

Abstract
For protein synthesis initiation in eukaryotes, eIF2B is the guanine-nucleotide exchange factor for eIF2. eIF2B is an essential multi-subunit factor and a major target for translational control in both yeast and mammalian cells. It was shown previously that the largest eIF2B subunit, eIF2Bε, is the only single subunit with catalytic function. Here we report the results of a molecular dissection of the yeast ε subunit encoded by GCD6 in which we have identified the catalytic domain. By analysis of a series of N-terminal deletions in vitro we find that the smallest catalytically active fragment contains residues 518–712 (termed Gcd6p518–712). Further deletion to position 581 (Gcd6p581–712) results in loss of nucleotide exchange function, but eIF2-binding activity is retained. C- terminal deletion of only 61 residues (Gcd6p1–651) results in loss of both functions. Thus Gcd6p518–712 contains two regions that together constitute the catalytic domain of eIF2B. Finally, we show that the catalytic domain can provide eIF2B biological function in vivo when elevated levels eIF2 and tRNAiMet are also present.
Keywords: eIF2/eIF2B/GCD6/guanine-nucleotide exchange/protein synthesis initiation
Introduction
Guanine-nucleotide binding (G) proteins control various key cellular processes. Many require accessory factors including nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) that control their nucleotide-bound state. While G proteins themselves share common consensus nucleotide-binding motif sequences and structures within their nucleotide binding domains, the GEFs in particular are structurally and mechanistically diverse (Sprang and Coleman, 1998; Cherfils and Chardin, 1999), suggesting they do not arise from a common evolutionary ancestor.
eIF2 is a G protein composed of three subunits that is required for the initiation of protein synthesis in all eukaryotic cells. It forms a ternary complex with GTP and aminoacylated initiator methionyl tRNA (Met-tRNAiMet) to deliver the latter to the 40S ribosomal subunit for each protein synthesis initiation event. Following mRNA selection and initiator codon recognition, eIF2-bound GTP is hydrolysed to release the Met-tRNAiMet at the P site of mRNA bound 40S complexes by the GAP eIF5. eIF2·GDP is released from the ribosome and 60S subunit joining, promoted by a second G protein eIF5B, completes the initiation process. As eIF2 has a higher affinity for GDP than GTP it requires the action of a GEF to reform eIF2·GTP in order to participate in further translation initiation events. GEF function is provided by eIF2B (reviewed in Hinnebusch, 2000; Dever, 2002).
eIF2B is a large protein complex composed of five subunits, α to ε (smallest to largest). Missense mutations have recently been identified in all subunits of human eIF2B that are causal in the rare inherited and fatal neurodegenerative brain disorder leucoencephalopathy with vanishing white matter (Leegwater et al., 2001; van der Knaap et al., 2002). This is the first genetic disorder known to affect translation factor genes directly.
eIF2B activity can be regulated in cells, and this has been shown to control both global and gene-specific translation initiation in yeast and mammalian cells. The most widely studied mechanism of eIF2B control involves phosphorylation of the α subunit of eIF2 in response to diverse cellular stresses by one of four protein kinases: HRI (or HCR) responds to haem deficiency in reticulocyte cells (Chen, 2000); PKR is part of the antiviral response and is activated by double-stranded RNAs (Kaufman, 2000); PERK (or PEK) responds to stress in the endoplasmic reticulum (Ron and Harding, 2000); and GCN2 responds to amino acid starvation (Hinnebusch, 2000). All four kinases phosphorylate eIF2 at a conserved serine at position 51 of the α subunit. This converts eIF2 from a substrate to a competitive inhibitor of eIF2B function. Reduction of eIF2B activity lowers ternary complex formation and can reduce global translation rates. In addition, translation of mRNAs that encode proteins required to respond to the cellular stress is elevated. The two best characterized examples of this are GCN4 in the yeast Saccharomyes cerevisiae and ATF4 in mammalian cells (Hinnebusch, 1996; Harding et al., 2000; Dever, 2002). In both cases, the presence of short open reading frames (ORFs) in the 5′ leaders of the mRNAs are required for translational control.
Previous genetic and biochemical experiments, mainly in yeast, have allowed progress in defining the roles for eIF2B subunits. The α, β and δ subunits share extensive sequence similarity with each other and can form a subcomplex in the absence of the other two subunits (Yang and Hinnebusch, 1996). Importantly, each plays a non-redundant role in sensing and binding eIF2α phosphorylated at Ser51, such that a single missense mutation in any one subunit can make eIF2B insensitive to the phosphorylation event (Vazquez de Aldana and Hinnebusch, 1994; Pavitt et al., 1997, 1998; Kimball et al., 1998; Krishnamoorthy et al., 2001).
Regulation of eIF2B function independent of eIF2 phosphorylation has also been reported. Phosphorylation of eIF2B itself on its ε subunit (eIF2Bε) by GSK-3 is reduced in response to insulin signalling, and this can regulate its activity in mammalian cells (Welsh and Proud, 1993). Recently, the sensitivity of yeast cells to fusel alcohols, such as butanol, was shown to be altered by a specific missense mutation in eIF2Bγ (Ashe et al., 2001). Together these findings underscore the importance of regulation of eIF2B function for control of gene expression.
Analyses of both yeast and mammalian factors have shown that eIF2Bε is the primary catalytic subunit: it is the only subunit that can catalyse release of guanine nucleotides from eIF2 in in vitro assays. However, the catalytic activity of both yeast and mammalian eIF2Bε alone is 10- to 40-fold lower than for the intact five-subunit complex (Fabian et al., 1997; Pavitt et al., 1998; Gomez and Pavitt, 2000). Analysis of the yeast factor suggests that eIF2Bγ will form a subcomplex with eIF2Bε and can enhance its rate of catalysis (Pavitt et al., 1998).
In this study, we report that a minimal C-terminal segment of yeast eIF2Bε (Gcd6p) composed of residues 518–712 is sufficient for eIF2B catalytic function in vitro, and can provide eIF2B biological function in vivo when sufficient eIF2 and tRNAiMet are also present. We find that a smaller fragment containing residues 581–712 is able to bind eIF2, but that binding alone is not sufficient to promote nucleotide exchange. We also show that the eIF2-binding domain found at the extreme C-terminus of the subunit is necessary for eIF2Bε function, as deletion of residues 652–712 eliminates stable eIF2 binding and nucleotide exchange when the ε subunit is analysed alone. In the context of the five-subunit complex we find that the other eIF2B subunits make a modest contribution to eIF2 binding when residues 651–712 are deleted, and this promotes a nucleotide exchange at a reduced rate. These results define yeast eIF2Bε residues 518–712 as the catalytic domain of eIF2B.
