

Abstract
p47phox is a key cytosolic subunit required for activation of phagocyte NADPH oxidase. The X-ray structure of the p47phox PX domain revealed two distinct basic pockets on the membrane-binding surface, each occupied by a sulfate. These two pockets have different specificities: one preferentially binds phosphatidylinositol 3,4-bisphosphate [PtdIns(3,4)P2] and is analogous to the phophatidylinositol 3-phosphate (PtdIns3P)-binding pocket of p40phox, while the other binds anionic phospholipids such as phosphatidic acid (PtdOH) or phosphatidylserine. The preference of this second site for PtdOH may be related to previously observed activation of NADPH oxidase by PtdOH. Simultaneous occupancy of the two phospholipid-binding pockets radically increases membrane affinity. Strikingly, measurements for full-length p47phox show that membrane interaction by the PX domain is masked by an intramolecular association with the C-terminal SH3 domain (C-SH3). Either a site-specific mutation in C-SH3 (W263R) or a mimic of the phosphorylated form of p47phox [Ser(303, 304, 328, 359, 370)Glu] cause a transition from a closed to an open conformation that binds membranes with a greater affinity than the isolated PX domain.
Keywords: NADPH oxidase/p40/phagocyte/phosphatidic acid/phosphoinositide/SH3 domains
Introduction
NADPH oxidase plays a decisive role in the ability of phagocytes to defend against bacterial infections. The enzyme catalyses the transfer of an electron from NADPH to oxygen to form superoxide that acts as a precursor to microbicidal oxidants. The NADPH oxidase activity also results in release of proteases within the phagosome (Reeves et al., 2002). The phagocyte oxidase consists of a membrane-bound, catalytic gp91phox–p22phox heterodimer known as flavocytochrome b558, and cytosolic regulatory subunits consisting of p40phox, p47phox, p67phox and Rac (reviewed in Babior, 1999). p40phox, p47phox and p67phox form a 1:1:1 complex in the resting state (Lapouge et al., 2002). Because of the potentially damaging reactions that can be initiated by the oxidase, the enzymatic activity is tightly regulated. Electron microscopy studies show that gp91phox and p22phox are located in the membranes of specific granules, gelatinase granules and secretory vesicles (reviewed in Seguchi and Kobayashi, 2002). Activation of the oxidase requires translocation of the cytosolic subunits to the membrane-bound flavocytochrome, yet the mechanism of this translocation is not clear. The p47phox subunit appears to have a key role in translocation. Studies of cells from chronic granulomatous disease (CGD) patients deficient in p47phox indicate that this subunit is required for translocation of p40phox and p67phox (Heyworth et al., 1991; Uhlinger et al., 1993; Dusi et al., 1996). Translocation of the cytosolic subunits to membranes could be the consequence of direct membrane interactions or interactions with membrane-localized proteins. Interaction between p47phox and the intrinsic membrane protein p22phox may play a role in translocating p47phox. Other interactions between NADPH oxidase cytosolic subunits and membrane-interacting proteins have been reported, but the significance of these for the activation of NADPH oxidase is not yet clear, e.g. both p47phox and p40phox interact with moesin, a protein with both membrane- and actin-binding domains (Wientjes et al., 2001), and p67phox interacts with JFC1, a protein that has a 3-phosphoinositide-binding domain (McAdara Berkowitz et al., 2001).
Recently, it was shown that both p47phox and p40phox can interact directly with membranes via their N-terminal PX domains (Ago et al., 2001; Ellson et al., 2001; Kanai et al., 2001). These direct membrane interactions may play a role in translocation, or they may be important for activation of the NADPH oxidase once the cytosolic subunits have already associated with the membrane. The PX domain of p47phox preferentially binds to vesicles containing phosphatidylinositol 3,4-bisphosphate [PtdIns(3,4)P2] (Kanai et al., 2001), whereas the p40phox-PX, like most other PX domains, has a strong preference for phosphatidylinositol 3-phosphate (PtdIns3P) (Ellson et al., 2001; Kanai et al., 2001; Yu and Lemmon, 2001). Binding of PtdIns3P to p40phox-PX stimulates NADPH oxidase activity in vitro (Ellson et al., 2001). We previously reported the structure of the p40phox PX domain in a complex with PtdIns3P (Bravo et al., 2001); however, the lipid-binding pocket in this structure was very different from the analogous pocket in the NMR structure of the p47phox-PX (Hiroaki et al., 2001). We report here a high-resolution X-ray structure of the p47phox PX domain showing two basic pockets occupied by sulfates that represent two potential phospholipid-binding sites. Using structure-guided mutagenesis and phospholipid-binding measurements, we show that these two sites strongly synergize in membrane binding. In the context of the full-length p47phox, membrane binding by both sites is masked by intramolecular interaction with the C-terminal SH3 domain (C-SH3) and unmasked in a phosphorylation mimic.
Results
Description of the overall structure
The X-ray structure of the p47phox PX domain was determined at 2.0 Å resolution by multiple anomalous dispersion (MAD) techniques for a crystal of selenomethionine-substituted protein (Table I). The overall fold consists of a three-stranded antiparallel β-sheet followed by a helical subdomain containing four α-helices and a polyproline II region (Figure 1). There are two sulfates bound on the same surface of the domain. One sulfate is in a basic pocket with a location analogous to the PtdIns3P-binding pocket of p40phox-PX (Bravo et al., 2001). The second sulfate is bound in a distinct shallow pocket that has no analogue in previously characterized PX domains (Figure 1).
Table I. Data collection, structure determination and refinement statistics.
| Data collection and MAD phasing statistics | ||
|---|---|---|
| Data set | Peakc | Inflectionc |
| Resolution (Å) |
2.0 |
2.0 |
| Observations/unique reflections |
83 922/24 307 |
84 280/24 356 |
| Completeness (last shell) % |
93.6 (74.1) |
93.7 (74.1) |
|
Rmergea |
0.050 |
0.044 |
| <I/σ> (last shell) |
20.1 (1.5) |
19.7 (1.3) |
| Phasing power (iso)b |
– |
0.74 |
| Phasing power (anom)b |
2.1 |
1.3 |
| Refinement statistics |
|
|
| Resolution (Å) |
38.9–2.0 |
|
| Protein atoms |
3018 |
|
| Heterogen atoms |
35 |
|
| Water molecules |
204 |
|
|
Rcrystd |
0.22 |
|
|
Rfreed (% data used) |
0.27 (5.0) |
|
| R.m.s.d. from idealitye |
|
|
| Bonds |
0.020 Å |
|
| Angles |
1.7° |
|
| Dihedrals | 25° | |
Overall figure of merit 0.43 before solvent flattening and 0.88 after solvent flattening.
aRmerge = ΣhklΣi |Ii(hkl) – <I(hkl)>|/ΣhklΣi Ii(hkl)
bThe phasing power is defined as the ratio of the r.m.s. value of the heavy atom structure factor amplitudes to the r.m.s. value of the lack-of-closure error.
cData sets were collected at ESRF beamline ID29.
dRcryst and Rfree = Σ |Fobs – Fcalc|/ΣFobs; Rfree calculated with the percentage of the data shown in parentheses.
eR.m.s. deviations for bond angles and lengths with regard to Engh and Huber parameters.

Fig. 1. The structure of the p47phox PX domain. The ribbon representation is coloured from red at the N-terminus to blue at the C-terminus. Selected residues are shown in stick representation. Residues in the phosphoinositide pocket are shown in red and those lining the second anion-binding pocket are coloured magenta. The sulfates bound in the two pockets are shown in CPK representation.
The Cα positions of the p47phox and p40phox PX domains closely superimpose throughout the domain except in the region from 59 to 86 (p47phox numbering) containing helix α1′, a distorted, left-handed PPII helix and the succeeding membrane insertion loop (residues 77–86). This 59–86 region also deviates greatly from what was observed in the NMR study of the p47phox-PX (Hiroaki et al., 2001). However, because part of this region forms much of the contact area between molecules in the asymmetric unit, some of these differences may arise due to crystal contacts. The membrane insertion loop is important for the shape of the phosphoinositide-binding pocket. Unlike the recently reported NMR structure of the unliganded Vam7p-PX, which has a PtdIns3P-binding pocket nearly identical to the p40phox-PX (Lu et al., 2002), the p47phox-PX has a phosphoinositide-binding pocket with considerable conformational differences. This conformational variation in the membrane insertion loop is not surprising given the sequence variation among PX domains in this region.
The structure of the phosphoinositide-binding pocket
Superimposing the p40phox PX domain on p47phox shows that the sulfate in the p47phox phosphoinositide-binding pocket is at the position of the 3-phosphate of the phosphoinositide bound to the p40phox-PX (Figure 2). The sulfate is hydrogen bonded to Arg43 and is within van der Waals contact distance of Arg90 (Figure 3). These two residues are equivalent to Arg58 and Arg105 of the p40phox PX domain, which are critical for PtdIns3P binding to the p40phox-PX and interact with the 3-phosphate and 4- and 5-OH groups, respectively. The most striking feature of the p47phox-PX phosphoinositide-binding pocket in comparison with p40phox-PX is the wide crevice between the C-terminal end of β2 and the N-terminal end of α2 (Figure 4).

