

Abstract
Phosphoinositides play a central role in the control of several cellular events including actin cytoskeleton organization. Here we show that, upon infection of epithelial cells with the Gram-negative pathogen Shigella flexneri, the virulence factor IpgD is translocated directly into eukaryotic cells and acts as a potent inositol 4-phosphatase that specifically dephosphorylates phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] into phosphatidylinositol 5-monophosphate [PtdIns(5)P] that then accumulates. Transfection experiments indicate that the transformation of PtdIns(4,5)P2 into PtdIns(5)P by IpgD is responsible for dramatic morphological changes of the host cell, leading to a decrease in membrane tether force associated with membrane blebbing and actin filament remodelling. These data provide the molecular basis for a new mechanism employed by a pathogenic bacterium to promote membrane ruffling at the entry site.
Keywords: bacterial entry/IpgD/phosphoinositides/S.flexneri
Introduction
Intracellular pathogens have evolved several mechanisms to hijack different cellular systems in the host to promote their entry, including modifications of the actin cytoskeleton and the exploitation of various signalling pathways (Galan and Bliska, 1996). A key factor of their virulence is invasion of enterocytes, which are cells that in normal circumstances are non-phagocytic. Two strategies, the so-called ‘zipper’ and ‘trigger’ mechanisms, are used by invasive bacteria to induce their uptake. Some pathogens, such as Listeria, express surface proteins that bind to eukaryotic receptors and lead to membrane zippering around the bacterium (Mengaud et al., 1996). Other pathogens, like Salmonella and Shigella, use a type III secretion system (Van Gijsegem et al., 1993) to inject effector proteins into the host cell and trigger their uptake via membrane ruffles in a process resembling macropinocytosis (Finlay and Falkow, 1990; Francis et al., 1993; Adam et al., 1995). The formation of these entry structures is the result of a complex cross-talk between injected bacterial proteins and components of the target cell that is not yet fully understood.
Shigella flexneri is a facultative intracellular pathogen that is the causative agent of bacillary dysentery in humans. Several proteins are secreted by the S.flexneri type III secretion apparatus during growth of bacteria in culture media. According to the current model of the type III secretion pathway, these proteins are potential effectors that might be translocated into epithelial cells upon contact of bacteria with the cell surface. However, because of the rapid uptake of S.flexneri by the cell and its intracytoplasmic lifestyle, no direct translocation of a secreted protein has been clearly demonstrated. Recently, we have shown that IpgD is one of the proteins potentially secreted by the S.flexneri type III secretion apparatus. IpgD is stored in the bacterial cytoplasm associated with a specific chaperone, IpgE. Although IpgD is not absolutely required for entry of bacteria into cultured cells (Allaoui et al., 1993), it is implicated in entry focus formation (Niebuhr et al., 2000). The sequence of IpgD exhibits two motifs that are present in the active site of mammalian inositol polyphosphate 4-phosphatase. Phosphoinositides, whose intracellular levels are controlled accurately by a complex set of kinases, phosphatases and phospholipases, play a key role in many processes including reorganization of the actin cytoskeleton (Toker, 1998; Czech, 2000; Sechi and Wehland, 2000; Payrastre et al., 2001) and plasma membrane–cytoskeleton linkage (Raucher et al., 2000). Here, we demonstrate that IpgD is translocated directly into the host cell, where it functions as a phosphoinositide phosphatase that dephosphorylates phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] to generate the novel lipid phosphatidylinositol 5-monophosphate [PtdIns(5)P]. Furthermore, we show that expression of IpgD in mammalian cells leads to a strong reduction in tether force that eventually causes membrane blebbing. Our results strongly suggest that IpgD uncouples the cellular plasma membrane from the actin cytoskeleton by locally reducing its adhesion energy through the transformation of PtdIns(4,5)P2 into PtdIns(5)P during S.flexneri entry. The system we describe is a novel strategy used by a pathogenic bacterium that dramatically and specifically disturbs a key element of phosphoinositide metabolism at the plasma membrane to increase its virulence.
Results
IpgD is a phosphoinositide phosphatase
The sequence of IpgD contains two motifs related to the active site of mammalian inositol polyphosphate 4-phosphatases (Norris et al., 1998), and it has been shown that Salmonella dublin and S.typhimurium proteins homologous to IpgD, SopB and SigD, respectively, are endowed with an inositol phosphate phosphatase activity (Norris et al., 1998; Marcus et al., 2001). To test the activity and the substrate specificity of IpgD, we used a purified GST–IpgD fusion protein expressed together with the chaperone IpgE. We found that it hydrolysed several phosphoinositides in vitro (Table I) and exhibited the greatest activity towards PtdIns(4,5)P2. Very weak or no activity was detected towards the inositol phosphates Ins(1,4,5)P3 and Ins(1,3,4,5)P4. This substrate specificity was not due to the presence of IpgE in the preparation of GST–IpgD since similar results were obtained when IpgD was produced without IpgE (data not shown). Replacement of the cysteine residue in the proposed catalytic domain of IpgD by a serine residue (C438S) led to a protein that had no activity against PtdIns(4,5)P2 (not shown) as classically observed with this phosphatase family (Norris et al., 1997). The substrate specificity of IpgD in vitro appears different from that of SopB or SigD, which have a preference for phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3] and phosphatidylinositol 3,4-bisphosphate [PtdIns(3,4)P2] (Norris et al., 1998). As previously noted for SopB (Norris et al., 1997), the specific activity of IpgD was relatively low compared with the activity of mammalian homologues, which may reflect the difficulty in setting appropriate conditions for in vitro lipid phosphatase assays for these proteins.
Table I. Phosphatase activity of recombinant IpgD.
| Substrate | nmol/min/mg protein |
|---|---|
| PtdIns(4)P |
10.0 ± 5 |
| PtdIns(3)P |
5.8 ± 3 |
| PtdIns(4,5)P2 |
61.0 ± 9 |
| PtdIns(3,4)P2 |
25.3 ± 2 |
| PtdIns(3,4,5)P3 |
32.1 ± 10 |
| Ins(1,4,5)P3 |
0.3 ± 0.1 |
| Ins(1,3,4,5)P4 | ND |
Assays were performed using exogenous lipids or inositol phosphates as indicated in Materials and methods. Results are the mean ± SEM from 3–4 independent experiments, except those for inositol phosphate phosphatase activity, which are from two experiments.
ND = not detectable.