Results
N-terminal deletions of GCD6 identify a minimal fragment of eIF2B that is active in vitro
Previous experiments with both yeast and mammalian eIF2B (Fabian et al., 1997; Pavitt et al., 1998; Anthony et al., 2000) have established that the ε subunit of eIF2B (eIF2Bε) is the primary catalytic subunit of the GEF eIF2B. Our analysis of missense and nonsense mutations in yeast eIF2Bε (Gcd6p) suggested that the C-terminal half of Gcd6p contained the catalytic domain (Gomez and Pavitt, 2000). To identify the minimal catalytically active region of Gcd6p, a series of N-terminal deletions of the GCD6 ORF was created by PCR and inserted downstream of the GAL1 promoter in a 2 µm yeast expression plasmid to fuse the GCD6 sequences to tandem N-terminal FLAG and His6 epitopes (FH6; see Supplementary data available at The EMBO Journal Online for details). Each deletion was designed to assess the role of a region of sequence conservation between GCD6 and its homologues from other eukaryotes, as shown schematically in Figure 1 (constructs 2–7). Previously, we showed that an in-frame internal deletion of residues 93–358 (Gcd6pΔ93–358; construct 3 in Figure 1) retained in vitro nucleotide exchange and eIF2 binding activity indistinguishable from the full-length isolated Gcd6p subunit (Gomez and Pavitt, 2000), so this construct was used as the starting point for the new series of deletions we made. As eIF2 and eIF2B co-purify at salt concentrations up to 500 mM, Gcd6p polypeptides were purified by immobilized metal affinity chromatography (IMAC) using Ni-NTA resin in the presence of 1 M KCl to ensure that eIF2 did not co-purify; this was confirmed by western blotting with antibodies specific for yeast eIF2α (Gomez and Pavitt, 2000; data not shown). Immunoblotting with anti FLAG M2 antibody allowed an accurate assessment of the integrity and relative molar concentrations of the purified proteins (Figure 2A and B). Immunoblot analysis with antibodies specific to the other four eIF2B subunits confirmed that the N-terminally deleted Gcd6p proteins were purified free of contaminating eIF2B subunits, while the purified full-length Gcd6p preparation contained low levels of full five-subunit eIF2B (Figure 2A, compare lanes 5–8 with lanes 1 and 2; data not shown). These results confirm that the N-terminal half of eIF2Bε functions in eIF2B complex formation.

Fig. 1. Summary of in vivo and in vitro results. Columns show: plasmid name; scheme of GCD6 fragment expressed; N-terminal tag F (FLAG-His6) or GST; relative expression level in cells following growth in 4% galactose + 2% raffinose (SCGR) media as determined by western blotting with anti-FLAG M2 antibody or anti-GST antibody (scale 0–4); activity and eIF2 binding in vitro of purified fragment indicated as + (yes), – (no) or nt (not tested); in vivo function in the presence of overexpressed eIF2 and tRNAiMet in strain GP4115 on agar containing 2% rafinose only (SCR, non-inducing conditions) or 4% galactose + 2% raffinose SCGR, (inducing conditions) where the rate of colony formation was assessed on a numerical scale 0–6, where ‘6’ is the rate of growth of wild-type GCD6 in this strain and ‘2’ is the rate of growth of the vector transformed strain on each medium.

Fig. 2. In vitro analysis of FH6-tagged fragments of eIF2Bε (Gcd6p). (A) Western blot analysis of IMAC partially purified Gcd6p polypeptides using M2 anti-FLAG antibody (top panel) to detect Gcd6p and antisera specific for the other four eIF2B subunits to detect low levels of co-purifying proteins, as indicated to the right of each panel. Loadings were designed for 2-fold increments and for the top panel only are: lane 1, 75 ng; lanes 2 and 3, 150 ng; lane 4, 300 ng; lanes 5 and 7, 82.5 ng; lanes 6 and 8, 165 ng. For the lower four panels 33× the protein concentration loaded for the top panel was loaded in each lane (i.e. lane 1, 2.5 µg, etc.) The band corresponding to Gcn3p is indicated in the lower panel. (B) Western blot comparison of Gcd6p518–712 with full-length Gcd6p as in (A) using the M2 anti-FLAG antibody. Amount loaded in each lane corresponds to 1.5-fold increments: lanes 1 and 4, 89 ng; lanes 2 and 5, 133 ng; lanes 3 and 6, 200 ng. (C) Nucleotide exchange activity of purified proteins. Amount of [3H]GDP dissociated (pmol) from 7 pmol eIF2·GDP complexes following 3 min incubation with 2.5 µg partially purified Gcd6p or an equivalent amount of the indicated Gcd6p fragments [as determined from western analysis, (A) and (B), and data not shown] at 10°C in our standard filter- binding assay. Buffer indicates the eIF2B independent dissociation of [3H]GDP under these assay conditions. Mean errors are <5% for each assay. (D) Western blots showing binding of eIF2 (20 nM) to FLAG M2 affinity gel (lanes 2–8) pre-incubated with FH6-Gcd6p proteins (lanes 3–8) or an equivalent concentration of FLAG peptide (200 nM, lane 2). Lane 1 (input) contains 6.25 ng of purified eIF2. Lanes 2–8 show the fraction of proteins remaining bound to resin after extensive washing, where 8.5% (top panel), 31% (middle panel) and 31% (bottom panel) of total bound material was loaded in each well. Membranes were probed with specific antiserum, indicated to the right of each panel. Ig indicates the heavy and light chains of the precipitating FLAG M2 antibody.
Equivalent molar amounts of each protein were assayed for guanine-nucleotide exchange activity in a standard filter-binding assay where the loss of [3H]GDP from eIF2·[3H]GDP binary complexes in the presence of a 100-fold excess of unlabelled GDP is assessed (Figure 2C). In these assays all N-terminal deletions assessed were as active as the wild type, including the smallest fragment tested, Gcd6p518–712. In addition, we assessed the ability of these proteins to bind to eIF2. Gcd6p proteins (∼200 nM) were incubated with anti-FLAG M2 affinity gel and then incubated with eIF2 (20 nM) in the presence of bovine serum albumin (BSA). The amount of eIF2 that remained bound to the resin after extensive washing was assessed by immunoblotting (Figure 2D). In accordance with the functional assay, all N-terminal deletions were active in this assay, including the smallest fragment tested, Gcd6p518–712.