Fig. 2. A comparison of the phosphoinositide-binding pockets of the p47phox and p40phox PX domains. The Cα atoms of the two domains were superimposed. The p47phox-PX is shown as a grey ribbon, while for p40phox the main chain ribbon (cyan) is shown only in the region of the phosphoinositide-binding pocket where the structures differ greatly. The side chains of residues that are critical for phosphoinositide binding are shown, with residue numbers for p40phox in parentheses. The side chain of Pro78 that protrudes into the binding pocket in p47phox is also illustrated. The bound PtdIns3P from the p40phox-PX structure is also shown (coloured yellow, red and cyan for carbon, oxygen and phosphorus, respectively). The sulfate in the phosphoinositide-binding pocket of p47phox-PX (grey bonds and magenta atoms) coincides with the location of the 3-phosphate of PtdIns3P. Arg42 that is associated with a CGD mutation points away from the binding site and has a role in stabilizing p47phox (Heyworth and Cross, 2002).
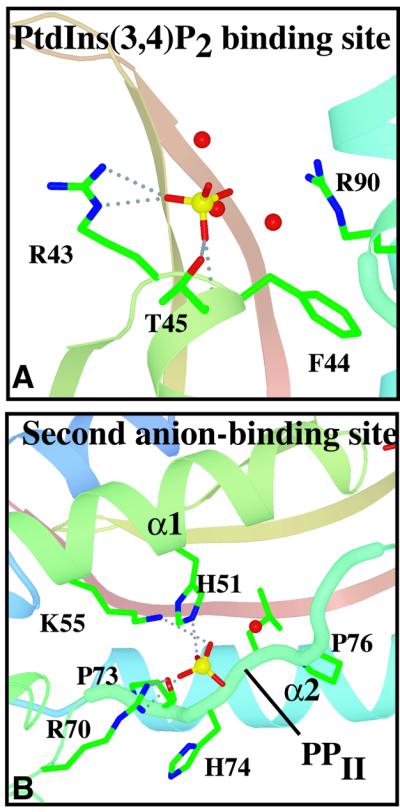
Fig. 3. Interactions with the sulfates bound in the phosphoinositide-binding and second anion-binding pockets of p47phox-PX. Dashed lines represent hydrogen bonding interactions with the protein. Ordered waters associated with the sulfates are shown as red spheres.

Fig. 4. Solid representations of the p40phox and p47phox PX domains. (A) View of the PtdIns3P (magenta) bound to the p40phox-PX. The principal ligands of the phosphoinositide, Arg58 and Arg105, are highlighted in blue, and Tyr59 at the bottom of the pocket is shown in yellow. (B) View of the membrane-binding surface of the p47phox-PX showing both phospholipid-binding sites with basic residues highlighted in blue. The sulfate in the phosphoinositide-binding site is shown in stick representation (coloured cyan). PtdIns3P from the superimposed structure of the p40phox-PX is illustrated (stick representation). The 3-phosphate of the PtdIns3P coincides with the bound sulfate in the p47phox phosphoinositide pocket. The Pro78 that sterically clashes with the PtdIns3P is highlighted in brown. The sulfate in the second anion-binding site is shown in CPK representation (coloured red and brown). (C) A model of a PtdIns(3,4)P2 bound in the p47phox-PX phosphoinositide-binding pocket. To avoid steric clashes, the inositide ring has been rotated ∼90° while maintaining the 3- and 4-phosphates in contact with the side chains of Arg43 and Arg90, respectively. The 4-phosphate (yellow) is extending into the crevice between β2 and α2. This crevice is filled by residues 97–100 in p40phox [as seen in (A)].
Within the p47phox-PX PtdIns(3,4)P2-binding site, Arg43 [the middle residue of the conserved (R/K)- (R/K)-(F/Y) motif located in the β3/α1 loop] superimposes closely with p40phox Arg58 and presumably has the same role as the principal 3-phosphate ligand (Figure 2). Arg90 (in the α2 helix) superimposes with p40phox Arg105 and, by analogy with p40phox, would be best placed to act as the ligand of the 4- and 5-positions of the phosphoinositide. However, if PtdIns(3,4)P2 were placed in the p47phox pocket in the same orientation as the PtdIns3P in p40phox, there would be a few steric clashes caused by differences in the shapes of the pockets. First, there is no room to accommodate the 4-phosphate because of the proximity of Arg90. Secondly, part of the pocket that binds PtdIns3P in p40phox is sterically hindered by the Pro78 side chain because the main chain of the region from 78 to 80 adopts a short 310 helix as opposed to the extended conformation of the analogous region, 90–94, in p40phox (Figures 2 and 4). In p47phox, Pro78 covers most of the Phe44 side chain. The residue analogous to Phe44 in p40phox (Tyr59) is essential for phospholipid binding and contacts one face of the inositide ring, suggesting either that phosphoinositides bind somewhat differently to these two PX domains or that binding to p47phox is accompanied by a conformational change that removes the occlusion imposed by Pro78. Given the steric clashes presented by both Arg90 and the 78–80 turn, it is most likely that the PtdIns(3,4)P2 headgroup would be rotated in the binding pocket so that the 3- and 4-phosphates interact with Arg43 and Arg90, respectively, and that the inositide ring would be shifted into the crevice created between the ends of β2 and α2 (Figure 4C). This crevice provides additional space that could accommodate the alternative phosphoinositide orientation in the p47phox-binding pocket relative to p40phox and is created by the very different conformation of the p47phox 83–85 turn. In the p40phox pocket, the analogous region (97–100) folds into the pocket and closes the crevice (Figure 4).
The structure of the second anion-binding pocket
Besides the bound sulfate in the putative phosphoinositide-binding pocket, the structure revealed another sulfate bound in a separate, shallower basic pocket. This secondary pocket is on the same surface of the domain as the phosphoinositide-binding pocket and is separated from it by a ridge made up of the membrane insertion loop (Figures 1 and 4). Walls of this pocket are formed by the side chains of Arg70, Lys55 and His51, giving it a basic character. The guanidinium group of Arg70 forms an extensive set of hydrogen bonding interactions both with the bound sulfate and with the backbone of Pro73 in the polyproline helix of the conserved PXXP motif (residues 73–76, Figures 1 and 3). The second anion-binding site is not present in either of the two PX domains whose structures were determined previously, p40phox-PX and Vam7p-PX. The presence of two basic residues analogous to Arg70 and Lys55 is an unusual feature among PX domain sequences, and only the PX domains from p47phox and phospholipase D (PLD) appear to have such a feature (Figure 5).

Fig. 5. Sequence alignment of several PX domains. The secondary structure elements and residue numbers of p47phox are shown above its sequence. The p47phox residues shown by mutagenesis to be involved in membrane binding and the p40phox residues that interact with the bound PtdIns3P are underlined. The basic residues in the second anion-binding pocket of p47phox are marked with asterisks. The regions containing the strongly conserved (R/K)-(R/K)-(Y/F) and PXXP motifs are boxed with thin lines. The portions of p40phox, p47phox and Vam7p that structurally superimpose the best are enclosed in boxes with thick lines. The sequences of PLD1 and PLD2 suggest that their PX domains, like p47phox, may have a second anion-binding pocket.
Arg70 is part of an (R/K)XXPXXP sequence motif that has been defined as a type I SH3-interacting motif (Mayer, 2001). A small subset of PX domains (besides p47phox and PLD) contain this motif, including the PX domains of the mammalian proteins FISH and sorting nexin 8, and the yeast proteins Mvp1 and Bem1, suggesting that these PX domains may, like p47phox-PX, bind to SH3 domains. In structures of complexes of SH3 domains with type I motif-containing peptides, the basic residue of the motif uses its side chain to interact with the negatively charged specificity pocket of the SH3 domain (Mayer, 2001), and the Cδ atoms of the prolines from the PXXP motif occupy adjacent hydrophobic pockets on the surface of the SH3 domain. In p47phox, only one proline (Pro76) from the PXXP motif is accessible for interaction, while the other (Pro73) is buried (Hiroaki et al., 2001; and Figure 1). In addition, the Arg70–Pro73 backbone interaction that we see in p47phox would preclude Arg70 binding in an SH3 domain specificity pocket. These observations suggest that if the proline-rich region of the p47phox-PX were to interact with the C-SH3 in a typical manner, p47phox-PX would have to undergo a conformational change.