IpgD hydrolyses PtdIns(4,5)P2 during infection of epithelial cells by S.flexneri
To investigate the role of IpgD on the cellular levels and/or turnover of the various phosphoinositides during infection of HeLa cells, we analysed the phospholipid content of cells labelled with 32Pi and infected with the wild-type strain M90T, the ipgD mutant and the non-invasive virulence plasmid cured strain BS176, over time. As soon as 15 min after contact between bacteria and host cells, cellular [32P]PtdIns(4,5)P2 levels dropped dramatically with a concomitant increase in [32P]PtdInsP (Figure 1A and C). In contrast, the levels of [32P] PtdIns(4,5)P2 and [32P]PtdInsP in cells infected with the ipgD mutant or with the non-invasive strain did not change, even after 30 min of infection (Figure 1B and C), demonstrating the absolute requirement for IpgD in the hydrolysis of [32P]PtdIns(4,5)P2. The levels of the major cellular [32P]phospholipids (i.e. phosphatidylcholine, -serine and -ethanolamine) (Figure 1A) as well as [32P]phosphatidylinositol (PtdIns) (data not shown) and PtdIns(3)P (see Figure 4) were not affected, indicating that IpgD phosphatase activity was directed towards [32P]PtdIns(4,5)P2 in vivo. Thus, IpgD appears as a S.flexneri effector that affects the level of specific host cell polyphosphoinositides during infection.


Fig. 1. IpgD-dependent PtdIns(4,5)P2 hydrolysis in HeLa cells infected with S.flexneri. HeLa cells were labelled with [32P]orthophosphate and infected with either the wild-type M90T (WT) (A) or the ipgD (ipgD) (B) strains. After infection, cells were washed with PBS and reactions were stopped by adding ice-cold HCl (2.4 M). Cells were recovered by scraping, and lipids were extracted and separated by TLC (left panel). The radiolabelled PtdInsP + PtdInsP2 were recovered by scraping the appropriate bands as indicated, and were deacylated and analysed by HPLC (right panels). MP, major phospholipids; CTS, counts per second. (C) Quantification of [32P]PtdInsP [PtdIns(4)P + PtdIns(5)P which co-elute with the classical HPLC technique] (diamonds) and [32P]PtdIns(4,5)P2 (squares) in HeLa cells infected either with WT, ipgD or the avirulant, non-invasive mutant BS176. Data shown are representative of 4–6 independent experiments with similar results.

Fig. 4. PI 3-kinase is activated during Shigella infection. HeLa cells were labelled with [32P]orthophosphate and infected with either the ipgD (A) or the wild-type M90T (WT) (B) strains for 30 min. Lipids were then extracted and analysed as described in Figure 1. The radiolabelled PtdInsP2 [PtdIns(3,5)P2, PtdIns(3,4)P2 and PtdIns(4,5)P2] + PtdIns(3,4,5)P3 were recovered by scraping the appropriate bands from the TLC plate, and were deacylated and analysed by HPLC (left panel). Peaks corresponding to PtdIns(3,5)P2 (1), PtdIns(3,4)P2 (2), PtdIns(4,5)P2 (3), ATP (4) and PtdIns(3,4,5)P3 (5) are indicated. (C) Quantification of [32P]D3 phosphoinositides in cells infected with either wild-type M90T (squares), ipgD mutant (diamonds) or the non-invasive mutant BS176 (circles). Data are from one experiment, representative of four.
PtdIns(5)P is the product of the IpgD-dependent PtdIns(4,5)P2 degradation in S.flexneri-infected cells
The increase of [32P]PtdInsP that was detected in parallel with the decrease of [32P]PtdIns(4,5)P2 suggested that IpgD specifically dephosphorylated one of the two positions of the inositol moiety leading to accumulation of the hydrolysis product. To identify the product of the enzymatic reaction, we analysed the nature of the [32P]PtdInsP produced in infected cells using an appropriate high-performance liquid chromatography (HPLC) technique allowing the separation of PtdIns(4)P and PtdIns(5)P (Tolias et al., 1998; Sbrissa et al., 1999). During the course of infection, the [32P]PtdIns(4)P levels remained unchanged whereas a second peak at the expected position for [32P]PtdIns(5)P was clearly detected (Figure 2). This recently discovered phosphoinositide has been shown to be quantitatively minor in several models (Rameh et al., 1997; Tolias et al., 1998) and indeed was undetectable by HPLC in resting HeLa cells.

Fig. 2. Production of PtdIns(5)P without changes in PtdIns(4)P levels during Shigella infection. HeLa cells were labelled with [32P]orthophosphate and infected with either ipgD mutant (ipgD) (A) or wild-type M90T (WT) (B) strains for 30 min. Lipids were then extracted and analysed as described in Figure 1. Radiolabelled PtdInsP was recovered by scraping the corresponding band from the TLC plate as indicated, and was then deacylated and analysed by an appropriate HPLC technique (Tolias et al., 1998) allowing the separation of PtdIns(4)P and PtdIns(5)P (right panel). Peaks corresponding to these phosphoinositides are indicated. Data are from one experiment, representative of three.
To confirm that PtdIns(5)P was accumulating in infected cells and to quantify its mass level, we performed a recently described mass assay (Morris et al., 2000). Recombinant PtdInsP–kinase IIα, whose major substrate is PtdIns(5)P, was used in the presence of [γ-32P]ATP to phosphorylate the PtdInsP fraction extracted from cells infected with the various strains. The PtdIns(5)P present in this fraction was transformed specifically to [32P]PtdIns(4,5)P2, which was quantified. As shown in Figure 3A, this assay allowed us to demonstrate unambiguously that the mass level of PtdIns(5)P dramatically increased during infection with the wild-type strain M90T (from 18.6 ± 6.9 to 280.5 ± 78.3 pmol/mg of HeLa cell proteins). Conversely, small amounts of PtdIns(5)P were detected in non-infected cells or in cells infected with either the ipgD mutant or BS176. To demonstrate further that IpgD produces PtdIns(5)P in mammalian cells, we measured the level of this phosphoinositide in HeLa cells transiently transfected by a green fluorescent protein (GFP)-tagged IpgD construct. As shown in Figure 3B, expression of IpgD induced the production of a significant amount of PtdIns(5)P. Altogether, these data demonstrated that, in vivo, IpgD specifically acts as a PtdIns(4,5)P2 4-phosphatase and leads to accumulation of PtdIns(5)P in the host cell. During S.flexneri infection, we also noticed an increase in PtdIns(3,4)P2, phosphatidylinositol 3,5-bisphosphate [PtdIns(3,5)P2] and PtdIns(3,4,5)P3, indicating a concomitant activation of a phosphatidylinositol 3-kinase (PI 3-kinase) (Figure 4). The increase in these phosphoinositides during infection highlights the in vivo speci ficity of this bacterial phosphatase towards cellular PtdIns(4,5)P2, which appears to be the only phosphoinositide to be hydrolysed. These results also indicate that the in vitro activity of IpgD towards PtdIns(3,4)P2 and PtdIns(3,4,5)P3 (Table I) does not appear to reflect its activity in infected cells. Experiments performed with the non-invasive mutant (BS176) or the ipgD mutant indicated that activation of the PI 3-kinase pathway was linked to invasion (Figure 4C), as was also suggested recently for Salmonella (Marcus et al., 2001). This activation of PI 3-kinase, however, was not required for S.flexneri entry since treatment of HeLa cells with the PI 3-kinase inhibitor wortmannin had no effect on the entry process (data not shown).