Thus our smallest tested segment (at this point in our analysis), an ∼200 amino acid segment, retained both eIF2 binding and nucleotide exchange activities. This segment contains two evolutionarily conserved regions of sequence similarity (Figure 3). The extreme C-terminal region (∼60 residues) contains two elements rich in aromatic and acidic residues and is similar to a region in eIF5. This region has been termed the AA box or eIF5C homology region, and has been implicated in mediating binding to the eIF2 β subunit (Asano et al., 1999; Das and Maitra, 2000). The N-terminus (residues 518–593) is also conserved, but less is known about this region. We previously identified mutations altering residues 552 and 576, both of which impaired the nucleotide exchange activity (Gomez and Pavitt, 2000; asterisks in Figure 3). In addition, mammalian eIF2Bε activity can be regulated by changes in phosphorylation by GSK-3 in this region (Wang et al., 2001; SRAGS underlined in rat sequence in Figure 3), suggesting that this first conserved region may be important for eIF2Bε function. An alternative possibility was that this region only indirectly modulated eIF2Bε activity and that a minimal eIF2-binding fragment would suffice. To test these possibilities, we made a further deletion construct, Gcd6p581–712, using the same expression system. Unfortunately, we could not detect expression of this Gcd6p fragment, suggesting that it was either not expressed efficiently or not stable once expressed (Figure 1, construct 7; data not shown).

Fig. 3. Alignment of Gcd6p518–712 with the C-termini of eIF2Bε sequences from other eukaryotes. eIF2Bε sequences from S.cerevisiae, Rattus norvegicus (rat), Drosophila melanogaster and Caenorhabditis elegans were aligned using Clustal_W. The region shown was selected and shaded with MacBoxshade v. 2.01. All identical residues are white type on black, three of four identical are shaded light grey, similar substitutions are shaded dark grey. The asterisks indicate previously mutated yeast residues. The GSK-3 phosphorylation site is underlined in the rat sequence (DDBJ/EMBL/GenBank accession numbers are, respectively, Z68195, Q64350, AL021086 and CAA91063.1).
To overcome the lack of expression of Gcd6p581–712, we reasoned that a longer N-terminal tag might stabilize the small protein. We therefore cloned the PCR product into an alternative galactose-inducible yeast expression plasmid that allowed the fusion the Gcd6p sequences to glutathione S-transferase (GST) from Schistosoma japonicum. As a positive control we also cloned the functional Gcd6p518–712 fragment into the same expression system. The expression of these constructs was assessed with anti-GST antibody and this demonstrated that both the resulting GST–Gcd6p518–712 and GST–Gcd6p581–712 proteins were efficiently expressed (Figure 1, constructs 10 and 11; data not shown). Proteins were purified using glutathione–Sepharose resin and eluted with reduced glutathione. Protein activities were assessed in both nucleotide exchange and in vitro binding assays, and identically purified GST alone was used as a negative control for all experiments (Figure 4). The nucleotide exchange assay clearly shows that this function is eliminated with the smaller Gcd6p581–712 fragment. As these new purified proteins lacked the FLAG epitope, the eIF2-binding assay was altered to employ glutathione– Sepharose resin. The results of this assay indicate that both GST–Gcd6p518–712 and GST–Gcd6p581–712 are able to bind to eIF2. While it is clear that the affinity of the smaller fragment is lower in this assay, it is significantly above the background binding observed for GST alone. Together these results suggest that two conserved regions of eIF2Bε are critical for its catalytic function. The C-terminal region is sufficient for eIF2 binding alone, while the region from 518–580 enhances eIF2 binding and additionally provides nucleotide exchange activity. We have designated this evolutionarily conserved region the ‘catalytic centre’ in Figure 1.

Fig. 4. In vitro analysis of GST-tagged fragments of eIF2Bε (Gcd6p). (A) Nucleotide exchange activity of GST fusion proteins shown as rate of removal of [3H]GDP from eIF2 binary complexes. Four micrograms of each protein was used. GST (diamonds) GST–Gcd6p518–712 (squares) and GST–Gcd6p581–712 (circles). Error bars show standard deviations, trendlines are exponential curve fits to the data, broken line is for GST alone. (B) Interaction of eIF2 (20 nM) with GST fusion proteins (4 µg each, ∼200 nM GST or ∼100 nM Gcd6p518–712) using glutathione–Sepharose chromatography. Proteins indicated on the left were assessed by western blotting using the antisera indicated on the right of each panel. Lanes 1–3, 0.25 µg input GST fusion proteins; lane 4, 1 µg input eIF2; lanes 5–10, upper panel, 10% of bound material loaded, lower panels, 55% of bound material loaded. The bottom panel is a longer exposure of the same experiment shown in the middle panel.
Deletion of the C-terminal 61 residues of Gcd6p eliminates in vitro function
Previous studies have implicated the C-terminal ∼60 residues of eIF2Bε as important in binding eIF2 (Asano et al., 1999; Wang et al., 2001). However, our above results suggested that the region 518–580 also contributed to the eIF2 binding affinity. To assess the role of Gcd6p residues 652–712 in our in vitro assays, we made a construct where these residues were deleted (Gcd6p1–651; Figure 1, construct 8). This construct was expressed and we were able to purify it intact. In our yeast expression system, like all constructs containing an intact eIF2Bε N-terminus including the wild type (Gomez and Pavitt, 2000), Gcd6p1–651 co-purified with low levels of the other four eIF2B subunits (Figure 2A). When nucleotide exchange activity was assessed, this construct was completely inactive; even when a 4-fold molar excess of protein was used [Figure 2C, Gcd6p1–651 (4×)]. Our protein–protein interaction assay confirmed that specific eIF2 binding was eliminated with this construct (Figure 2D, lane 8). These results demonstrate the critical importance of residues 652–712 to eIF2Bε function.
We have shown previously that the eIF2Bα, β and δ subunits act together to sense and bind to eIF2 when the latter is phosphorylated on Ser51 of its α subunit by a regulatory kinase (see Introduction). Therefore, regions of the five-subunit eIF2B complex other than residues 618–712 of eIF2Bε may contribute to the binding surface with non-phosphorylated eIF2. It was therefore of interest to determine whether eIF2Bε residues 652–712 are as critical for five-subunit eIF2B function as they are in the isolated eIF2Bε subunit, or whether other determinants for eIF2 binding provided by other subunits can compensate. We therefore altered our protein expression system to purify five-subunit wild-type eIF2B and mutant eIF2B lacking Gcd6p residues 652–712 (eIF2Bε1–651). In this expression system, the eIF2Bγ subunit (Gcd1p) is FLAG- and His6-tagged at its C-terminus (see Materials and methods). Assays for nucleotide exchange activity and eIF2 binding showed that both functions were severely impaired (Figure 5). When the exchange activity of 2 µg of each protein was compared, the mutant activity was ∼15% that of wild-type eIF2B in these assays. Increasing the protein concentration above 3 µg did not result in any enhanced rate of nucleotide exchange (data not shown), suggesting that one or more kinetic parameters for the mutant may be altered. As stable eIF2 binding was virtually eliminated in our assay we can conclude that residues 652–712 of Gcd6p provide the major eIF2-binding region.