Phosphoinositide specificity of the p47phox PX domain
In order to determine the roles of the two apparent phospholipid-binding sites suggested by the structure and to elucidate the mechanism of membrane binding, we measured the vesicle binding of wild-type and a series of site-specific mutants by surface plasmon resonance (SPR) and sedimentation measurements.
We first measured the binding of wild-type p47phox-PX to various phosphoinositide-containing lipid vesicles immobilized to the sensor surface by kinetic SPR analysis to determine the effects of phosphoinositides on membrane binding of p47phox-PX. In the absence of phosphoinositide, p47phox-PX has extremely low affinity for 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)/1-palmitoyl-2-oleoyl-sn-glycero-phosphoethanolamine (POPE) (80:20) vesicles (Kd >20 µM). Increasing the concentration of PtdIns(3,4)P2 in the vesicles from 0 to 3% dramatically increases the affinity (Kd = 38 nM) (Table II). To validate the Kd value determined from the kinetic SPR analysis, we also determined the Kd for p47phox-PX by equilibrium SPR analysis and by a sedimentation assay using lipid-coated beads (Kim et al., 1997) and radiolabelled PX domain. As shown in Figure 6, the Kd values (38 ± 0.1 and 30 ± 7 nM) calculated from the equilibrium binding isotherms agree well with the Kd determined from the kinetic analysis. Table II shows that p47phox-PX has 18- to 26-fold greater affinity for PtdIns(3,4)P2 than for any other tested phosphoinositide, confirming the reported specificity of the p47phox-PX for PtdIns(3,4)P2 (Kanai et al., 2001).
Table II. Effect of phosphoinositides on membrane binding of p47phox-PX determined by SPR analysis.
| Phosphoinositide | ka (M–1 s–1) | kd (s–1) | Kd (M) | Fold increase in Kda |
|---|---|---|---|---|
| PtdIns(3,4)P2 |
(1.6 ± 0.3) × 104 |
(6.1 ± 0.7) × 10–4 |
(3.8 ± 0.8) × 10–8 |
1 |
| PtdIns(4,5)P2 |
(9.0 ± 1.1) × 103 |
(8.5 ± 0.8) × 10–3 |
(9.5 ± 3.4) × 10–7 |
25 |
| PtdIns(3,4,5)P2 |
(8.8 ± 0.9) × 103 |
(7.3 ± 0.7) × 10–3 |
(8.3 ± 1.2) × 10–7 |
22 |
| PtdIns3P |
(7.8 ± 0.8) × 103 |
(7.1 ± 0.7) × 10–3 |
(9.1 ± 1.3) × 10–7 |
24 |
| PtdIns4P |
(7.8 ± 0.8) × 103 |
(8.1 ± 0.9) × 10–3 |
(1.0 ± 1.5) × 10–6 |
26 |
| PtdIns5P | (9.8 ± 1.2) × 103 | (6.9 ± 0.6) × 10–3 | (7.0 ± 1.1) × 10–7 | 18 |
Values represent the mean and standard deviation from five determinations. All measurements were performed with POPC/POPE/phosphoinositide vesicles (77:20:3) in 20 mM Tris–HCl pH 7.4 containing 0.1 M KCl.
aIncrease in Kd relative to binding to POPC/POPE/PtdIns(3,4)P2 vesicles.

Fig. 6. Determination of the Kd for p47phox-PX by equilibrium binding measurements. (A) Binding isotherms for p47phox-PX and POPC/POPE/PtdIns(3,4)P2 (77:20:3) vesicles from equilibrium SPR measurements. A solid line represents a theoretical curve constructed from Rmax (45 ± 0.1) and Kd (38 ± 0.1 nM) values determined by non-linear least-squares analysis of the isotherm using an equation, Req = Rmax/(1 + Kd/P0). (B) Binding isotherm for p47phox-PX and beads coated with POPC/POPE/PtdIns(3,4)P2 (77:20:3). Phospholipid-coated beads (1.5 µM bulk concentration) were incubated with 14C-labelled p47phox-PX (10–400 nM) in 20 mM Tris–HCl buffer pH 7.4 containing 0.1 M KCl and 1 µM BSA. A solid line represents a theoretical curve constructed from n (19 ± 1) and Kd (30 ± 7 nM) values determined by non-linear least-squares analysis of the isotherm using Equation 1.
The enhanced membrane affinity of the p47phox-PX domain due to 3 mol% PtdIns(3,4)P2 is almost entirely due to a decrease in the kd, with the ka remaining nearly constant. This is consistent with our previous finding that short-range specific interactions increase membrane affinity mainly by decreasing the kd (Stahelin and Cho, 2001). The only other phosphoinositide-binding domain known to bind specifically PtdIns(3,4)P2 is the C-terminal PH domain of TAPP1 (TAPP1-PHCT) (Dowler et al., 2000; Thomas et al., 2001; Kimber et al., 2002). Using SPR, we compared the affinities of the p47phox PX domain and the TAPP1 PHCT domain for POPC/POPE/PtdIns(3,4)P2 vesicles. As previously reported (Dowler et al., 2000), the TAPP1-PHCT has a high affinity for PtdIns(3,4)P2, and this necessitated measurements with vesicles containing only 0.5% PtdIns(3,4)P2. Under these conditions, the affinity of the TAPP1 PHCT domain is 1.5 nM as compared with 2.6 µM for the p47phox PX domain (Table III). In addition to the difference in affinities, there is a difference in the modes of lipid interaction. The TAPP1-PHCT affinity is dictated largely by the PtdIns(3,4)P2 headgroup binding (Ferguson et al., 2000), whereas the p47phox-PX binds the soluble headgroup only very weakly (affinity >50 µM as measured by isothermal titration calorimetry, data not shown). These differences are likely to manifest themselves in the behaviours of the two domains in cells.
Table III. Binding parameters for p47phox-PX and mutants determined from SPR analysis.
| Proteins | ka (M–1 s–1) | kd (s–1) | Kd (M) | Fold increase in Kda |
|---|---|---|---|---|
| POPC/POPE/PtdIns(3,4)P2 (77:20:3) |
|
|
|
|
| p47phox-PX |
(1.6 ± 0.3) × 104 |
(6.1 ± 0.7) × 10–4 |
(3.8 ± 0.8) × 10–8 |
1 |
| R43Q |
(7.0 ± 0.6) × 103 |
(6.5 ± 0.7) × 10–2 |
(9.3 ± 1.3) × 10–6 |
245 |
| K52A |
(1.5 ± 0.2) × 104 |
(6.0 ± 0.8) × 10–4 |
(4.0 ± 0.8) × 10–8 |
1 |
| I65A |
(1.3 ± 0.2) × 104 |
(2.8 ± 0.4) × 10–3 |
(2.2 ± 0.5) × 10–7 |
6 |
| R70Q |
(1.0 ± 0.2) × 104 |
(6.0 ± 0.7) × 10–4 |
(6.0 ± 1.4) × 10–8 |
2 |
| K79A |
(9.0 ± 1.0) × 103 |
(2.1 ± 0.4) × 10–3 |
(2.3 ± 0.5) × 10–7 |
6 |
| W80A |
(1.7 ± 0.3) × 105 |
(2.0 ± 0.3) × 10–3 |
(1.1 ± 0.3) × 10–6 |
29 |
| R90A |
(8.4 ± 0.7) × 103 |
(2.7 ± 0.3) × 10–2 |
(3.2 ± 0.4) × 10–6 |
84 |
| POPC/POPE/PtdIns(3,4)P2 (79.5:20:0.5) |
|
|
|
|
| TAPP1-PHCT |
(1.6 ± 0.4) × 105 |
(2.4 ± 0.5) ×10–4 |
(1.5 ± 0.5) × 10–9 |
1 |
| p47phox-PX |
(1.0 ± 0.5) ×104 |
(2.6 ± 0.4) × 10–2 |
(2.6 ± 1.4) × 10–6 |
1700b |
| POPC/POPE/POPS/PtdIns(3,4)P2 (49.5:20:30:0.5) |
|
|
|
|
| p47phox-PX |
(1.1 ± 0.3) × 105 |
(5.5 ± 0.6) × 10–4 |
(5.0 ± 1.5) × 10–9 |
|
| POPC/POPE/POPS (50:20:30) |
|
|
|
|
| p47phox-PX |
(1.2 ± 0.3) × 105 |
(6.0 ± 0.5) × 10–3 |
(5.0 ± 1.3) × 10–8 |
1 |
| R70Q |
(2.3 ± 0.4) × 104 |
(2.1 ± 0.2) × 10–2 |
(9.1 ± 1.8) × 10–7 |
18 |
| R90A |
(1.1 ± 0.2) × 105 |
(7.0 ± 0.8) × 10–3 |
(6.3 ± 1.4) × 10–8 |
1 |
| POPC/POPE/POPS/PtdIns(3,4)P2 (74:20:3:3) |
|
|
|
|
| p47phox-PX |
(9.6 ± 0.9) × 104 |
(1.4 ± 0.2) × 10–4 |
(1.5 ± 0.3) × 10–9 |
1 |
| R43Q |
(8.4 ± 0.7) × 104 |
(1.1 ± 0.1) × 10–2 |
(1.3 ± 0.2) × 10–7 |
87 |
| K52A |
(5.0 ± 0.6) × 104 |
(1.8 ± 0.2) × 10–4 |
(3.6 ± 0.6) × 10–9 |
2 |
| I65A |
(8.6 ± 0.9) × 104 |
(6.1 ± 0.5) × 10–4 |
(7.1 ± 0.9) × 10–9 |
5 |
| R70Q |
(8.5 ± 0.8) × 104 |
(3.1 ± 0.5) × 10–3 |
(3.6 ± 0.7) × 10–8 |
24 |
| K79A |
(8.0 ± 0.7) × 104 |
(3.8 ± 0.4) × 10–4 |
(4.8 ± 0.7) × 10–9 |
3 |
| R90A |
(7.1 ± 0.8) × 104 |
(3.9 ± 0.3) × 10–3 |
(5.5 ± 0.8) × 10–8 |
37 |
| POPC/POPE/POPA/PtdIns(3,4)P2 (74:20:3:3) |
|
|
|
|
| p47phox-PX |
(1.5 ± 0.3) × 105 |