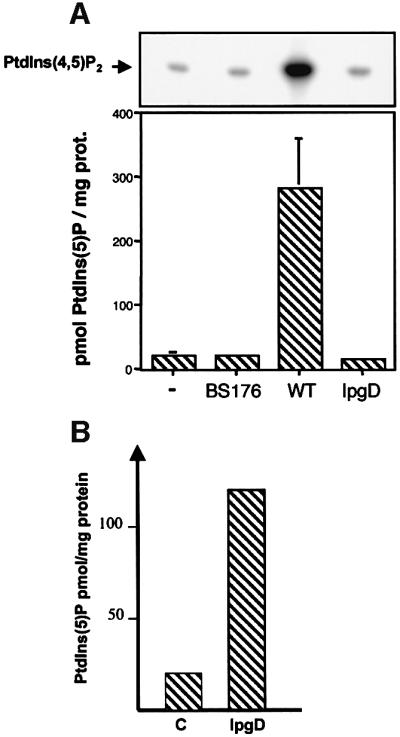
Fig. 3. Quantification of IpgD-dependent production of PtdIns(5)P by mass assay. (A) The mass level of PtdIns(5)P was measured in non- infected cells (–) or in cells infected with either M90T, ipgD or the avirulant, non-invasive mutant BS176 for 20 min using a specific mass assay as previously described by Morris et al. (2000). Recombinant PIPkinase IIα was used in the presence of [γ-32P]ATP to phosphorylate PtdIns(5)P present in the PtdInsP fraction extracted from cells infected with the different strains. The PtdIns(5)P was transformed specifically to [32P]PtdIns(4,5)P2, which was quantified. A representative TLC illustrating the production of [32P]PtdIns(4,5)P2 from PtdIns(5)P present in the PtdInsP fraction extracted from cells infected with various strains is shown (top panel). Results are also expressed as pmol PtdIns(5)P/mg of HeLa cell proteins and are the mean ± SEM of three independent experiments (lower panel). (B) The mass level of PtdIns(5)P was measured in HeLa cells transfected with GFP (C) or GFP-tagged IpgD (IpgD) after 24 h. Results are representative of three independent experiments.
IpgD is translocated into the eukaryotic cell by extracellular bacteria
The rapid degradation of cellular PtdIns(4,5)P2 upon contact with S.flexneri suggested that this mechanism might be an early event during the invasion process, preceding the entry of bacteria into host cells. To determine whether IpgD was injected into the host cell cytoplasm by extracellular bacteria or whether it was secreted by bacteria that had already reached the cytoplasm, HeLa cells were treated with cytochalasin D to prevent the actin rearrangements required for bacterial uptake (Clerc and Sansonetti, 1987). During infection in the presence of 0.5 µg/ml cytochalasin D, the entry of S.flexneri was blocked by 100%, whereas an important production of PtdIns(5)P was still observed (201.8 ± 50 pmol/mg of HeLa cell proteins in 0.5 µg/ml cytochalasin D-treated cells versus 280.5 ± 78.3 in non-treated cells). Accordingly, a drop in cellular [32P]PtdIns(4,5)P2 was also detected (30 ± 9% decrease). These results demonstrated that IpgD is injected into the host cell by extracellular bacteria as a part of the initial step of the infection process.
IpgD causes membrane blebbing and cell rounding when expressed in HeLa cells
A comparison between the wild-type and the ipgD mutant strains showed that the mutant elicited entry structures with a highly altered morphology (Niebuhr et al., 2000). In contrast to the wild-type strain, which caused prominent cell surface and actin rearrangements around its attachment sites, the ipgD mutant induced dense, cup-like actin structures beneath adherent bacteria that resembled focal adhesion plaques. To examine the effect of IpgD on eukaryotic cells in the absence of other bacterial factors, we transiently expressed a myc-tagged IpgD in HeLa cells. Transfected cells were analysed by SDS–PAGE, and IpgD was detected with antibodies against the tag or the protein itself, to confirm that IpgD was expressed as a protein of the expected size (not shown). Moreover, after 24 h of expression of IpgD in HeLa cells, the production of PtdIns(5)P was clearly detected (20 pmol/mg protein in non-transfected versus 122 pmol/mg protein in transfected cells), indicating that the transfected phosphatase was active. Analysis of transfected cells by confocal microscopy revealed that expression of IpgD affected cell morphology. After a period of 8–24 h, HeLa cells started to form membrane blebs that protruded up to 15 µm above the cell surface, while the actin stress fibres began to disappear (Figure 5). Expression of the inactive myc-tagged IpgD-C438S did not induce these morphological changes (not shown). Furthermore, expression of GFP-tagged IpgD in NIH-3T3 cells also affected their morphology. Again, after 8–24 h, these cells formed membrane blebs and the actin stress fibres disappeared (Figure 5C and E). As a control, expression of the inactive GFP-tagged IpgD-C438S mutant in NIH-3T3 cells did not significantly affect their morphology (Figure 5D and F).
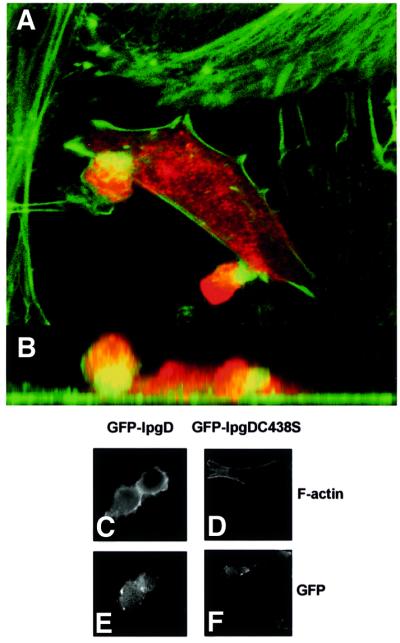
Fig. 5. Expression of IpgD causes formation of membrane blebs. Confocal laser scan analysis of the surface structures elicited on HeLa cells transfected with myc-tagged IpgD after 24 h. For immunofluorescence, transfected cells were visualized using an anti-myc antibody (red), and filamentous actin was stained using FITC-coupled phalloidin. The blebs observed on such cells protruded up to 15 µm above the cells. (A) The sum of all optical sections. (B) The corresponding side view (z-projection). Alternatively, GFP-tagged IpgD (C and E) and GFP-tagged IpgDC438S mutant (D and F) were transfected in NIH-3T3 cells. After 24 h, the filamentous actin was stained using rhodamine-coupled phalloidin (C and D) and the preparations were observed by fluorescence microscopy, using a Zeiss Axioskop microscope equipped with a 63× objective and a Princeton microMAX camera. The data shown are representative of four independent experiments with similar results.