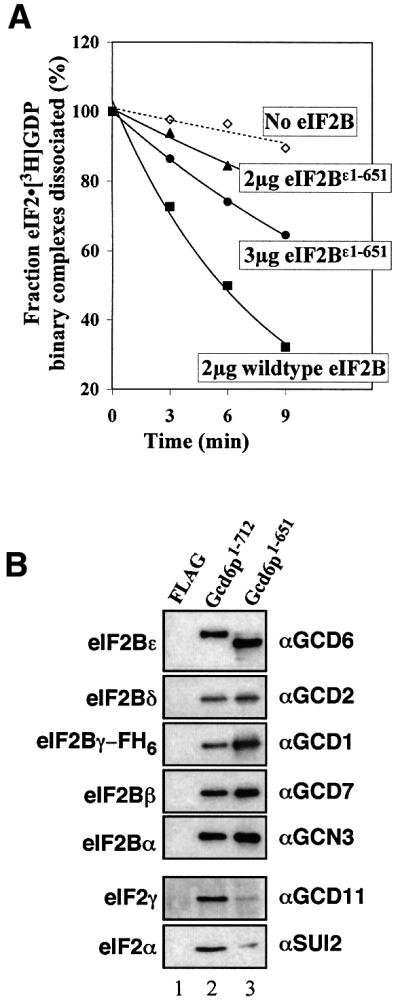
Fig. 5. Impaired activities of eIF2B complex lacking the C-terminal 61 residues of the epsilon subunit. (A) Nucleotide exchange assay with indicated amounts of wild-type eIF2B (squares) eIF2Bε1–651 (triangles, circles) or buffer control (diamonds).Trendlines are exponential curve fits to the data, broken line is for buffer control. (B) Interaction of eIF2 (5 nM) with five-subunit complexes of wild-type eIF2B (lane 2), eIF2Bε1–651 (lane 3) or an equivalent amount of FLAG peptide (100 nM, lane 1) on anti-FLAG M2 resin. eIF2 and eIF2B subunits indicated on the left were probed with the specific antiserum indicated on the right, where 8.5% (eIF2B panels) and 31% (eIF2 panels) of total bound material was loaded in each lane.
Gcd6p518–712 provides eIF2B biological activity in vivo
We wished to obtain in vivo results of biological activity to complement our in vitro biochemical studies. Four of the five genes encoding eIF2B subunits are essential in yeast (all but GCN3, encoding eIF2Bα). As Gcd6p518–712 and larger fragments are catalytically active in vitro, but possess reduced activity compared with five-subunit eIF2B, we reasoned that elevated expression of these fragments may provide eIF2B function in vivo. Initially we asked whether overexpression of catalytically active Gcd6p fragments from galactose-inducible expression plasmids were able to complement a deletion of GCD6 on galactose-containing medium. All results were negative (data not shown).
Hannig and colleagues recently showed that they could delete the genes encoding all five subunits of eIF2B, providing they co-overexpressed all three subunits of eIF2 and one of the four genes encoding tRNAiMet (Erickson et al., 2001). The resulting yeast strain was severely slow growing. We constructed a similar strain by transforming a gcd6Δ yeast strain bearing GCD6 on a low-copy plasmid with a multicopy plasmid co-overexpressing eIF2 and tRNAiMet (the latter from the IMT4 gene). Plasmid shuffling with 5-fluoro-orotic acid (5-FOA)-containing media was then used to select for cells that had segregated off the GCD6 plasmid. As expected from the previous report, all the resulting 5-FOA-resistant colonies were extremely slow-growing on rich media (Figure 6A) with a doubling time of ∼6 h in exponential phase at 30°C (cf. 90 min for the parent strain, data not shown). Three independent 5-FOA-resistant, slow-growing colonies were assessed by immunoblotting for the presence of Gcd6p. All showed that Gcd6p was no longer present, confirming that the GCD6 plasmid had been lost from these cells (Figure 6B, lanes 2–4). Thus, in our strain only the catalytic subunit of eIF2B is deleted, but this strain has a growth rate similar to the Hannig laboratory strain where all five eIF2B subunits were deleted. One strain (called GP4115) was used for further analysis.

Fig. 6. Minimal eIF2Bε518–712 fragment provides nucleotide exchange activity in vivo. (A) growth of strains GP4113 (left) and GP4115 (right) on YPD agar. Photographed after 5 days at 30°C (B). Western blot analysis of GP4113 (lane 1) and three 5-FOA-resistant colonies (lanes 2–4, where GP4115 is lane 2). Fifteen micrograms of total soluble cell protein was loaded in each lane. Blot was probed with the specific antisera indicated to the right of each panel. (C) Growth of indicated transformants of strain GP4115 on SCGR minus uracil, leucine, isoleucine and valine (SCGR-ULIV) media. ‘Vector’ indicates plasmid pRS316 (Sikorski and Hieter, 1989) and ‘GCD6’ the low copy GCD6 URA3 plasmid pJB85 (Bushman et al., 1993); other plasmids are indicated in Figure 1. Times listed are doubling times ± SD (σn) for each strain in liquid SCGR-ULIV during exponential growth phase. (D) Growth of indicated transformants of strain GP4181 on SCGR-uracil media. ‘GCD2’ indicates the low copy GCD2 URA3 plasmid pCP62 (Pavitt et al., 1997).
We reasoned that in this strain the overexpressed eIF2 and tRNAiMet provided barely sufficient ternary complex formation rates in the absence of eIF2B function for cell growth; therefore, if catalytically active Gcd6p fragments were also expressed in these cells the rate of ternary complex formation and hence growth would be enhanced. Accordingly, strain GP4115 was transformed with plasmids to express different fragments of Gcd6p and growth rates were assessed on media containing 2% raffinose media, where the GAL1 promoter is not induced and on the same media also containing 4% galactose (Figures 1, columns SCR and SCGR, respectively, and 6C). In complete agreement with the in vitro analysis, the results show that all Gcd6p that were catalytically active in our in vitro biochemical assays improve the rate of growth of yeast in vivo. The smallest biologically active Gcd6p fragment in this in vivo assay is Gcd6p518–712. When the growth rates were compared quantitatively by monitoring A600 in liquid SCGR media, both control vectors and the non-catalytic Gcd6p581–712 fragment expressing cells grew with a doubling time of ∼360–370 min. This was improved to ∼250 min in cells expressing the Gcd6p518–712 catalytic fragment and to ∼190 min for cells expressing native eIF2B (Figure 6C; data not shown).
