummelkamp T.R., Bernards,R. and Agami,R. (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science, 296, 550–553. [DOI] [PubMed] [Google Scholar]
- Condie B.G., Brivanlou,A.H. and Harland,R.M. (1990) Most of the homeobox-containing Xhox 36 transcripts in early Xenopus embryos cannot encode a homeodomain protein. Mol. Cell. Biol., 10, 3376–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello P.S., Walters,A.E., Mee,P.J., Turner,M., Reynolds,L.F., Prisco,A., Sarner,N., Zamoyska,R. and Tybulewicz,V.L. (1999) The Rho-family GTP exchange factor Vav is a critical transducer of T cell receptor signals to the calcium, ERK and NF-κB pathways. Proc. Natl Acad. Sci. USA, 96, 3035–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitratos S.D., Woods,D.F., Stathakis,D.G. and Bryant,P.J. (1999) Signaling pathways are focused at specialized regions of the plasma membrane by scaffolding proteins of the MAGUK family. BioEssays, 21, 912–921. [DOI] [PubMed] [Google Scholar]
- Doi T.S., Takahashi,T., Taguchi,O., Azuma,T. and Obata,Y. (1997) NF-κB RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J. Exp. Med., 185, 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T., Shibuya,H., Hotta,H., Yamanishi,K. and Taniguchi,T. (1987) Interferon-β gene regulation: tandemly repeated sequences of a synthetic 6 bp oligomer function as a virus-inducible enhancer. Cell, 49, 357–367. [DOI] [PubMed] [Google Scholar]
- Gaide O., Martinon,F., Micheau,O., Bonnet,D., Thome,M. and Tschopp,J. (2001) Carma1, a CARD-containing binding partner of Bcl10, induces Bcl10 phosphorylation and NF-κB activation. FEBS Lett., 496, 121–127. [DOI] [PubMed] [Google Scholar]
- Gerondakis S., Grossmann,M., Nakamura,Y., Pohl,T. and Grumont,R. (1999) Genetic approaches in mice to understand Rel/NF-κB and IκB function: transgenics and knockouts. Oncogene, 18, 6888–6895. [DOI] [PubMed] [Google Scholar]
- Ghosh S., May,M.J. and Kopp,E.B. (1998) NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol., 16, 225–260. [DOI] [PubMed] [Google Scholar]
- Herndon T.M., Shan,X.C., Tsokos,G.C. and Wange,R.L. (2001) ZAP-70 and SLP-76 regulate protein kinase CΘ and NF-κB activation in response to engagement of CD3 and CD28. J. Immunol., 166, 5654–5664. [DOI] [PubMed] [Google Scholar]
- Hettmann T. and Leiden,J.M. (2000) NF-κB is required for the positive selection of CD8+ thymocytes. J. Immunol., 165, 5004–5010. [DOI] [PubMed] [Google Scholar]
- Kaech S.M., Whitfield,C.W. and Kim,S.K. (1998) The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C.elegans EGF receptor LET-23 in vulval epithelial cells. Cell, 94, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. and Ben-Neriah,Y. (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol., 18, 621–663. [DOI] [PubMed] [Google Scholar]
- Kontgen F., Grumont,R.J., Strasser,A., Metcalf,D., Li,R., Tarlinton,D. and Gerondakis,S. (1995) Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity and interleukin-2 expression. Genes Dev., 9, 1965–1977. [DOI] [PubMed] [Google Scholar]
- Koseki T., Inohara,N., Chen,S., Carrio,R., Merino,J., Hottiger,M.O., Nabel,G.J. and Nunez,G. (1999) CIPER, a novel NF-κB-activating protein containing a caspase recruitment domain with homology to Herpesvirus-2 protein E10. J. Biol. Chem., 274, 9955–9961. [DOI] [PubMed] [Google Scholar]
- Lucas P.C., Yonezumi,M., Inohara,N., McAllister-Lucas,L.M., Abazeed,M.E., Chen,F.F., Yamaoka,S., Seto,M. and Nunez,G. (2001) Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-κB signaling pathway. J. Biol. Chem., 276, 19012–19019. [DOI] [PubMed] [Google Scholar]
- McAllister-Lucas L.M. et al. (2001) Bimp1, a MAGUK family member linking protein kinase C activation to Bcl10-mediated NF-κB induction. J. Biol. Chem., 276, 30589–30597. [DOI] [PubMed] [Google Scholar]
- Morgan J.A. et al. (1999) Breakpoints of the t(11;18)(q21;q21) in mucosa-associated lymphoid tissue (MALT) lymphoma lie within or near the previously undescribed gene MALT1 in chromosome 18. Cancer Res., 59, 6205–6213. [PubMed] [Google Scholar]
- Naviaux R.K., Costanzi,E., Haas,M. and Verma,I.M. (1996) The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol., 70, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz J.L. and Baltimore,D. (1999) NF-κB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J., 18, 6694–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed J.C. (2000) Mechanisms of apoptosis. Am. J. Pathol., 157, 1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe M., Sarma,V., Dixit,V.M. and Goeddel,D.V. (1995) TRAF2-mediated activation of NF-κB by TNF receptor 2 and CD40. Science, 269, 1424–1427. [DOI] [PubMed] [Google Scholar]
- Ruland J. et al. (2001) Bcl10 is a positive regulator of antigen receptor-induced activation of NF-κB and neural tube closure. Cell, 104, 33–42. [DOI] [PubMed] [Google Scholar]
- Senftleben U., Li,Z.W., Baud,V. and Karin,M. (2001) IKKβ is essential for protecting T cells from TNFα-induced apoptosis. Immunity, 14, 217–230. [DOI] [PubMed] [Google Scholar]
- Sheng M. (1996) PDZs and receptor/channel clustering: rounding up the latest suspects. Neuron, 17, 575–578. [DOI] [PubMed] [Google Scholar]
- Sun Z. et al. (2000) PKCΘ is required for TCR-induced NF-κB activation in mature but not immature T lymphocytes. Nature, 404, 402–407. [DOI] [PubMed] [Google Scholar]
- Voll R.E., Jimi,E., Phillips,R.J., Barber,D.F., Rincon,M., Hayday,A.C., Flavell,R.A. and Ghosh,S. (2000) NF-κB activation by the pre-T cell receptor serves as a selective survival signal in T lymphocyte development. Immunity, 13, 677–689. [DOI] [PubMed] [Google Scholar]
- Wang L. et al. (2001) Card10 is a novel caspase recruitment domain/membrane-associated guanylate kinase family member that interacts with BCL10 and activates NF-κB. J. Biol. Chem., 276, 21405–21409. [DOI] [PubMed] [Google Scholar]
- Willis T.G. et al. (1999) Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell, 96, 35–45. [DOI] [PubMed] [Google Scholar]
- Woronicz J.D., Gao,X., Cao,Z., Rothe,M. and Goeddel,D.V. (1997) IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science, 278, 866–869. [DOI] [PubMed] [Google Scholar]
- Zhang Q. et al. (1999) Inactivating mutations and overexpression of BCL10, a caspase recruitment domain-containing gene, in MALT lymphoma with t(1;14)(p22;q32). Nat. Genet., 22, 63–68. [DOI] [PubMed] [Google Scholar]