IpgD decreases cytoskeletal–membrane adhesion when expressed in NIH-3T3 cells
Tether force measurement was coupled with fluorescence quantification in NIH-3T3 cells that had been transfected with 0.5 µg of a GFP-tagged IpgD construct. Experiments were performed 8 h post-transfection. As shown in Figure 6A and B, cells expressing increasing levels of GFP-tagged IpgD molecules have an inversely proportional decrease in tether force. Transfected NIH-3T3 cells exhibit dynamic lamellar structures similar to those of transfected HeLa cells at lower concentrations and relatively uniform fluorescence of the fusion protein, as seen in Figure 6C. A blebbing phenotype was observed at concentrations >2 × 105 molecules of GFP-tagged IpgD. After calibration of the laser tweezers (Kuo and Sheetz, 1993), >5 tether force measurements (i.e. the force needed to hold a tethered bead at a constant distance from the trap) were performed in the course of three experiments. As shown in Figure 6A and B, tether force was inversely proportional to the amount of GFP-tagged IpgD fusion expressed in the relevant cell, and a significant decrease in membrane tension was observed even with low levels of expression of GFP-tagged IpgD.

Fig. 6. Membrane cytoskeleton adhesion energy is decreased, as seen by tether force in cells expressing IpgD–GFP fusion protein. After calibration of the laser tweezers, the displacement of the bead from the centre of the trap was converted to tether force (i.e. the force needed to hold a tether at a constant length). (A and B) Increasing levels of IpgD–GFP create an inversely proportional decrease in tether force. An expression level of 3.5 × 105 molecules of IpgD decreases the force by half. (C) NIH-3T3 cells transfected with IpgD–GFP show relatively uniform fluorescence with random punctate spots. Hazy fluorescence is due to the high turnover rate of active lamellae. A standardized bead was used to quantitate the number of molecules of IpgD–GFP. Bar = 7.4 µm.
IpgD enhances the effect of Cdc42 and Rac agonists on cytoskeletal remodelling
Cdc42 and Rac are essential regulators governing production of the filopodial and lamellipodial structures that form the entry focus of Shigella. It was shown previously that these structures were much shorter during entry of a Shigella ipgD mutant (Niebuhr et al., 2000). As shown in Figure 7, expression of IpgD by transfected HEK cells amplified and altered the structure of the cytoskeletal projections induced by bradykinin, an agonist of Cdc42 (Kozma et al., 1995), and by epidermal growth factor (EGF), an agonist of Rac (Azuma et al., 1998). Figure 7 shows the ‘hairless’ aspect of HEK cells and the lack of a significant effect of GFP expression on these cells (Figure 7A), whereas expression of the GFP-tagged IpgD fusion altered cytoskeletal structures, leading to a significant rounding of the cells and a concentration of the actin cytoskeleton at the periphery of these rounded cells (Figure 7D, arrows). The duration of transfection was such that cells did not reach the blebbing stage. In the presence of bradykinin, activation of Cdc42 led to the formation of thin but numerous filopodial structures at the cellular surface (Figure 7B, arrows), whereas those with bradykinin-induced filopodia were significantly extended in the cells expressing the GFP-tagged IpgD construct (Figure 7C, arrows). In the presence of EGF, activation of Rac led to the formation of lamellipodial structures at the cell periphery (Figure 7C, arrows). These lamellipodia were not only extended, but also exhibited modified morphology producing gigantic extensions (Figure 7F, arrows). These results are very consistent with the small size of entry foci induced by the Shigella ipgD mutant (Niebuhr et al., 2000).

Fig. 7. IpgD enhances the effect of Cdc42 and Rac agonists on cytoskeletal remodelling. HEK cells were transfected with GFP (A–C) or GFP-tagged IpgD (D–F). The duration of transfection was such that cells did not reach the blebbing stage. Cells were then stimulated (B, C, E and F) or not (A and D) with 0.1 µg/ml bradykinin for 20 min (B and E) or 5 nM EGF for 5 min (C and F), and filamentous actin was stained using rhodamine-coupled phalloidin. The preparations were observed by confocal microscopy. Data shown are representative of three independent experiments.
Discussion
Seven different polyphosphoinositides have been identified so far, and most of them play central roles in the control of fundamental cell functions, such as spatio-temporal organization of key signalling pathways, reorganization of the actin cytoskeleton or intracellular membrane trafficking (Toker, 1998; Czech, 2000; Odorizzi et al., 2000). A complex set of phosphoinositide-metabolizing enzymes, such as kinases and phosphatases, accurately regulates the level of these quantitatively minor lipid molecules. These enzymes, which seem to be targeted specifically to various intracellular membrane domains, can also control locally the complex interconversions between the different phosphoinositides (Payrastre et al., 2001). Phosphoinositides can exert their functions either as precursors of second messengers, or directly on themselves, or both. For instance, besides its well documented role as a precursor of inositol 1,4,5-trisphosphate (InsP3) and diacylglycerol in response to agonist-dependent activation of specific phospholipase C (PLC), PtdIns(4,5)P2 is involved directly in several key cellular processes (Hinchliffe et al., 1998; Toker, 1998; Czech, 2000; Hinchliffe, 2000). A number of reports provide compelling evidence for a central role for PtdIns(4,5)P2 in the actin cytoskeleton remodelling by regulating the activities of proteins that sequester actin monomers, bind or cross-link actin filaments, and cap and/or sever these filaments (Toker, 1998; Sechi and Wehland, 2000; Payrastre et al., 2001). Recently, it was shown that plasma membrane PtdIns(4,5)P2 concentrations control the adhesion energy of the cortical cytoskeleton to the plasma membrane and thereby regulate dynamic membrane functions and cell shape (Raucher et al., 2000). It is likely that such a membrane–cytoskeleton interaction is an important component of the strategy that invasive pathogens apply to facilitate their entry into non-phagocytic cells.
In eukaryotic cells, it is believed classically that PtdIns(4)P and PtdIns(4,5)P2 are kept at a steady-state level in the inner leaflet of the plasma membrane, due to the continuous sequential phosphorylation/dephosphorylation reactions by specific kinases and phosphatases that have not yet been fully identified (Toker, 1998; Payrastre et al., 2001). By specifically dephosphorylating PtdIns (4,5)P2 into the recently identified phosphoinositide, PtdIns(5)P, as an early step during the invasion process, S.flexneri alters one of the central switches of phosphoinositide metabolism. This function of IpgD probably allows a local detachment of the plasma membrane from the cytoskeleton to facilitate extension of membrane filopodia and ruffles, which also depend on Cdc42- and Rac-mediated actin cytoskeleton reorganization, upon S.flexneri entry. In the absence of Shigella, continuous ectopic expression of IpgD in the cell leads to increasing membrane detachment, thus causing cell blebbing. This interpretation is supported by several findings. First, IpgD transfection into eukaryotic cells leads to a dramatic reduction of membrane tether force and cell surface rearrangements that eventually led to blebbing. This result supports the role of PtdIns(4,5)P2 as a regulator of the adhesion energy directly by reducing or adding local binding interactions between the cytoskeleton and the membrane (Raucher et al., 2000). This is in agreement with the fact that microinjection of GST–IpgD but not of the inactive GST–IpgD (C438S) mutant induced local morphological changes similar to membrane blebbing (not shown). Secondly, an ipgD mutant of S.flexneri showed an altered phenotype with respect to the morphology of its entry structures. The wild-type strain triggered massive actin-rich, membraneous cell surface rearrangements around its attachment sites, whereas the ipgD mutant induced entry structures with smaller membrane ruffles and actin rearrangements that were focused mostly around adherent bacteria (Niebuhr et al., 2000).