In the analysis described above, we asked for complementation of gcd6Δ only. It was therefore possible that the rescue of growth rate we observed in this assay was either due to the action of the overexpressed Gcd6p polypeptide alone, or was due to complex formation of the expressed Gcd6p fragments with the other four eIF2B subunits still present in this strain. From our previous analyses (see Figure 2A; Gomez and Pavitt, 2000) and that of others (Anthony et al., 2000), we believe that an intact eIF2Bε N-terminus is required for eIF2B complex formation. This would suggest that Gcd6p518–712 acts independently of the other four eIF2B subunits present in this strain to promote eIF2B biological activity. To provide an additional test of this interpretation, we repeated the growth rescue assay using a strain deleted for GCD2, which encodes the eIF2Bδ subunit. In a gcd2Δ strain transformed with Gcd6p fragments, no five-subunit eIF2B can form, and therefore any growth rescue must be due to the overexpressed Gcd6p fragments acting alone. This new strain (GP4181) was made in an identical manner to GP4115 and the series of transformation experiments was repeated with comparable results (Figure 6D; data not shown). Together, these data demonstrate that Gcd6p518–712 comprises the minimal catalytically and biologically active domain of eIF2B.
Discussion
In this study, we used a combination of in vitro functional assays and in vivo growth assays to determine the minimal fragment of eIF2B capable of catalysing nucleotide exchange. We find that a small ∼200 residue, ∼23 kDa region at the C-terminus of the largest subunit, eIF2Bε (Gcd6p in yeast), contains the sequences necessary for both eIF2 binding and exchange of guanine nucleotides. The γ subunit of eIF2B shares extensive sequence similarity over its entire length with the N-terminal 467 residues of eIF2Bε (Figure 1). These conserved regions appear to be required for protein–protein interactions between eIF2B subunits and are not required for minimal catalytic function of eIF2Bε (Figure 2; Gomez and Pavitt, 2000). The level of activity observed for this fragment was indistinguishable from the activity of the full-length isolated Gcd6p subunit (Figure 2; data not shown). This represents a reduction of at least 10-fold in the rate of exchange when compared in similar assays with the full five-subunit factor (Gomez and Pavitt, 2000). It was therefore not surprising that this fragment could not support the growth of a deletion of GCD6 without some additional help. Remarkably, Hannig and co-workers observed that cells with excess eIF2 and tRNAiMet are able to survive without eIF2B function, but have a severe growth defect (Erickson et al., 2001; Figure 6). This suggested that the rate of eIF2B-independent eIF2·GTP· Met-tRNAiMet ternary complex formation in these strains is severely growth limiting. We have shown here that expression of the Gcd6p518–712 catalytic fragment greatly enhanced the rate of growth of strains deleted for eIF2B subunits that are normally essential proteins. Together these findings provide strong evidence that Gcd6p518–712 is the minimal catalytic segment of eIF2B.
The C-terminal ∼60 residues provide the major domain for interaction with non-phosphorylated eIF2
When the sequence of Gcd6p518–712 is compared with the equivalent region from other eukaryotes, two obvious regions of relatively high sequence similarity are evident (top and bottom row in Figure 3), which are separated by a poorly conserved region. The sequences of the extreme C-terminal region of eIF2Bε are conserved with eIF5, which binds eIF2·GTP·Met-tRNAiMet to activate GTP hydrolysis during translation initiation. Asano et al. (1999) found that mutation of seven or 12 conserved acidic or aromatic residues within this region of yeast eIF2Bε to alanines (termed AA boxes 1 and 2) reduced binding of eIF2Bε to eIF2β. Similar results were reported for equivalent mutations in eIF5. Wang et al. (2001) provided evidence that this region was also important in mammalian eIF2B. Mutation of two serine residues (S706 and S707 in the rat sequence), both substrates for casein kinase II phosphorylation, reduced both eIF2 interaction and activity in vitro. While these studies implicated this region of eIF2B in eIF2 binding it was possible that the missense mutations altered the folding of this region of the molecule and that this indirectly weakens eIF2–eIF2B interaction. However, our results demonstrate that this region alone is sufficient for eIF2 binding activity (Figure 4B; construct GST–Gcd6p581–712).
We also found that deletion of these residues in construct Gcd6p1–651 eliminates both stable eIF2 binding and nucleotide exchange activity of the isolated eIF2Bε subunit (Figure 2), demonstrating that this region is both necessary and sufficient for binding eIF2. When the purified mutant five-subunit complex (eIF2Bε1–651) was analysed, we found a substantial reduction in function in both assays (Figure 5). Therefore, in contrast to the results with the single subunit where eIF2 binding and GDP exchange functions were completely eliminated, some residual activity remained. These findings suggest that the other four eIF2B subunits can provide some eIF2-binding activity, at least in vitro.
Residues 518–580 contain the eIF2B catalytic centre
The region from residues 518–580 of yeast eIF2Bε is essential for its catalytic activity, and enhances eIF2 binding (Figure 4). This region is also conserved in other eIF2Bε sequences (Figure 3, top row). These results strongly suggest that residues required for catalysis are within this region of eIF2B. We previously identified missense mutations within this region at T552 and S576 that reduced the catalytic activity of Gcd6p and led to a slow-growth phenotype in vivo (Gomez and Pavitt, 2000). These findings are consistent with this region being important for eIF2B function and suggest that this conserved region contains catalytic residues of eIF2B. We have therefore termed this region the ‘catalytic centre’. It is interesting that phosphorylation of mammalian eIF2B by GSK-3 in this region (the rat sequence is underlined in Figure 3) can reduce mammalian eIF2B activity in a similar way to the yeast mutations at T552 and S576. We suggest that all these modifications to the structure in this region directly impair the catalytic function of eIF2B.
Residues within the catalytic centre region that are conserved through all eIF2Bε proteins are good candidates to be directly involved in catalysis. It is noteworthy that residues with charged and/or bulky hydrophobic side chains have been implicated in catalysis of nucleotide exchange for other GEF proteins where G protein·GEF co-crystal structures have been determined. Several of the conserved eIF2Bε residues have similar side chains. However, subsequent mutational analysis has not always borne out these predictions. For example, in translation elongation factors Escherichia coli EF-Ts in complex with EF-Tu, EF-Ts residues D80 and F81 in the conserved TDFV motif were implicated in mediating nucleotide exchange (Kawashima et al., 1996). However, mutation of these two residues affected EF-Ts function only 2- to 3-fold each, or 10-fold when both were altered (Zhang et al., 1996). Instead, mutations altering EF-Ts interaction with EF-Tu had a more pronounced effect on rates of nucleotide exchange (Zhang et al., 1998; Wieden et al., 2002). In contrast to this, K205 of S.cerevisiae eEF1Bα in its complex with eEF1A has a catalytic role (Andersen et al., 2001). Therefore, in conclusion, directed mutagenesis of conserved residues within the catalytic centre region may provide clues to which residues of eIF2Bε are directly involved in catalysis of nucleotide exchange and future work will address this.