(9.0 ± 0.8) × 10–5 |
(6.0 ± 0.1) × 10–10 |
1 |
| R43Q |
(4.0 ± 0.5) × 104 |
(1.9 ± 0.3) × 10–3 |
(4.8 ± 1.0) × 10–8 |
80 |
| K52A |
(6.4 ± 0.7) × 104 |
(1.0 ± 0.2) × 10–4 |
(1.7 ± 0.4) × 10–9 |
3 |
| I65A |
(8.8 ± 1.0) × 104 |
(4.3 ± 0.5) × 10–4 |
(4.1 ± 0.7) × 10–9 |
7 |
| R70Q |
(3.8 ± 0.3) × 104 |
(5.0 ± 0.4) × 10–4 |
(1.3 ± 0.1) × 10–8 |
22 |
| K79A |
(9.0 ± 0.8) × 104 |
(2.4 ± 0.4) × 10–4 |
(2.6 ± 0.5) × 10–9 |
4 |
| W80A |
(6.6 ± 0.7) × 104 |
(2.0 ± 0.4) × 10–3 |
(3.0 ± 0.7) × 10–8 |
50 |
| R90A | (5.8 ± 0.6) × 104 | (1.4 ± 0.3) × 10–3 | (1.6 ± 0.4) × 10–8 | 27 |
Values represent the mean and standard deviation from five determinations. All measurements were performed in 20 mM Tris–HCl pH 7.4 containing 0.1 M KCl.
aIncrease in Kd relative to the wild-type binding to the same vesicles.
bIncrease in Kd relative to the TAPP1-PHCT binding to the same vesicles.
Distinct specificities of the two phospholipid-binding pockets in the p47phox PX domain
The roles of the two phospholipid-binding pockets were clarified by measuring the binding of a series of site-specific mutants to vesicles containing PtdIns(3,4)P2 and different anionic lipids. We first measured the binding of mutants to POPC/POPE/PtdIns(3,4)P2 (77:20:3) vesicles by SPR analysis. Results are summarized in Table III. Clearly, mutations in the putative phosphoinositide- binding pocket strongly reduce binding to PtdIns(3,4)P2-containing vesicles. Arg43, the putative 3-phosphate ligand, has the most decisive role in recognizing PtdIns(3,4)P2. The R43Q mutation decreased the affinity for POPC/POPE/PtdIns(3,4)P2 vesicles by 245-fold. The putative 4-phosphate ligand, Arg90, also has a substantial role in binding. The R90A mutation decreased the affinity for POPC/POPE/PtdIns(3,4)P2 vesicles by 84-fold. The decreased membrane affinities of the R43Q and R90A mutants are due mostly to large increases in the kd, consistent with our finding that specific phosphoinositide headgroup interactions primarily result in longer membrane residence time (Stahelin et al., 2002). The p47phox-PX binding to PtdIns(3,4)P2 and the importance of Arg90 was also apparent in semi-quantitative multilamellar vesicle (MLV) sedimentation assays (Figure 7). When compared with R43Q and R90A mutations, the mutation of a putative 1-phosphate ligand, K79A, showed a smaller impact on PtdIns(3,4)P2 binding (a 6-fold reduction) (Table III). In contrast to mutations in the phosphoinositide-binding pocket, mutation of Arg70, which is the principal sulfate ligand in the second anion-binding pocket, resulted in only a 2-fold decrease in affinity for POPC/POPE/PtdIns(3,4)P2 vesicles, indicating that PtdIns(3,4)P2 does not interact productively with the second anion-binding pocket. Lastly, Lys52, which is remote from both sulfate-binding pockets, also has no influence on PtdIns(3,4)P2 binding.

Fig. 7. Binding of p47phox constructs to MLVs. (A) The proteins were incubated with MLVs containing 1 mM total lipid of POPC:POPE (50:50), POPC:POPE:POPA (47.5:47.5:5), POPC:POPE:PtdIns(3,4)P2 (47.5:47.5:5) and POPE:POPE:POPA:PtdIns(3,4)P2 (45:45:5:5). P and S indicate ‘pellet’ and ‘supernatant’ fractions after centrifugation. Samples were analysed by SDS–PAGE, and three representative gels for the p47phox-PX, p47phox full-length wild-type, and p47phox full-length with the W263R mutation in the C-SH3 are shown. (B) The intensities of the stained ‘pellet’ bands were quantitated by densitometry, and the percentage of protein bound was determined. The values represent at least four independent measurements. (C) SDS–PAGE analysis of His6-p47phox constructs used in the binding studies. An estimated 0.5 µg of each protein was loaded on a 4–20% gradient SDS–polyacrylamide gel. The gel was stained with SimplyBlue SafeStain. All proteins were expressed with a His6 affinity tag, and purified as described.
Given that the second sulfate-binding pocket is shallow, we thought that it might bind an anionic lipid with a smaller headgroup such as phosphatidic acid (PtdOH) or phosphatidylserine (PtdSer). To test this notion, we measured the binding of mutants to vesicles contain ing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidic acid (POPA) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS). Increasing POPA or POPS composition to 3 mol% in POPC/POPE/POPA(S) vesicles (i.e. 77:20:3) did not significantly enhance the affinity of p47phox-PX (data not shown). However, 30 mol% of POPS in the vesicles was nearly as effective as 3 mol% PtdIns(3,4)P2 in enhancing vesicle affinity of the p47phox-PX (see Table III). Furthermore, 30 mol% POPA had an even more pronounced effect: the Kd for the p47phox-PX for POPC/POPE/POPA (50:20:30) vesicles was 1.2 ± 0.3 nM. To assess the roles of the two binding pockets in PtdOH or PtdSer binding, we first measured the effects of critical mutants in each of the pockets on POPC/POPE/POPS (50:20:30) vesicles in the absence of phosphoinositide. As shown in Table III, a mutation in the phosphoinositide pocket (R90A) did not affect POPS-dependent binding. However, a mutation in the second anion-binding pocket (R70Q) decreased binding by 18-fold. This shows that only the second pocket binds POPS.
In order to determine whether both pockets can be occupied simultaneously by their preferred lipids, we carried out measurements using POPC/POPE/POPS/PtdIns(3,4)P2 or POPC/POPE/POPA/PtdIns(3,4)P2 (74: 20:3:3) vesicles. When compared with POPC/POPE/PtdIns(3,4)P2 (77:20:3) vesicles, the affinity of p47phox-PX increased 63-fold when 3% POPA was included in the PtdIns(3,4)P2-containing vesicles and 25-fold for 3% POPS (Table III). This indicates that simultaneous occupation of the two lipid-binding sites synergistically increases the membrane binding of p47phox-PX [note that 3 mol% of POPA or POPS had little effect on the affinity of p47phox-PX for POPC/POPE (80:20) vesicles]. The synergistic effect of PtdIns(3,4)P2 and POPS increases the membrane affinity of the p47phox-PX to about the same as the TAPP1-PHCT affinity (Table III).
The p47phox-PX mutants R43Q and R90A that showed 245- and 84-fold decreases, respectively, in affinity for POPC/POPE/PtdIns(3,4)P2 (77:20:3) vesicles exhibited less pronounced 87- and 37-fold reductions, respectively, in affinity for POPC/POPE/POPS/PtdIns(3,4)P2 (74:20: 3:3) vesicles. A similar trend was seen with POPC/POPE/POPA/PtdIns(3,4)P2 (74:20:3:3) vesicles. Thus, the occupation of the second anion site by POPA or POPS lessens the deleterious effect of mutations in the phosphoinositide-binding site, underscoring the complementary nature of the two binding sites. In contrast, R70Q, which has 50% of wild-type affinity for POPC/POPE/PtdIns(3,4)P2 (77:20:3) vesicles, has >20-fold reduced affinity for POPC/POPE/POPS (or POPA)/PtdIns(3,4)P2 (74:20:3:3) vesicles. Together, these data indicate that POPA or POPS show a strong preference for the second anion-binding site and that PtdIns(3,4)P2 does not interact effectively with this anion-binding site.