In order to make sense of this difference, we studied whether expression of IpgD in eukaryotic cells was able to facilitate the development of cytoskeleton rearrangement triggered by physiological agonists such as bradykinin and EGF, which activate Cdc42 and Rac, respectively. Such a potentiation indeed was clearly demonstrated. Degradation of PtdIns(4,5)P2 or appearance of PtdIns(5)P may directly potentiate Cdc42 and/or Rac activation, although preliminary observations suggest that expression of IpgD does not activate Rho GTPases per se. The release of membrane tension may also allow a fuller development of the cytoskeletal rearrangements, thus explaining the formation of massive ruffling in the presence of these agonists. Similarly, in the process of S.flexneri entry, the signalling pathway that is known to involve Cdc42 and Rac (Mounier et al., 1999; Tran Van Nieu et al., 1999) expresses its full potential of cytoskeleton extensions thanks to the membrane tension release induced by IpgD. These data strongly suggest that a decrease in PtdIns(4,5)P2 levels has a much stronger impact on the promotion of cytoskeleton extension via the release of membrane tension than on the stabilization of the length of actin filaments that can be expected based on the known regulation of capping proteins by PtdIns(4,5)P2 (Sechi and Wehland, 2000). However, this does not take into account the activation of PI 3-kinase observed during S.flexneri entry, which uses a pool of PtdIns(4,5)P2 spared by the phosphatase activity of IpgD, and may also contribute to the reorganization of the cytoskeleton. In addition, it may not only be the decrease of PtdIns(4,5)P2 that affects the cell, since the accumulation of the product of the enzymatic reaction might also play a role during infection. Indeed, the mass level of PtdIns(5)P, which is only found in trace amounts in resting cells, increased up to 280 pmol/mg of HeLa cell protein during infection. This value is in the expected range of the drop in PtdIns(4,5)P2. The physiological role of this phospholipid that, until 1997, was thought not to exist in vivo (Rameh et al., 1997) is still unknown. The amount of PtdIns(5)P in resting cells is considered very low compared with PtdIns(4)P. Recently, a thrombin-dependent increase in PtdIns(5)P was observed in platelets (Morris et al., 2000), suggesting that this lipid might have a physiological function and that its synthesis is tightly controlled through enzymes regulated by physiological stimuli. The route of synthesis of PtdIns(5)P in vivo is also still unknown, but it may involve either a PtdIns 5-kinase acting on PtdIns (Tolias et al., 1998; Sbrissa et al., 1999) or, as performed by IpgD, 4-dephosphorylation of PtdIns(4,5)P2. It is tempting to speculate that the role of IpgD is not only to uncouple the cellular plasma membrane from the cytoskeleton to facilitate the formation of entry structures, but also to generate a novel messenger molecule to stimulate further or interfere with host cellular functions for the benefit of the pathogen. Whether PtdIns(5)P is involved in the typical actin reorganization at the site of entry, or rather is involved in the inflammatory response, remains to be established. In any case, this model provides some insights into the function of this new phosphoinositide in the eukaryotic cell.
Proteins homologous to IpgD have been described in S.dublin (SopB) (Galyov et al., 1997) and S.typhimurium (SigD) (Hong and Miller, 1998). The two motifs that are related to sequences in mammalian inositol polyphosphate 4-phosphatases are conserved in all these bacterial proteins (Norris et al., 1998; Marcus et al., 2001). However, characterization of the enzymatic activity of recombinant GST–SopB revealed a specificity different from that of IpgD because its major in vitro substrates are PtdIns(3,4,5)P3, PtdIns(3,4)P2 and PtdIns(3)P (Norris et al., 1998). In contrast, the major IpgD substrate in vivo and in vitro, PtdIns(4,5)P2, was not hydrolysed by SopB or SigD (Marcus et al., 2001) in in vitro assays. Moreover, in vivo, SopB was found to induce a significant production of Ins(1,4,5,6)P4 upon S.dublin infection (Eckmann et al., 1997; Norris et al., 1998), whereas this inositol phosphate was produced only weakly during S.flexneri infection (Eckmann et al., 1997). Although the mode of entry of S.flexneri and S.dublin into epithelial cells is very similar, their intracellular lifestyles are very distinct. Whereas S.flexneri rapidly lyses the phagosomal membrane, multiplies freely in the cytosol and uses the actin cytoskeleton to spread to neighbouring cells, S.dublin resides in a specialized vacuole. These observations suggest that distinct disruption of phosphoinositide metabolism might contribute to different cellular responses against S.flexneri and S.dublin.
The fact that a pathogenic bacterium manipulates a very specific step of phosphoinositide metabolism during infection highlights the critical role of these lipids in controlling highly dynamic mechanisms of cell regulation. In this respect, the S.flexneri effector, IpgD, provides a useful tool to investigate the exact role of PtdIns(5)P in cellular functions. Furthermore, this work stresses the importance of investigating in more detail the role of phosphoinositides during the infection process.
Materials and methods
Cell infections and phospholipid analysis
HeLa cells were incubated in phosphate-free minimal essential medium (MEM) containing 1 mCi [32P]orthophosphate/dish (200 µCi/ml) for 6 h before infection in order to reach the isotopic equilibrium. Derivatives of the S.flexneri wild-type strain M90T, the ipgD mutant or the non-invasive strain BS176, each expressing the AfaE adhesin (Garcia et al., 1994), were grown to exponential growth phase and, at t0, 5 × 108 bacteria were added per dish and incubated at 37°C. Lipids from infected cells were extracted, separated on thin-layer chromatography (TLC), scraped off, deacylated and analysed by HPLC as described previously (Ireton et al., 1996). [32P]lipids separated by TLC or HPLC were identified using standards. The separation of PtdIns(4)P and PtdIns(5)P was performed by an appropriate HPLC technique as described previously (Tolias et al., 1998; Sbrissa et al., 1999).
PtdIns(5)P mass assay
Quantification of the PtdIns(5)P level was performed as described by Morris et al. (2000), except that the PtdInsP fraction was purified by TLC after total lipid extraction according to a modified Bligh and Dyer procedure (Bligh and Dyer, 1959; Ireton et al., 1996) from HeLa cells infected or not by the different bacterial strains or transfected by GFP-tagged IpgD.