Models for action of the minimal catalytic domain
The results presented and discussed here are consistent with a two-step binding model for guanine-nucleotide release catalysed by eIF2B. In this model (shown in Figure 7A) the C-terminal AA box domain of eIF2Bε makes contact with the lysine-rich domain of eIF2β (Asano et al., 1999). This interaction then allows binding of the catalytic centre region and nucleotide exchange to proceed. Where the catalytic centre interacts with eIF2 is not known. As all known GEFs bind directly to the G protein subunit to mediate nucleotide exchange, this seems a likely option for eIF2. In contrast, Gcd6p581–712 can only make the AA box–eIF2β interaction (Figure 7B). However, as no direct interaction between eIF2B and eIF2γ has been detected to date, it is possible that another mechanism may operate. Perhaps binding of the eIF2B catalytic centre to eIF2β induces a conformational change that is transmitted from eIF2β to eIF2γ to induce nucleotide exchange (Figure 7C).

Fig. 7. A model for catalysis of GDP release from eIF2 by Gcd6p. (A) A two-step model is proposed. Gcd6p518–712 binds through contacts between AA box motifs and eIF2β. The catalytic centre then binds eIF2γ catalysing GDP release. (B) Gcd6p581–712 is able to bind eIF2β only and GDP is not released. (C) Alternative model where all Gcd6p518–712 contacts are via eIF2β. A conformational change in eIF2β induced upon binding Gcd6p518–712 is transmitted to eIF2γ (arrow), triggering nucleotide release.
Models very similar to these for eIF2B function could apply to eIF5 action. eIF5 stimulates GTP hydrolysis within 40S ribosome-bound eIF2·GTP·Met-tRNAiMet ternary complexes. eIF5 shares a similar C-terminal AA box eIF2β-binding domain with eIF2Bε, and it was recently demonstrated that a separate conserved N-terminal domain is required for eIF5 to stimulate GTPase activity (Das et al., 2001; Paulin et al., 2001). Therefore, the arrangement of similar C-terminal eIF2β binding domains (AA boxes) and separate catalytic domains that promote GEF and GAP function appears to be common to these eIF2-controlling translation factors. However, eIF2B functions with eIF2·GDP while eIF5 acts on eIF2·GTP·Met-tRNAiMet; therefore, although the AA box–eIF2β interaction is necessary for both proteins to interact with eIF2, these interactions alone are unlikely to determine the specificity of binding. For eIF2B this specificity may be due to the catalytic centre. Our results suggest that the remainder of the ∼294 kDa five-subunit eIF2B complex functions to stimulate and regulate this activity to control the rate of translation, either globally or for specific mRNAs.
Materials and methods
Yeast cell culture
Standard yeast media were used throughout (Rose et al., 1990): YPD (2% yeast extract, 1% peptone, 2% dextrose); SC (synthetic complete with 2% dextrose); SCR (synthetic complete with 2% raffinose); SCGR (synthetic complete with 4% galactose and 2% raffinose). Amino acids and/or uracil were omitted from SC media to maintain plasmid selection as necessary.
Yeast strains and genetic methods
Standard genetic methods were used to construct yeast strains (Guthrie and Fink, 1991). Transformation of yeast strains with plasmids was by the lithium acetate method (Gietz et al., 1995). Plasmid shuffling employing 5-FOA was carried out as described previously (Boeke et al., 1987). Yeast strains used are described in Table I. PEP4 was disrupted in GP3667 using a pep4::LEU2 allele as described previously (Pavitt et al., 1998) to create GP3889. Deletion of PEP4 enhanced expression levels and reduced proteolysis of many of the Gcd6p fragments made (data not shown).
Table I. Yeast strains used in this study.
| Strain | Genotype | Construction/reference |
|---|---|---|
| GP3040 |
MATa ura3-52 trp1-63 leu2-3 leu2-112 gcd2Δ pCP62[URA3 CEN4 GCD2] |
Pavitt et al. (1997) |
| GP3511 |
MATa leu2-3 leu2-112 ura3-52::his4-lacZ ino1 gcn2Δ pep4::LEU2 sui2Δ pAV1089[SUI2 SUI3 GCD11-6×His 2 µm URA3] |
Pavitt et al. (1998) |
| KAY16/GP3750 |
MATa leu2-3 leu2-112 ura3-52::his4-lacZ ino1 gcd6Δ gcn2Δ::hisG pJB85[GCD6 CEN6 URA3] |
Asano et al. (1999) |
| GP3667 |
MATα trp1-63 ura3-52 leu2-3 leu2-112 GAL2 gcn2Δ |
Gomez and Pavitt (2000) |
| GP3889 |
MATa trp1-63 ura3-52 leu2-3 leu2-112 GAL2 gcn2Δ pep4::LEU2 |
pep4::LEU2 in GP3667; this study |
| GP4113 |
KAY16/ + pAV1732[h.c. eIF2 IMT4 2 µm LEU2] |
Transformation of KAY16; this study |
| GP4115 |
MATa leu2-3 leu2-112 ura3-52 ino1 gcd6Δ gcn2Δ::hisG[HIS4-lacZ::ura3-52] pAV1732[SUI2 SUI3 GCD11-6×His IMT4 2 µm LEU2] |
GP4113 following 5-FOA growth; this study |
| GP4173 |
MATa ura3-52 trp1-63 leu2-3 leu2-112 gcd2Δ pCP62[URA3 CEN4 GCD2] pAV1732[hc eIF2 IMT4 LEU2] |
Transformation of GP4040; this study |
| GP4181 | MATa ura3-52 trp1-63 leu2-3 leu2-112 gcd2Δ pAV1732[h.c. eIF2 IMT4 LEU2] | GP4173 following 5-FOA growth; this study |
Plasmid constructions
Standard methods were used to construct all plasmids (Sambrook and Russell, 2001). Details can be found in the Supplementary data.
Protein expression and purification from yeast
His6-tagged yeast eIF2 was purified from strain GP3511 by immobilized metal affinity chromatography (IMAC) using Ni-NTA resin (Qiagen) followed by heparin–Sepharose chromatography (Amersham) as described previously (Pavitt et al., 1998). As strain GP3511 is deleted for the sole yeast eIF2α kinase (gcn2Δ), eIF2 preparations are entirely non-phosphorylated at the regulatory Ser51 site. Expression of FH6-tagged Gcd6p fragments in strains GP3667 or GP3889 and their purification by IMAC using Ni-NTA resin was as described previously (Gomez and Pavitt, 2000). Purification of FH6-tagged wild-type five-subunit eIF2B overexpressed in strain GP3667 was also by IMAC using Ni-NTA resin as described previously (Gomez and Pavitt, 2000). Purification of FH6 five-subunit eIF2Bε1–651 was identical to that for wild type except that pAV1687 (2 µm URA3 FH6-GCD1 gcd6-P652*) was transformed into strain GP3889 in place of pAV1533 (2 µm URA3 FH6-GCD1 GCD6) with pAV1494 (2 µm LEU2 GCN3 GCD7 GCD2).