Hydrophobic residues surrounding the lipid-binding pockets contribute to membrane affinity
Both the PtdIns(3,4)P2 and the secondary anion-binding pockets are flanked by exposed hydrophobic residues that protrude from the putative membrane-binding surface. Mutation of Trp80, which is in the membrane insertion loop between the two lipid-binding pockets, reduces the affinity 29- or 50-fold depending on the vesicle composition (Table III). Mutation of Ile65 (I65A), which is adjacent to the anionic-lipid-binding pocket, reduced the affinity 5- to 7-fold (Table III). This suggests that these hydrophobic residues have a role in membrane penetration during binding.
The PX domain lipid binding is masked in the full-length p47phox
It was reported previously that the C-SH3 of p47phox interacts with the PXXP motif of the PX domain (Hiroaki et al., 2001). In order to determine the effect of this interaction on membrane binding, we compared binding of the full-length p47phox with binding by the isolated PX domain. Remarkably, the full-length protein shows no significant interaction with PtdIns(3,4)P2 in MLV sedimentation assays (Figure 7). SPR measurements show that the affinity of the full-length p47phox for POPC/POPE/PtdIns(3,4)P2 (77:20:3) vesicles is 34-fold lower than that of the isolated PX domain (Table IV). However, the W263R mutation in the C-SH3 that was reported to abrogate binding to p47phox-PX resulted in a huge increase in binding (540-fold) compared with the full-length wild-type protein, suggesting that the mutation unmasks the lipid-binding surface of the PX domain. Lipid binding by p47phox-1–286, a construct containing the PX domain and the two SH3 domains but lacking the C-terminal tail, is also unmasked only in the W263R variant (as judged by MLV binding, Figure 7), but the binding was somewhat weaker than that of the full-length W263R. A mutation in the PtdIns(3,4)P2-binding pocket (R43Q) of the unmasked full-length protein (R43Q/W263R) reduced binding to approximately the same level as the masked, wild-type form (Table IV). Consistent with this, a construct lacking the PX domain either with or without the W263R mutation shows no measurable binding in the MLV assay (data not shown). These results indicate that high affinity lipid binding of the full-length p47phox depends on the PX domain interacting with phospholipids, although other regions of the protein also contribute. The observation that the 1–286 W263R p47phox has a somewhat weaker membrane binding than the full-length W263R p47phox suggests that the C-terminal tail (287–390) accounts for some of these additional interactions.
Table IV. Binding parameters for p47phox and p47phox-PX determined from SPR analysis.
| Proteins | ka (M–1 s–1) | kd (s–1) | Kd (M) | Fold increase in Kda |
|---|---|---|---|---|
| POPC/POPE/PtdIns(3,4)P2 (77:20:3) |
|
|
|
|
| p47phox-PX |
(1.6 ± 0.3) ×104 |
(6.1 ± 0.7) × 10–4 |
(3.8 ± 0.8) × 10–8 |
1 |
| p47phox |
(8.7 ± 0.7) × 103 |
(1.1 ± 0.2) × 10–2 |
(1.3 ± 0.3) × 10–6 |
34 |
| W263R p47phox |
(4.2 ± 0.4) × 104 |
(1.0 ± 0.1) × 10–4 |
(2.4 ± 0.3) × 10–9 |
0.06 |
| R43Q/W263R p47phox |
(1.6 ± 0.2) × 104 |
(1.8 ± 0.2) × 10–2 |
(1.1 ± 0.2) × 10–6 |
29 |
| 5SE p47phox [S(303,304,328,359,370)E] |
(3.8 ± 0.7) × 104 |
(5.0 ± 0.4) × 10–4 |
(1.3 ± 0.3) × 10–8 |
0.34 |
| R43Q/5SE p47phox | (1.3 ± 0.4) × 104 | (1.3 ± 0.2) × 10–2 | (1.0 ± 0.3) × 10–6 | 26 |
Values represent the mean and standard deviation from five determinations. All measurements were performed in 20 mM Tris–HCl pH 7.4 containing 0.1 M KCl.
aIncrease in Kd relative to the binding of the p47phox PX domain to the same vesicles.
A mimic of the phosphorylated state of p47 is ‘open’ and capable of binding PtdIns(3,4)P2
The p47phox subunit is phosphorylated on up to nine sites in the C-terminal tail. Functionally important sites of phosphorylation include serines 303, 304, 328, 359 and 370. Mimicking phosphorylation by replacing serines 303, 304 and 328 with glutamates led to an open conformation of p47phox that was able to bind to p22phox (Ago et al., 1999). Replacement of serines 359 and 370 with alanines results in a p47phox that has dramatically reduced phosphorylation and fails to translocate to membranes, but mutation of these residues into glutamates results in a p47phox subunit for which normal phosphorylation and translocation are observed (Johnson et al., 1998). To examine the effect of phosphorylation on phosphoinositide binding, we made a quintuple mutant S303E/S304E/S228E/S359E/S370E (p47phox-5SE), which mimics the phosphorylated form. The p47phox-5SE mutant binds PtdIns(3,4)P2 with an affinity that is ∼100-fold greater than that of the wild-type p47phox and only ∼5-fold weaker than that of the W263R mutant (Table IV). Like the W263R mutant of the C-SH3, this phosphorylation mimic appears to have an ‘open’ conformation with an unmasked PX domain, in contrast to the full-length wild-type p47phox, even though none of the sites of pseudo-phosphorylation are within the C-SH3.
Discussion
Synergistic membrane binding of the PX domain using two distinct lipid-binding pockets
The structure of the p47phox PX domain and the lipid-binding studies of the wild-type and site-specific mutants reveal that the domain has two distinct lipid-binding sites. Simultaneous occupancy of the phosphoinositide site by PtdIns(3,4)P2 and the second anion-binding site by PtdOH (or less preferentially PtdSer) leads to a great synergistic increase in membrane affinity. The ability of PtdOH to increase p47phox membrane binding substantially is consistent with reports that PtdOH can interact directly with cytosolic subunits of NADPH oxidase and activate it via a kinase-independent mechanism (Erickson et al., 1999; Palicz et al., 2001). One interesting question is whether PX domains of p40phox and p47phox can synergize in membrane binding in the context of the p40phox–p47phox–p67phox heterotrimer and, if so, are the specific ligands of the two domains [PtdIns(3,4)P2, PtdIns3P and PtdOH] present at the same time and place.
Masking of the lipid-binding surface of the p47phox PX domain by intramolecular interactions
The p47phox subunit contains an N-terminal PX domain followed by two SH3 domains (N-SH3 and C-SH3) and a C-terminal tail. The N-SH3 interacts with a proline-rich region in the C-terminus of p22phox and is necessary for both translocation of the cytosolic complex to the membrane and activation of the NADPH oxidase (Leto et al., 1994; Sumimoto et al., 1994, 1996; Leusen et al., 1996; Huang and Kleinberg, 1999). However, in the context of full-length, unphosphorylated p47phox, the N-SH3 is masked by interaction with the p47phox C-terminal tail. The phosphorylation of p47phox causes a conformational change (Swain et al., 1997) that releases the C-terminal tail from its association with the tandem SH3 domains, freeing the N-SH3 to bind the flavocytochrome (Ago et al., 1999; Huang and Kleinberg, 1999). It has been shown that the C-SH3 can interact with the p47phox PX domain via the PXXP motif (Hiroaki et al., 2001). However, because the C-SH3 does not interact with the flavocytochrome b558, it was not clear what might be the consequence of this interaction for the function of the oxidase. A study using a cell-free assay suggested that the role of the PX domain is not simply to mask the C-SH3 because a p47phox construct lacking the PX domain was not able to activate the oxidase (Hata et al., 1998).
We demonstrate here that the full-length p47phox in its basal, ‘closed’ form does not bind phosphoinositides due to a masking of the PX domain by the C-SH3. A site-specific mutation (W263R) in the C-SH3 of p47phox unmasks the lipid-binding surface of the PX domain. However, this mutant has an even higher membrane affinity than the isolated PX, suggesting that additional sites of membrane interaction, probably in the C-terminal tail, are exposed in the full-length protein.