In vitro measurements of IpgD activity
The 32P-labelled substrates were prepared as described previously (Blondeau et al., 2000). Briefly, human platelets (3 × 109 cells) were incubated for 1 h at 37°C in the presence of 350 µCi/ml [32P]orthophosphate (Amersham Pharmacia Biotech, Orsay, France). Lipids were then extracted and TLC was performed in order to separate PtdIns(4)P and PtdIns(4,5)P2. They were then scraped and extracted from the silica. 32P-labelled D3-phosphoinositides were produced and purified as indicated previously (Payrastre et al., 1994).
TLC-purified [32P]phosphoinositides and phosphatidylserine, as a carrier lipid, were dried under a stream of nitrogen and suspended in 50 µl of 20 mM MES pH 6.5, 0.6% octylglucoside, 2.5 mM EDTA, 100 mM KCl, and sonicated for 30 s in a bath sonicator. The phosphatase assay was performed on labelled PtdIns(3)P, PtdIns(3,4)P2, PtdIns(4)P, PtdIns(4,5)P2 and PtdIns(3,4,5)P3 whose concentrations were made up to 10 µM with dipalmitoyl PtdIns(3)P and PtdIns(3,4)P2 (Echelon Research Labs, Salt Lake City, UT) and 1-stearoyl 2-arachidonoyl PtdIns(4)P and PtdIns(4,5)P2 (Sigma, St Louis, MO) and PtdIns(3,4,5)P3 (Alexis Corporation, San Diego, CA), respectively. Reactions were started by addition of 50 µl of recombinant GST–IpgD (1–3 µg) in 50 mM Tris pH 7.5 containing 50 mM glutathione and 100 mM NaCl and incubated at 37°C under gentle shaking for 15 min. The reaction mixture contained 10 µM phosphoinositides and 20 µM phosphatidylserine. The reaction was stopped by adding CHCl3:CH3OH (v/v), and lipids were extracted, separated by TLC using CHCL3:CH3COCH3:CH3OH:CH3COOH:H2O (80:30:26:24:14, v/v), visualized by a PhosphorImager 445SI (Molecular Dynamics, Inc., Sunnyvale, CA), scraped off and quantified by liquid scintillation counting.
A 10 µM concentration of Ins(1,4,5)P3 (Sigma) and 1500 c.p.m. of [3H]Ins(1,4,5)P3 (NEN) or 1 µM Ins(1,3,4,5)P4 (Boehringer) and 1500 c.p.m. of [3H]Ins(1,3,4,5)P3 (NEN) were used as substrate to measure the inositol phosphate phosphatase activity of recombinant GST–IpgD (1–3 µg). Reactions were performed at 37°C for 10 min in a 50 µl final volume containing 50 mM HEPES pH 7.4, 0.1% bovine serum albumin (BSA), 2 mM MgCl2, and 0.033% β-mercaptoethanol as described previously (Erneux et al., 1989). The reaction was stopped by addition of 5 ml of 0.4 or 0.7 M ammonium formate for Ins(1,4,5)P3 and Ins(1,3,4,5)P4, respectively. These concentrations of formate allow the elution of InsP, InsP2 and InsP3 on silica dowex chromatography.
Expression constructs and transient transfections
Expression and purification of GST-tagged IpgD were as described (Niebuhr et al., 2000). The IpgDC438S mutant was constructed by site-directed mutagenesis (Stratagene, La Jolla, CA) that converted TGT (C438) to TCG (S), also creating an EcoRI site in the mutant allele. Plasmid pKNE4, harbouring GST–IpgD and expressing IpgE (Niebuhr et al., 2000), was used as template for the PCR mutagenesis using primers IpgDmut1 (TGTACCTTGCTGGAATTCGAAGAGTGGGAAGGACA) and IpgDmut2 (TGTCCTTCCCACTCTTCGAATTCCAGCAAGGTA CA). For transfection experiments, the ipgD coding sequence was cloned into the BamHI–EcoRI site of the eukaryotic expression vector pRK5 to allow expression of a myc-tagged IpgD protein. To construct plasmid pKN16 that expressed a GFP-tagged IpgD recombinant protein, a PCR fragment encompassing the IpgD and IpgE genes was amplified using oligonucleotides IpgD3 (CCACGCTCCGGAATGCACATAACTAATT TGGGA) and IpgE1 (CCACCGGTCGACTTAATACCCCTTCATTC TTCG) and cloned between the BspEI and SalI sites of pEGFP-C1 (Clontech Laboratories, Palo Alto, CA). The GFP-tagged IpgDC438S mutant was constructed by site-directed mutagenesis that converted TGT (C438) to TCG (S). Sequences were confirmed by nucleotide sequencing analyses.
Semi-confluent HeLa cells or NIH-3T3 cells grown on coverslips in 12-well plates were transfected with 1 µg of plasmid DNA mixed with 3 µl of Fugene transfection reagent (Boehringer). After 8–24 h, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min and subsequently permeabilized with 0.2% Triton X-100 and processed for immunofluorescence using a monoclonal antibody against IpgD and phalloidin–fluorescein isothiocyanate (FITC).
Measurement of adhesion energy
NIH-3T3 cells were grown in Dulbeco’s modified Eagle’s medium (DMEM; Gibco, Invitrogen Corporation, Carlsbad, CA) supplemented with 10% newborn calf serum (NCS; Gibco), 100 µg/ml streptomycin, 100 U/ml penicillin, 200 mM l-glutamine and 7.5 mM HEPES (Gibco) at 37°C in 5% CO2. Cells were removed from flasks by brief treatment with trypsin-EDTA 0.25% (Gibco) and plated onto glass coverslips. Cells were transfected 1 day after plating with 1 µg of the plasmid pKN16 expressing the GFP-tagged IpgD construct and Lipofectamine Plus (Gibco). Experiments were performed at 8 h post-transfection in DMEM (10% NCS) without phenol red.
Bead preparation. To prepare concanavalin A (ConA)-coated beads, ConA (Sigma, St Louis, MO) was solubilized at a concentration of 10 mg/ml in PBS with Ca2+ and Mg2+ added (Gibco). A 100 µg aliquot of silica microspheres (0.9 µM diameter, Bangs Labs, SS03N) was activated using a hydroxyl-modified covalent coupling method (Bangs Labs, Tech Note 204). A 100 µl aliquot of a ConA solution was added and incubated overnight at 4°C. The beads were pelleted and resuspended with 40 mM ethanolamine (30 min at room temperature) to quench unreacted CNBr. A final pelleting and resuspension was done in 1 ml of PBS with Ca2+ and Mg2+. For experiments, the bead solution was diluted 1:1000 in DMEM without phenol red.