GST fusion protein expression and purification
Plasmids for GST and GST fusion protein expression were transformed into GP3889 and grown in SC-uracil, leucine, isoleucine and valine (SC-ULIV) to saturation. Cells from these cultures were subcultured into SCGal-ULIV (0.4% w/v glucose + 2% w/v galactose) and grown to A600 3–5. All subsequent steps were performed at 0–4°C. Cells were harvested by centrifugation at 5000 g for 15 min, washed and resuspended in 2 ml/g wet wt ice-cold GST lysis buffer [20 mM Tris–HCl pH 7.5, 100 mM KCl, 1 mM EDTA, 1 mM MgCl2, 1 mM dithiothreitol, 1× complete EDTA-free protease inhibitor tablet (Roche)] and cells were lysed by vortexing with glass beads. The resulting cell lysate was clarified by sequential centrifugation at 5000 g for 5 min and 100 000 g for 1 h. Supernatants were applied to a Hi trap GST 1 ml column, pre-equilibrated with the same buffer. After extensive washing, the bound proteins were eluted with reduced glutathione (Sigma) in elution buffer (100 mM Tris–HCl pH 8.8, 120 mM KCl, 0.1% Triton, 20 mM reduced glutathione). Protein-containing fractions were combined, dialysed using (10 kDa mol. wt cut off) dialysis cassettes (Pierce) into storage buffer (100 mM Tris–HCl pH 8.0, 100 mM KCl, 3 mM MgCl2, 5% glycerol, 5 mM β-mercaptoethanol) and stored in aliquots at –80°C. Protein concentrations were determined by the Bradford assay (Bio-Rad) using BSA as a standard (Bradford, 1976).
In vitro nucleotide exchange assays
In vitro nucleotide exchange assays using eIF2·[3H]GDP substrate and 100-fold excess unlabelled GDP and partially purified eIF2B or eIF2Bε (Gcd6p) fragments were performed as described previously (Pavitt et al., 1998; Gomez and Pavitt, 2000).
In vitro protein–protein interaction assays
Assays for interaction between eIF2 and FH6-tagged Gcd6p proteins were performed using FLAG M2 affinity resin as described previously (Gomez and Pavitt, 2000). Assays where GST-tagged Gcd6p fragments replaced FH6-tagged Gcd6p were performed similarly, but using glutathione– Sepharose 4B resin (Amersham Bioscience) in place of FLAG M2 resin. Purified GST or GST–Gcd6p fusion proteins (∼4 µg) were bound to prewashed glutathione–Sepharose resin (40 µl of 50% slurry) made up to 400 µl with binding buffer (20 mM Tris–HCl pH 7.5, 100 mM KCl, 2 mM MgCl2, 5 mM β-mercaptoethanol containing 2.5 mg/ml BSA) at 4°C for 2 h on a blood tube rotator. Resin was pelleted by low-speed centrifugation (2000 g for 2 min) and washed three times to remove any unbound protein, and 1 µg eIF2 was added with fresh binding buffer. Incubation was continued for 2 h and the washing steps were repeated. Proteins remaining bound to the resin were eluted into 20 µl 1× Laemmli sample buffer (Sambrook and Russell, 2001).
Immunoblotting
Proteins and protein:protein interaction assays were analysed by SDS–PAGE on 12% PAGE gels. Proteins were visualized by immunoblotting using specific antisera: mouse monoclonal: αFLAG M2 (Sigma); rabbit polyclonal: αGST (Sigma). eIF2B and eIF2 subunit-specific rabbit polyclonal antisera have been described previously (Dever et al., 1995). Detection was by horseradish peroxidase-linked anti-rabbit/mouse secondary antibodies and enhanced chemiluminescence (Amersham Biosciences).
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank members of the Pavitt, Ashe and Grant laboratories for useful discussions during the course of these experiments, and Mark Ashe for critical reading of the manuscript. This work was supported by a career development award from the UK Medical Research Council to G.D.P. and by a UMIST Life Sciences Initiative studentship to S.S.M.
References
- Andersen G.R., Valente,L., Pedersen,L., Kinzy,T.G. and Nyborg,J. (2001) Crystal structures of nucleotide exchange intermediates in the eEF1A–eEF1Bα complex. Nat. Struct. Biol., 8, 531–534. [DOI] [PubMed] [Google Scholar]
- Anthony T.G., Fabian,J.R., Kimball,S.R. and Jefferson,L.S. (2000) Identification of domains within the epsilon-subunit of the translation initiation factor eIF2B that are necessary for guanine nucleotide exchange activity and eIF2B holoprotein formation. Biochim. Biophys. Acta, 1492, 56–62. [DOI] [PubMed] [Google Scholar]
- Asano K., Krishnamoorthy,T., Phan,L., Pavitt,G.D. and Hinnebusch,A.G. (1999) Conserved bipartite motifs in yeast eIF5 and eIF2Bε, GTPase-activating and GDP–GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J., 18, 1673–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe M.P., Slaven,J.W., De Long,S.K., Ibrahimo,S. and Sachs,A.B. (2001) A novel eIF2B-dependent mechanism of translational control in yeast as a response to fusel alcohols. EMBO J., 20, 6464–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke J.D., Trueheart,J., Natsoulis,G. and Fink,G.R. (1987) 5-fluoroorotic acid as a selective agent in yeast molecular genes. Methods Enzymol., 154, 164–175. [DOI] [PubMed] [Google Scholar]
- Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Bushman J.L., Asuru,A.I., Matts,R.L. and Hinnebusch,A.G. (1993) Evidence that GCD6 and GCD7, translational regulators of GCN4 are subunits of the guanine nucleotide exchange factor for eIF-2 in Saccharomyces cerevisiae. Mol. Cell. Biol., 13, 1920–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.-J. (2000) Heme-regulated eIF2α kinase. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 529–546.