Phosphorylation of p47phox leads to an open conformation with an unmasked PX domain
Formation of the active oxidase complex is critically dependent on phosphorylation of p47phox (Heyworth et al., 1989; Rotrosen and Leto, 1990; El Benna et al., 1994; Johnson et al., 1998). Our binding study of a quintuple phosphorylation mimic suggests that the phosphorylation-induced ‘open’ conformation of p47phox that enables interaction of the N-SH3 with p22phox also enables interaction of the p47phox PX domain with PtdIns(3,4)P2, as illustrated schematically in Figure 8. This would be consistent with observations that phosphorylation of p47phox results in its translocation to the plasma membrane in vivo (Leto et al., 1994; Sumimoto et al., 1994, 1996; Leusen et al., 1996; Huang and Kleinberg, 1999).

Fig. 8. Activation-dependent membrane binding of p47phox. In the resting state, p47phox is locked in a closed state by an intramolecular interaction between the C-SH3 and the PXXP motif of the PX domain, preventing phospholipid binding. Phosphorylation of sites in the C-terminal tail releases the lock, freeing the PX domain to bind to membranes with both PX phospholipid-binding pockets and a basic region in the C-terminal tail.
Conclusions
The functional role of the two phospholipid-binding sites of the p47phox PX domain in vivo needs to be determined. The non-overlapping specificities of the phospholipid-binding pockets may affect the types of membranes to which p47phox binds or the timing and duration of NADPH oxidase activation in relation to levels of PtdIns(3,4)P2 or PtdOH. A previous report had shown that the p47phox-PX could be visualized in membrane ruffles that form in response to insulin-like growth factor 1 (Zhan et al., 2002). However, the study provided no quantification to indicate that the domain was enriched in these structures. Although it is uncertain whether it is the driving force in translocation to plasma membranes, the p47phox-PX may be important for the regulatory complex achieving a productive orientation on the membrane to enable activation of the oxidase. The results presented here establishing the structure, binding characteristics and phosphorylation-dependent regulation of two distinct phospholipid-binding sites in the p47phox PX domain provide a foundation upon which further studies aimed at correlating membrane binding and NADPH oxidase activity in vivo can build.
Materials and methods
Materials
POPC, POPS, POPE, egg l-α phosphatidic acid, brain l-α phosphatidylcholine and brain l-α phosphatidylethanolamine were from Avanti Polar Lipids (Alabaster, AL). 1,2-dipalmitoyl derivatives of PtdIns3P, phosphatidylinositol 4-phosphate (PtdIns4P), phosphatidylinositol-5-phosphate (PtdIns5P), PtdIns(3,4)P2, phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2], and phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3] used in SPR measurements were a generous gift of Dr Karol Bruzik of the University of Illinois at Chicago. For MLV sedimentation assays, di-C16PtdIns(3,4)P2 and di-C16PtdIns3P were purchased from Echelon Research Labs (P-3416 and P-3016). For SPR studies, phospholipid concentrations were determined by phosphate analysis (Kates, 1986). For MLV sedimentation assays, lipid concentrations were based on the masses supplied by the manufacturer. The Liposofast microextruder and 100 nm polycarbonate filters were from Avestin (Ottawa, Ontario, Canada). Fatty acid-free bovine serum albumin (BSA) was from Bayer, Inc. (Kankakee, IL). [3-(3-cholamidopropyl)dimethylammonio]-1-propane-sulfonate and octyl glucoside were from Sigma and Fisher Scientific, respectively. Pioneer L1 sensor chip was from Biacore AB (Piscataway, NJ).
Protein expression and purification
A PCR product encoding residues 1–123 of human p47phox was cloned into vector pJL using NdeI–HindIII sites. The vector codes for an MAH6 affinity tag immediately preceding residue Met1. Protein was expressed in Escherichia coli strain C41(DE3), grown in 2× TY/Amp at 37°C to a density of 1 OD600, then induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 22°C for 5 h. The protein for crystallography was purified by Ni2+ affinity, heparin–Sepharose and gel filtration chromatography. For mutants used in binding studies, the heparin chromatography was omitted. The gel filtration was run in a buffer consisting of 20 mM Tris pH 7.4 (4°C), 100 mM NaCl. The protein was concentrated to 4.5 mg/ml and frozen in liquid nitrogen. Seleno methionine-substituted (Se-Met) protein was grown in the methionine-requiring auxotrophic strain 834(DE3) using a medium consisting of 2× M9 minimal medium supplemented with 1 mg/l riboflavin, 1 mg/l niacinamide, 1 mg/l thiamine, 0.1 mg/l pyridoxine monohydrate, 0.4% d(+)-glucose, 2 mM MgSO4, 25 mg/l FeSO4, 40 mg/l of each amino acid except methionine, 80 mg/l seleno-d,l-methionine and 0.1 g/l ampicillin. Mutations in the p47phox-PX were constructed using the overlap extension method. All of the mutants except I65A were cloned in the pJL vector. The I65A mutant was subcloned into the pET21a vector containing a His6 tag. The full-length p47phox, the 1–286 deletion variant, the 124–390 deletion variant and mutations of these constructs were cloned with the same N-terminal tag as the p47phox-PX (MAH6). The TAPP1-PHCT was cleaved from a GST fusion and purified as previously described (Thomas et al., 2001). All constructs were checked by DNA sequencing. Protein concentration was determined by OD280 using calculated molar extinction coefficients (for crystallography and MLV sedimentation assays) or using the Pierce BCA method (for SPR studies).
Protein refolding
The R70Q mutant in the p47phox-PX was expressed as an inclusion body and thus needed to be refolded. The inclusion body pellet was resuspended in 25 ml Tris–HCl pH 8.0 containing 50 mM NaCl, 0.8% (v/v) Triton X-100 and 0.8% (w/v) sodium deoxycholate. After centrifugation at 48 000 g for 15 min, the pellet was resuspended in 25 ml of 50 mM Tris–HCl pH 8.0 containing 5 M urea. The pellet was stirred at room temperature for 1 h, and then the suspension was centrifuged at 100 000 g for 25 min. The supernatant was collected and dialysed against 50 mM Tris–HCl pH 8.0 and 1.5 M urea for 16 h, and then against 50 mM Tris–HCl pH 8.0 for 24 h. The refolded R70Q mutation was purified by Ni2+ affinity and gel filtration chromatography.
Crystallization
The protein was crystallized in sitting drops using the vapour diffusion method. The drops contained 1 µl of protein mixed with 1 µl of reservoir solution. Initial crystals were obtained from Hampton Crystal Screen II, condition 42. The optimized reservoir solution consisted of 1.0 M NH2SO4, 0.1 M Tris pH 8.5 (4°C) and 12% glycerol. Crystals used for data collection were grown by hair seeding. The Se-Met protein crystallized in space group I222 with cell dimensions a = 74.2 Å, b = 92.2 Å, c = 144.0 Å.
Diffraction data collection
For data collection, crystals were cryo-protected in paratone-N (Hampton). Data for the Se-Met-substituted crystals were collected at wavelengths corresponding to the peak and inflection point of the fluorescence spectrum for Se (12.664 and 12.662 keV, respectively). Data were processed with MOSFLM (Leslie, 1992) and CCP4 (1994). Se sites were located and refined using autoSHARP (C.Vonrhein, E.Blanc, P.Roversi and G.Bricogne, in preparation) and SHARP (de La Fortelle and Bricogne, 1997). Solvent flattening was carried out with a solvent content of 44.6%. The asymmetric unit of the crystal consists of three molecules related to each other by a 3-fold rotation axis. The overall fold of the p47phox-PX from the crystal structure agrees with the fold that was determined by NMR (Hiroaki et al., 2001); however, the NMR structure was not sufficiently similar to be useful as a model for molecular replacement. The models were built independently for the three chains in the asymmetric unit then refined using REFMAC (Murshudov et al., 1997) with isotropic B-factors and wARP (Perrakis et al., 1999). All residues in chain A of the trimer are visible in the electron density, while the three N-terminal residues are disordered in chains B and C. The N-terminal MAH6 tag was disordered for all three chains. The loop between β1 and β2 (residues 16–22) is disordered in chain C. Non-crystallographic symmetry (NCS) was tightly restrained for residues not involved in crystal contacts during the initial stages of refinement. In the later stages of refinement, the NCS restraints were relaxed and finally removed. At each stage of relaxing the NCS restraints, the free R-factor decreased. Table I gives statistics for data collection and refinement.