Optical laser trap manipulation. The NIH-3T3 fibroblasts were viewed by a video-enhanced differential interference contrast (DIC) microscope (Zeiss Axiovert S1OO TV) equipped with laser optical tweezers (Choquet et al., 1997). Beads were trapped with 950 mW of laser power, attached to the cell membrane and pulled away from the cell surface to create a tether. The tether force was calculated from the displacement of the bead from the centre of the trap during tether formation and multiplied by the trap stiffness (calculations were done using Innovision software; Dai and Sheetz, 1995).
Fluorescence quantification. The transfected NIH-3T3 cells were visualized using a GFP filter set (Chroma Technology) at 448λ. Images were obtained using a Roper Coolsnap Fx Cooled-CCD camera at 1 s exposure. Standardized 7.4 mm polystyrene beads containing 43 907 MESF of FITC (Bangs Labs, CO-122096) were diluted 1:1000 to give 1–2 beads per field of view. Fluorescence quantification was performed using public domain software image J 1.28g (http//rsb.info.nih.gov/ij/). The number of molecules of IpgD–GFP fusion protein are calculated using beads standarized for GFP in the following calculation:
Total No. of pe of cell (W) = [Ac × (Ic – Ibk)]
Total No. of pe of bead (U) = [Abd × (Ibd – Ibk)]
W/U = M (ratio of bead equivalents)
Q = (No. of molecules of EGFP/bead) factor
Q*M = Kbd (No. of molecules of EGFP fusion)
Where Ic is the intensity in photoelectrons (pe) per pixel for the cell, Ibk is the pe per pixel for background, Ibd is the pe per pixel for the bead, Ac is the area of the cell in pixels, and Abd is the area of the bead in pixels. Q is standardized against a stable pEGFP population of 293 cells to obtain the number of molecules of EGFP per standard bead. Total intensity in pe was calculated for each transiently transfected cell and divided by the total pe of the beads in the viewing area. This is then multiplied by our factor (number of molecules of EGFP/bead) and results are given in number of molecules of IpgD–EGFP.
Effect of IpgD on cytoskeletal remodelling mediated by Cdc42 and Rac agonists
Semi-confluent HEK cells in DMEM–10% fetal calf serum (FCS) were cultured on coverslips in 6-well plates.
After washings in DMEM, without FCS, the cells were cultured in the same medium. A mixture of 300 µl of DMEM and 25 µl of Fugene reagent was incubated for 5 min at room temperature before addition of 75 µl (4.5 µg) of the plasmid pKN16 expressing the GFP-tagged IpgD construct, GFP or GFP-tagged IpgD DNA. After 15 min, 120 µl of this mixture was added to the cells, and the incubation was performed for 8 h at 37°C and 5% CO2. The cells were then cultured in the absence or presence of EGF (5 nM, 5 min) or with bradykinin (0.1 µg/ml, 20 min). After washings with PBS, the cells were fixed with 2.5% paraformaldehyde (PFA) for 20 min and then washed in PBS. After permeabilization by 0.1% Triton X-100 in PBS for 5 min, the cells were incubated in BSA (1% in PBS, 20 min), then incubated for 1 h with rhodamine-coupled phalloidin (1/100 in PBS–BSA). After washings, the coverslips were mounted with Mowiol, and the preparations were observed by confocal microscopy.
Acknowledgments
Acknowledgements
We thank N.Divecha for the cDNA for typeII PIPkinase, C.Erneux and V.Dewaste for help with the inositol phosphate phosphatase assay, V.Sallé and H.Tronchère for the construction of the GFP-tagged IpgDC438S mutant, F.Capilla for technical assistance, and T.Adam for helpful discussions. This work was partly supported by a grant from Association pour la Recherche Contre le Cancer (ARECA-Toulouse). K.N. was supported by an EMBO long-term postdoctoral fellowship and a grant from the GIP-HMR (Hoechst-Marion-Roussel), and D.J.P. was supported by a fellowship from the Canadian Institute of Health Research. P.J.S. is a Howard Hughes Medical Institute Scholar.
References
- Adam T., Arpin,M., Prévost,M.-C., Gounon,P. and Sansonetti,P.J. (1995) Cytoskeletal rearrangements and the functional role for T-plastin during entry of Shigella flexneri into HeLa cells. J. Cell Biol., 129, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaoui A., Ménard,R., Sansonetti,P.J. and Parsot,C. (1993) Characterization of the Shigella flexneri ipgD and ipgF genes, which are located in the proximal part of the mxi locus. Infect. Immun., 61, 1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma T., Witke,W., Stossel,T.P., Hartwig J.H. and Kwiatkowski,D.J. (1998) Gelsolin is a downstream effector of rac for fibroblast motility. EMBO J., 17, 1362–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh E.G. and Dyer,W.J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol., 37, 911–919. [DOI] [PubMed] [Google Scholar]
- Blondeau F., Laporte,J., Bodin,S., Superti-Furga,G., Payrastre,B. and Mandel,J.L. (2000) Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum. Mol. Genet., 9, 2223–2229. [DOI] [PubMed] [Google Scholar]
- Choquet D., Felsenfeld,D.P. and Sheetz,M.P. (1997) Extracellular matrix rigidity causes strengthening of integrin–cytoskeleton linkages. Cell, 88, 39–48. [DOI] [PubMed] [Google Scholar]
- Clerc P. and Sansonetti,P.J. (1987) Entry of Shigella flexneri into HeLa cells: evidence for directed phagocytosis involving actin polymer ization and myosin accumulation. Infect. Immun., 55, 2681–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech M.P. (2000) PIP2 and PIP3: complex roles at the cell surface. Cell, 100, 603–606. [DOI] [PubMed] [Google Scholar]
- Dai J. and Sheetz,M.P. (1995) Mechanical properties of neuronal growth cone membranes studied by tether formation with laser optical tweezers. Biophys. J., 68, 988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann L. et al. (1997) d-Myo-inositol 1,4,5,6-tetrakisphosphate produced in human intestinal epithelial cells in response to Salmonella invasion inhibits phosphoinositide 3-kinase signaling pathways. Proc. Natl Acad. Sci. USA, 94, 14456–14460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erneux C., Lemos,M., Verjans,B., Vanderhaeghen,P., Delvaux,A. and Dumont,J.E. (1989) Soluble and particulate Ins(1,4,5)P3/Ins(1,3,4,5)P4 5-phosphatase in bovine brain. Eur. J. Biochem., 181, 317–322. [DOI] [PubMed] [Google Scholar]