- Cherfils J. and Chardin,P. (1999) GEFs: structural basis for their activation of small GTP-binding proteins. Trends Biochem. Sci., 24, 306–311. [DOI] [PubMed] [Google Scholar]
- Christianson T.W., Sikorski,R.S., Dante,M., Shero,J.H. and Hieter,P. (1992) Multifunctional yeast high-copy-number shuttle vectors. Gene, 110, 119–122. [DOI] [PubMed] [Google Scholar]
- Das S. and Maitra,U. (2000) Mutational analysis of mammalian translation initiation factor 5 (eIF5): role of interaction between the beta subunit of eIF2 and eIF5 in eIF5 function in vitro and in vivo. Mol. Cell. Biol., 20, 3942–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S., Ghosh,R. and Maitra,U. (2001) Eukaryotic translation initiation factor 5 functions as a GTPase-activating protein. J. Biol. Chem., 276, 6720–6726. [DOI] [PubMed] [Google Scholar]
- Dever T.E. (2002) Gene-specific regulation by general translation factors. Cell, 108, 545–556. [DOI] [PubMed] [Google Scholar]
- Dever T.E., Yang,W., Astrom,S., Bystrom,A.S. and Hinnebusch,A.G. (1995) Modulation of tRNAiMet, eIF-2 and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2·GTP·Met-tRNAiMet ternary complexes. Mol. Cell. Biol., 15, 6351–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson F.L., Nika,J., Rippel,S. and Hannig,E.M. (2001) Minimum requirements for the function of eukaryotic translation initiation factor 2. Genetics, 158, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian J.R., Kimball,S.R., Heinzinger,N.K. and Jefferson,L.S. (1997) Subunit assembly and guanine nucleotide exchange activity of eukaryotic initiation factor-2B expressed in Sf9 cells. J. Biol. Chem., 272, 12359–12365. [DOI] [PubMed] [Google Scholar]
- Gietz R.D., Willems,A.R. and Woods,R.A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast, 11, 355–560. [DOI] [PubMed] [Google Scholar]
- Gomez E. and Pavitt,G.D. (2000) Identification of domains and residues within the epsilon subunit of eukaryotic translation initiation factor 2B (eIF2Bε) required for guanine nucleotide exchange reveals a novel activation function promoted by eIF2B complex formation. Mol. Cell. Biol., 20, 3965–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C. and Fink,G.R. (eds) (1991) Guide to Yeast Genetics and Molecular Biology. Academic Press, Inc., San Diego, CA.
- Harding H.P., Novoa,I., Zhang,Y., Zeng,H., Wek,R., Schapira,M. and Ron,D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell, 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Hinnebusch A.G. (1996) Translational control of GCN4: gene-specific regulation by phosphorylation of eIF2. In Hershey,J.W.B., Mathews,M.B. and Sonenberg,N. (eds), Translational Control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 199–244.
- Hinnebusch A.G. (2000) Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 185–243.
- Kaufman R.J. (2000) Double-stranded RNA activated protein kinase PKR. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 503–527.
- Kawashima T., Berthet-Colominas,C., Wulff,M., Cusack,S. and Leberman,R. (1996) The structure of the Escherichia coli EF-Tu·EF-Ts complex at 2.5Å resolution. Nature, 379, 511–518. [DOI] [PubMed] [Google Scholar]
- Kimball S.R., Fabian,J.R., Pavitt,G.D., Hinnebusch,A.G. and Jefferson,L.S. (1998) Regulation of guanine nucleotide exchange through phosphorylation of eukaryotic initiation factor eIF2α. Role of the α- and δ-subunits of eIF2B. J. Biol. Chem., 273, 12841–12845. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy T., Pavitt,G.D., Zhang,F., Dever,T.E. and Hinnebusch,A.G. (2001) Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2α) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol., 21, 5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leegwater P.A. et al. (2001) Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat. Genet., 29, 383–388. [DOI] [PubMed] [Google Scholar]
- Mitchell D.A., Marshall,T.K. and Deschenes,R.J. (1993) Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast, 9, 715–723. [DOI] [PubMed] [Google Scholar]
- Paulin F.E., Campbell,L.E., O’Brien,K., Loughlin,J. and Proud,C.G. (2001) Eukaryotic translation initiation factor 5 (eIF5) acts as a classical GTPase-activator protein. Curr. Biol., 11, 55–59. [DOI] [PubMed] [Google Scholar]
- Pavitt G.D., Yang,W. and Hinnebusch,A.G. (1997) Homologous segments in three subunits of the guanine nucleotide exchange factor eIF2B mediate translational regulation by phosphorylation of eIF2. Mol. Cell. Biol., 17, 1298–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavitt G.D., Ramaiah,K.V., Kimball,S.R. and Hinnebusch,A.G. (1998) eIF2 independently binds two distinct eIF2B subcomplexes that catalyze and regulate guanine-nucleotide exchange. Genes Dev., 12, 514–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. and Harding,H.P. (2000) PERK and translational control by stress in the endoplasmic reticulum. In Sonenberg,N., Hershey,J.W.B. and Mathews,M.B. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 547–560.
- Rose M.D., Winston,F. and Hieter,P. (1990) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sambrook J. and Russell,D.W. (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprang S.R. and Coleman,D.E. (1998) Invasion of the nucleotide snatchers: structural insights into the mechanism of G protein GEFs. Cell, 95, 155–158. [DOI] [PubMed] [Google Scholar]
- van der Knaap M.S., Leegwater,P.A., Konst,A.A., Visser,A., Naidu,S., Oudejans,C.B., Schutgens,R.B. and Pronk,J.C. (2002) Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann. Neurol., 51, 264–270. [DOI] [PubMed] [Google Scholar]
- Vazquez de Aldana C.R. and Hinnebusch,A.G. (1994) Mutations in the GCD7 subunit of yeast guanine nucleotide exchange factor eIF-2B overcome the inhibitory effects of phosphorylated eIF-2 on translation initiation. Mol. Cell. Biol., 14, 3208–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Paulin,F.E., Campbell,L.E., Gomez,E., O’Brien,K., Morrice,N. and Proud,C.G. (2001) Eukaryotic initiation factor 2B: identification of multiple phosphorylation sites in the ε-subunit and their functions in vivo. EMBO J., 20, 4349–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh G.I. and Proud,C.G. (1993) Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem. J., 294, 625–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieden H.J., Gromadski,K., Rodnin,D. and Rodnina,M.V. (2002) Mechanism of elongation factor (EF)-Ts-catalyzed nucleotide exchange in EF-Tu. Contribution of contacts at the guanine base. J. Biol. Chem., 227, 6032–6036. [DOI] [PubMed] [Google Scholar]
- Yang W. and Hinnebusch,A.G. (1996) Identification of a regulatory subcomplex in the guanine nucleotide exchange factor eIF2B that mediates inhibition by phosphorylated eIF2. Mol. Cell. Biol., 16, 6603–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Li,X. and Spremulli,L.L. (1996) Role of the conserved aspartate and phenylalanine residues in prokaryotic and mitochondrial elongation factor Ts in guanine nucleotide exchange. FEBS Lett., 391, 330–332. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Yu,N.J. and Spremulli,L.L. (1998) Mutational analysis of the roles of residues in Escherichia coli elongation factor Ts in the interaction with elongation factor Tu. J. Biol. Chem., 273, 4556–4562. [DOI] [PubMed] [Google Scholar]