MLV sedimentation binding assay
Protonation of di-C16PtdIns(3,4)P2 was essential for obtaining reproducible lipid binding. Prior to use in the assay, solid di-C16PtdIns(3,4)P2 was resuspended in CHCl3:MeOH:1 M HCl (volume ratios 2:1:0.01), dried with argon, then vacuum desiccated for 1 h, followed by three cycles of chloroform resuspension and desiccation and finally storage in chloroform at –70°C. Phospholipids in chloroform were mixed, dried and desiccated under vacuum for 1 h. The phospholipids were then resuspended in 500 µl of binding buffer [20 mM Tris pH 7.4, 100 mM NaCl, 1 mM dithiothreitol (DTT)] and rehydrated on ice for 2 h. The lipid mixture was then vortexed to produce MLVs. Protein stock and MLVs were mixed in 100 µl reactions to yield solutions containing 1 mM total lipid and 5 µM protein. The reactions were incubated for 15 min (20°C), then centrifuged for 10 min at 14 000 r.p.m. in a microfuge at room temperature. Supernatants were removed carefully and the pellets were resuspended in SDS sample buffer. Both the pellets and the supernatants were analysed by SDS–PAGE. Gels were stained in SimplyBlue SafeStain (Invitrogen), and quantitated with a ChemiGenius densitometer (SynGene) using GeneTools analysis software.
SPR experiments
A detailed protocol for coating the L1 sensor chip has been described elsewhere (Bittova et al., 2001; Stahelin and Cho, 2001). Briefly, after washing the sensor chip surface, 90 µl of 0.5 mM vesicles of a given composition [e.g. POPC/POPE/PtdIns(3,4)P2 = 77:20:3)] were injected at 5 µl/min to give a response of 4000 RU. Similarly, a control surface was coated with the same vesicles minus the phosphoinositide of interest (e.g. POPC/POPE = 80:20) to give the same RU response as the active binding surface. This control surface was selected over the bare chip surface to circumvent any potential artefacts caused by the hydrophobic nature of the L1 chip. No binding was detected to this surface beyond the refractive index change for p47phox-PX and other proteins used. Each lipid layer was stabilized by injecting 10 µl of 50 mM NaOH three times at 100 µl/min, which showed no decrease in lipid signal after the second injection. The coating efficiency of each surface was tested using a 25 µl injection of 0.1 mg/ml BSA. The BSA response was ∼100 RU compared with 1200 RU of BSA for an uncoated chip surface. Control experiments with vesicles containing 5-carboxyfluorescein confirmed that vesicles remained intact on the chip. After stabilization of signal (i.e. <0.3 RU/min drift), experiments were peformed at 60 µl/min. A 90 µl aliquot of protein in 20 mM Tris pH 7.4 containing 0.1 M KCl was injected to give an association time of 90 s, while the dissociation was monitored for 500 s. The protein concentrations used were within a 10-fold range above and below the Kd values listed in Tables II–IV. After sensorgrams were obtained for five or more concentrations of each protein and corrected for refractive index changes, the association and dissociation phases of all sensorgrams were globally fit to a 1:1 Langmuir binding model: protein + (protein-binding site on vesicle) ↔ (complex), using BIAevaluation 3.0 software (Biacore). The association phase was analysed using the equation:
R = [kaC/(kaC + kd)] Rmax (1 – e–(kaC + kd)(t – t0)) + RI
where RI = refractive index change, Rmax is the theoretical binding capacity, C is analyte concentration, ka is the association rate constant, and t0 is the time at start of fit data. The dissociation phase was analysed using an equation:
R = R0e–kd(t – t0)
where kd is the dissociation rate constant and R0 is the response at the start of fit data. The dissociation constant (Kd) was then calculated from the equation, Kd = kd/ka. It should be noted that in our SPR analysis, Kd is defined in terms of not the molarity of phospholipids but the molarity of protein-binding sites on the vesicle. Thus, if each protein-binding site on the vesicle is composed of n lipids, nKd is the dissociation constant in terms of molarity of lipid monomer (Cho et al., 2001). Due to difficulty involved in accurate determination of the concentration of lipids coated on the sensor chip, however, only Kd was determined in our SPR analysis and the relative affinity was calculated as a ratio of Kd values assuming that n values are essentially the same for wild-type and mutants. The 1:1 Langmuir binding model gives a good fit to the data with random residuals. Each measurement was repeated three or more times to calculate a standard deviation.
To verify the Kd determined from kinetic measurements for p47phox-PX binding to POPC/POPE/PtdIns(3,4)P2 (77:20:3) vesicles, we also performed equilibrium SPR measurements. For this purpose, the flow rate was reduced to 1 µl/min, and 90 µl of p47phox-PX (10–600 nM) was injected and bound to a saturating value. These saturating (equilibrium) values (Req) were then plotted versus protein concentration ([P]0) and the Kd value was determined by a non-linear least-squares analysis of the binding isotherm using the equation:
Req = Rmax/(1 + Kd/([P]0))
where Rmax is a maximal Req value. Furthermore, n and Kd values were determined for the binding of radiolabelled p47phox-PX to hydrophobic beads coated with POPC/POPE/PtdIns(3,4)P2 (77:20:3) as described previously (Cho et al., 2001). Radiolabelling of the p47phox-PX was performed as described (Stahelin et al., 2002). Parameters n and Kd were determined by non-linear least-squares analysis of the bound ([P]b) versus [P]0 plot using a standard binding equation, Equation 1 (Cho et al., 2001):
![]()
where [PL]o represents total phospholipid concentration. The Kd value for p47phox-PX determined from non-linear least square analysis of the binding isotherms (30 ± 7 nM) is comparable with that determined from the kinetic SPR analysis under the same condition (38 nM). The n value (19 ± 1) for p47phox-PX suggests that each molecule interacts with ∼19 phospholipid molecules. Thus the apparent dissociation constant in terms of total lipid concentration (nKd) is ∼0.6 µM for the p47phox-PX.
Acknowledgments
Acknowledgements
We would like to thank Karol S.Bruzik for generous gift of phosphoinositides for SPR studies, Aura Burian for preparing the I65A mutant, Dario Alessi for providing a TAPP1 plasmid, Gordon Leonard and Raimond Ravelli for their assistance with data collection at ESRF beamlines ID29 and ID14-4, and Phil Hawkins, Len Stephens and Chris Ellson for valuable discussions. The work was supported by a grant from the British Heart Foundation (to R.L.W.) and a National Institutes of Health grant GM53987 (to W.C.).
References
- Ago T., Nunoi,H., Ito,T. and Sumimoto,H. (1999) Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47phox. J. Biol. Chem., 274, 33644–33653. [DOI] [PubMed] [Google Scholar]
- Ago T., Takeya,R., Hiroaki,H., Kuribayashi,F., Ito,T., Kohda,D. and Sumimoto,H. (2001) The PX domain as a novel phosphoinositide-binding module. Biochem. Biophys. Res. Commun., 287, 733–738. [DOI] [PubMed] [Google Scholar]
- Babior B.M. (1999) NADPH oxidase: an update. Blood, 93, 1464–1476. [PubMed] [Google Scholar]
- Bittova L., Stahelin,R.V. and Cho,W. (2001) Roles of ionic residues of the C1 domain in protein kinase C-α activation and the origin of phosphatidylserine specificity. J. Biol. Chem., 276, 4218–4226. [DOI] [PubMed] [Google Scholar]
- Bravo J. et al. (2001) The crystal structure of the PX domain from p40phox bound to phosphatidylinositol 3-phosphate. Mol. Cell, 8, 829–839. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) Collaborative Computing Project Number 4: a suite of programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cho W., Bittova,L. and Stahelin,R.V. (2001) Membrane binding assays for peripheral proteins. Anal. Biochem., 296, 153–161. [DOI] [PubMed] [Google Scholar]
- de La Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Dowler S., Currie,R.A., Campbell,D.G., Deak,M., Kular,G., Downes,C.P. and Alessi,D.R. (2000) Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem. J., 351, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusi S., Donini,M. and Rossi,F. (1996) Mechanisms of NADPH oxidase activation: translocation of p40phox, Rac1 and Rac2 from the cytosol to the membranes in human neutrophils lacking p47phox or p67phox. Biochem. J., 314, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Benna J., Faust,L.P. and Babior,B.M. (1994) The phosphorylation of the respiratory burst oxidase component p47phox during neutrophil activation. J. Biol. Chem., 269, 23431–23436. [PubMed] [Google Scholar]
- Ellson C.D. et al. (2001) PtdIns(3)P regulates the neutrophil oxidase complex by binding to the PX domain of p40phox. Nat. Cell Biol., 3, 679–682. [DOI] [PubMed] [Google Scholar]
- Erickson R.W., Langel-Peveri,P., Traynor-Kaplan,A.E., Heyworth,P.G. and Curnutte,J.T. (1999) Activation of human neutrophil NADPH oxidase by phosphatidic acid or diacylglycerol in a cell-free system. Activity of diacylglycerol is dependent on its conversion to phosphatidic acid. J. Biol. Chem., 274, 22243–22250. [DOI] [PubMed] [Google Scholar]
- Ferguson K.M., Kavran,J.M., Sankaran,V.G., Fournier,E., Isakoff,S.J., Skolnik,E.Y. and Lemmon,M.A. (2000) Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol. Cell, 6, 373–384. [DOI] [PubMed] [Google Scholar]
- Hata K., Ito,T., Takeshige,K. and Sumimoto,H. (1998) Anionic amphiphile-independent activation of the phagocyte NADPH oxidase in a cell-free system by p47phox and p67phox, both in C terminally truncated forms. J. Biol. Chem., 273, 4232–4236. [DOI] [PubMed] [Google Scholar]
- Heyworth P.G. and Cross,A.R. (2002) Chronic granulomatous disease mutations and the PX domain. Nat. Cell Biol., 4, E110. [DOI] [PubMed] [Google Scholar]
- Heyworth P.G., Shrimpton,C.F. and Segal,A.W. (1989) Localization of the 47 kDa phosphoprotein involved in the respiratory-burst NADPH oxidase of phagocytic cells. Biochem. J., 260, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyworth P.G., Curnutte,J.T., Nauseef,W.M., Volpp,B.D., Pearson,D.W., Rosen,H. and Clark,R.A. (1991) Neutrophil nicotinamide adenine dinucleotide phosphate oxidase assembly. Translocation of p47-phox and p67-phox requires interaction between p47-phox and cytochrome b558. J. Clin. Invest., 87, 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroaki H., Ago,T., Ito,T., Sumimoto,H. and Kohda,D. (2001) Solution structure of the PX domain, a target of the SH3 domain. Nat. Struct. Biol., 8, 526–530. [DOI] [PubMed] [Google Scholar]
- Huang J. and Kleinberg,M.E. (1999) Activation of the phagocyte NADPH oxidase protein p47(phox). Phosphorylation controls SH3 domain-dependent binding to p22(phox). J. Biol. Chem., 274, 19731–19737. [DOI] [PubMed] [Google Scholar]
- Johnson J.L., Park,J.W., El Benna,J., Faust,L.P., Inanami,O. and Babior,B.M. (1998) Activation of p47(PHOX), a cytosolic subunit of the leukocyte NADPH oxidase—phosphorylation of Ser-359 or Ser-370 precedes phosphorylation at other sites and is required for activity. J. Biol. Chem., 273, 35147–35152. [DOI] [PubMed] [Google Scholar]
- Kanai F., Liu,H., Field,S.J., Akbary,H., Matsuo,T., Brown,G.E., Cantley,L.C. and Yaffe,M.B. (2001) The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat. Cell Biol., 3, 675–678. [DOI] [PubMed] [Google Scholar]
- Kates M. (1986) Techniques of Lipidology. Elsevier, Amsterdam, pp. 114–115.