- Finlay B.B. and Falkow,S. (1990) Salmonella interactions with polarized human intestinal Caco-2 epithelial cells. J. Infect. Dis., 162, 1096–1106. [DOI] [PubMed] [Google Scholar]
- Francis C.L., Ryan,T.A., Jones,B.D., Smith,S.J. and Falkow,S. (1993) Ruffles induced by Salmonella and other stimuli direct macropinocytosis of bacteria. Nature, 364, 639–642. [DOI] [PubMed] [Google Scholar]
- Galan J.E. and Bliska,J.B. (1996) Cross talks between bacterial pathogens and their host cells. Annu. Rev. Cell. Dev. Biol., 12, 221–255. [DOI] [PubMed] [Google Scholar]
- Galyov E.E., Wood,M.W., Rosquist,R., Mullan,P.B., Watson,P.R., Hedges,S. and Wallis,T.S. (1997) A secreted effector protein of Salmonella dublin is translocated into eukaryotic cells and mediates inflammation and fluid secretion in infected ileal mucosa. Mol. Microbiol., 25, 903–912. [DOI] [PubMed] [Google Scholar]
- Garcia M.I., Labigne,A. and Le Bouguenec,C. (1994) Nucleotide sequence of the afimbrial-adhesin-encoding afa-3 gene cluster and its translocation via flanking IS1 insertion sequences. J. Bacteriol., 176, 7601–7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchliffe K.A. (2000) Intracellular signalling: is PIP2 a messenger too ? Curr. Biol., 10, R104–R105. [DOI] [PubMed] [Google Scholar]
- Hinchliffe K.A., Ciruela,A. and Irvine,R.F. (1998) PIPkins, their substrates and their products: new functions for old enzymes. Biochim. Biophys. Acta, 1436, 87–104. [DOI] [PubMed] [Google Scholar]
- Hong K.H. and Miller,V.L. (1998) Identification of a novel Salmonella invasion locus homologous to Shigella ipgDE. J. Bacteriol., 180, 1793–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireton K., Payrastre,B., Chap,H., Ogawa,W., Sakaue,H., Kasuga,M. and Cossart,P. (1996) A role for phosphoinositide 3-kinase in bacterial invasion. Science, 274, 780–782. [DOI] [PubMed] [Google Scholar]
- Kozma R., Ahmed,S., Best,A. and Lim,L. (1995) The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol., 15, 1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo S.C. and Sheetz,M.P. (1993) Force of single kinesin molecules measured with optical tweezers. Science, 260, 232–234. [DOI] [PubMed] [Google Scholar]
- Marcus S.L., Wenk,M.R., Steele-Mortimer,O. and Finlay,B.B. (2001) A synaptogamin-homologous region of Salmonella typhimurium SigD is essential for inositol phosphatase activity and Akt activation. FEBS Lett., 494, 201–207. [DOI] [PubMed] [Google Scholar]
- Mengaud J., Ohayon,H., Gounon,P., Mège,R.-M. and Cossart,P. (1996) E-cadherin is the receptor for internalin, a surface protein required for entry of Listeria monocytogenes into epithelial cells. Cell, 84, 923–932. [DOI] [PubMed] [Google Scholar]
- Morris J.B., Hinchliffe,K.A., Ciruela,A., Letcher,A.J. and Irvine,R.F. (2000) Thrombin stimulation of platelets causes an increase in phosphatidylinositol 5-phosphate revealed by mass assay. FEBS Lett., 475, 57–60. [DOI] [PubMed] [Google Scholar]
- Mounier J., Laurent,V., Hall,A., Fort,P., Carlier,F., Sansonetti,P.J. and Egile,C. (1999) Rho family GTPases control entry of Shigella flexneri into epithelial cells but not intracellular motility. J. Cell Sci., 112, 2069–2080. [DOI] [PubMed] [Google Scholar]
- Niebuhr K., Jouihri,N., Allaoui,A., Gounon,P., Sansonetti,P. and Parsot,C. (2000) IpgD, a protein secreted by the type III secretion machinery of Shigella flexneri, is chaperoned by IpgE and implicated in entry focus formation. Mol. Microbiol., 38, 8–19. [DOI] [PubMed] [Google Scholar]
- Norris F.A., Atkin R.C. and Majerus,P.W. (1997) The cDNA cloning and characterization of inositol polyphosphate 4-phosphatase type II. Evidence for conserved alternative splicing in the 4-phosphatase family. J. Biol. Chem., 272, 23859–23864. [DOI] [PubMed] [Google Scholar]
- Norris F.A., Wilson,M.P., Wallis,T.S., Galyov,E.E. and Majerus,P.W. (1998) SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc. Natl Acad. Sci. USA, 95, 14057–14059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi G., Babst,M. and Emr,S.D. (2000) Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem. Sci., 25, 229–235. [DOI] [PubMed] [Google Scholar]
- Payrastre B., Gironcel,D., Plantavid,M., Mauco,G., Breton,M. and Chap,H. (1994) Phosphoinositide 3-phosphatase segregates from phosphatidylinositol 3-kinase in EGF-stimulated A431 cells and fails to in vitro hydrolyze phosphatidylinositol (3,4,5)trisphosphate. FEBS Lett., 341, 113–118. [DOI] [PubMed] [Google Scholar]
- Payrastre B., Missy,K., Giuriato,S., Bodin,S., Plantavid,M. and Gratacap,M.P. (2001) Phosphoinositides: key players in cell signalling, in time and space. Cell Signal., 13, 377–387. [DOI] [PubMed] [Google Scholar]
- Rameh L.E., Tolias,K.F., Duckworth,B.C. and Cantley,L.C. (1997) A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature, 390, 192–196. [DOI] [PubMed] [Google Scholar]
- Raucher D., Stauffer,T., Chen,W., Shen,K., Guo,S., York,J.D., Sheetz,M.P. and Meyer,T. (2000) Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton–plasma membrane adhesion. Cell, 100, 221–228. [DOI] [PubMed] [Google Scholar]
- Sbrissa D., Ikonomov,O.C. and Shisheva,A. (1999) PIKfyve, a mammalian ortholog of yeast Fab1p lipid kinase, synthesizes 5-phosphoinositides. J. Biol. Chem., 274, 21589–21597. [DOI] [PubMed] [Google Scholar]
- Sechi A.S. and Wehland,J. (2000) The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P2 influences cytoskeletal protein activity at the plasma membrane. J. Cell Sci., 113, 3685–3695. [DOI] [PubMed] [Google Scholar]
- Toker A. (1998) The synthesis and cellular roles of phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Cell Biol., 10, 254–261. [DOI] [PubMed] [Google Scholar]
- Tolias K.F., Rameh,L.E., Ishihara,H., Shibasaki,Y., Chen,J., Prestwich,G.D., Cantley,L.C. and Carpenter,C.L. (1998) Type I phosphatidyl inositol-4-phosphate 5-kinases synthesize the novel lipids phosphatidylinositol 3,5-bisphosphate and phosphatidylinositol 5-phosphate. J. Biol. Chem., 273, 18040–18046. [DOI] [PubMed] [Google Scholar]
- Tran Van Nhieu G., Caron,E., Hall,A. and Sansonetti,P.J. (1999) IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J., 18, 3249–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gijsegem F., Genin,S. and Boucher,C. (1993) Conservation of secretion pathways for pathogenicity determinants of plant and animal bacteria. Trends Microbiol., 1, 175–180. [DOI] [PubMed] [Google Scholar]