- Kim Y., Lichtenbergova,L., Snitko,Y. and Cho,W. (1997) A phospholipase A2 kinetic and binding assay using phospholipid-coated hydrophobic beads. Anal. Biochem., 250, 109–116. [DOI] [PubMed] [Google Scholar]
- Kimber W.A. et al. (2002) Evidence that the tandem-pleckstrin-homology-domain-containing protein TAPP1 interacts with Ptd(3,4)P2 and the multi-PDZ-domain-containing protein MUPP1 in vivo. Biochem. J., 361, 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge K., Smith,S.J., Groemping,Y. and Rittinger,K. (2002) Architecture of the p40–p47–p67phox complex in the resting state of the NADPH oxidase: a central role for p67phox. J. Biol. Chem., 277, 10121–10128. [DOI] [PubMed] [Google Scholar]
- Leslie A.G.W. (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography, Daresbury Laboratory, Warrington, UK, Vol. 26.
- Leto T.L., Adams,A.G. and de Mendez,I. (1994) Assembly of the phagocyte NADPH oxidase: binding of Src homology 3 domains to proline-rich targets. Proc. Natl Acad. Sci. USA, 91, 10650–10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leusen J.H.W., Verhoeven,A.J. and Roos,D. (1996) Interactions between the components of the human NADPH oxidase: a review about the intrigues in the phox family. Frontiers Biosci., 1, d72–d90. [DOI] [PubMed] [Google Scholar]
- Lu J., Garcia,J., Dulubova,I., Sudhof,T.C. and Rizo,J. (2002) Solution structure of the Vam7p PX domain. Biochemistry, 41, 5956–5962. [DOI] [PubMed] [Google Scholar]
- Mayer B.J. (2001) SH3 domains: complexity in moderation. J. Cell Sci., 114, 1253–1263. [DOI] [PubMed] [Google Scholar]
- McAdara Berkowitz J.K., Catz,S.D., Johnson,J.L., Ruedi,J.M., Thon,V. and Babior,B.M. (2001) JFC1, a novel tandem C2 domain-containing protein associated with the leukocyte NADPH oxidase. J. Biol. Chem., 276, 18855–18862. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin,A.A. and Dodson,E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Palicz A., Foubert,T.R., Jesaitis,A.J., Marodi,L. and McPhail,L.C. (2001) Phosphatidic acid and diacylglycerol directly activate NADPH oxidase by interacting with enzyme components. J. Biol. Chem., 276, 3090–3097. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris,R. and Lamzin,V.S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol., 6, 458–463. [DOI] [PubMed] [Google Scholar]
- Reeves E.P. et al. (2002) Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature, 416, 291–297. [DOI] [PubMed] [Google Scholar]
- Rotrosen D. and Leto,T.L. (1990) Phosphorylation of neutrophil 47-kDa cytosolic oxidase factor. J. Biol. Chem., 265, 19910–19915. [PubMed] [Google Scholar]
- Seguchi H. and Kobayashi,T. (2002) Study of NADPH oxidase-activated sites in human neutrophils. J. Electron Microsc., 51, 87–91. [DOI] [PubMed] [Google Scholar]
- Stahelin R.V. and Cho,W. (2001) Differential roles of ionic, aliphatic and aromatic residues in membrane–protein interactions: a surface plasmon resonance study on phospholipases A2. Biochemistry, 40, 4672–4678. [DOI] [PubMed] [Google Scholar]
- Stahelin R.V., Long,F., Diraviyam,K., Bruzik,K.S., Murray,D. and Cho,W. (2002) Phosphatidylinositol-3-phosphate induces the membrane penetration of the FYVE domains of Vps27p and Hrs. J. Biol. Chem., 277, 26379–26388. [DOI] [PubMed] [Google Scholar]
- Sumimoto H., Kage,Y., Nunoi,H., Sasaki,H., Nose,T., Fukumaki,Y., Ohno,M., Minakami,S. and Takeshige,K. (1994) Role of Src homology 3 domains in assembly and activation of the phagocyte NADPH oxidase. Proc. Natl Acad. Sci. USA, 91, 5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumimoto H., Hata,K., Mizuki,K., Ito,T., Kage,Y., Sakaki,Y., Fukumaki,Y., Nakamura,M. and Takeshige,K. (1996) Assembly and activation of the phagocyte NADPH oxidase—specific interaction of the N-terminal Src homology 3 domain of p47(phox) with p22(phox) is required for activation of the NADPH oxidase. J. Biol. Chem., 271, 22152–22158. [DOI] [PubMed] [Google Scholar]
- Swain S.D., Helgerson,S.L., Davis,A.R., Nelson,L.K. and Quinn,M.T. (1997) Analysis of activation-induced conformational changes in p47phox using tryptophan fluorescence spectroscopy. J. Biol. Chem., 272, 29502–29510. [DOI] [PubMed] [Google Scholar]
- Thomas C.C., Dowler,S., Deak,M., Alessi,D.R. and van Aalten,D.M. (2001) Crystal structure of the phosphatidylinositol 3,4-bisphosphate-binding pleckstrin homology (PH) domain of tandem PH-domain-containing protein 1 (TAPP1): molecular basis of lipid specificity. Biochem. J., 358, 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlinger D.J., Tyagi,S.R., Inge,K.L. and Lambeth,J.D. (1993) The respiratory burst oxidase of human neutrophils. Guanine nucleotides and arachidonate regulate the assembly of a multicomponent complex in a semirecombinant cell-free system. J. Biol. Chem., 268, 8624–8631. [PubMed] [Google Scholar]
- Wientjes F.B., Reeves,E.P., Soskic,V., Furthmayr,H. and Segal,A.W. (2001) The NADPH oxidase components p47(phox) and p40(phox) bind to moesin through their PX domain. Biochem. Biophys. Res. Commun., 289, 382–388. [DOI] [PubMed] [Google Scholar]
- Yu J.W. and Lemmon,M.A. (2001) All phox homology (PX) domains from Saccharomyces cerevisiae specifically recognize phosphatidyl inositol 3-phosphate. J. Biol. Chem., 276, 44179–44184. [DOI] [PubMed] [Google Scholar]
- Zhan Y., Virbasius,J.V., Song,X., Pomerleau,D.P. and Zhou,G.W. (2002) The p40phox and p47phox PX domains of NADPH oxidase target cell membranes via direct and indirect recruitment by phosphoinositides. J. Biol. Chem., 277, 4512–4518. [DOI] [PubMed] [Google Scholar]