

Abstract
In this study, using heat shock factor 1 (Hsf1) knockout mice as a model, we tested the hypothesis that HSF1-dependent regulation of heat shock proteins (Hsps) is required to maintain redox state and attenuate oxidative damage in the normal heart. Here we report that, in mice, HSF1 deficiency reduces cardiac expression of Hsp25, αB-crystallin and Hsp70, but not Hsp60 and Hsp90. Consistent with the downregulation of Hsp25, for example, a significantly lower glutathione (GSH)/glutathione disulfate (GSSG) ratio was associated with the decreased activity, but not protein content, of glucose 6-phosphate dehydrogenase. Con sequently, superoxide was generated at a higher rate, and several mitochondrial proteins, including adenine nucleotide translocase 1 (ANT1), were more oxidized by HSF1 deficiency in vivo. Oxidative damage to ANT1 protein, a structural component of the mitochondrial permeability transition pore (MPTP), decreases its catalytic activity and increases MPTP opening, respectively. Taken together, our results indicate for the first time that constitutive expression of HSP chaperones requires HSF1 activity, and that such HSF1-dependent requirements are directly and functionally linked to maintain redox homeostasis and antioxidative defenses at normal (37°C) temperature.
Keywords: HSF1/Hsps/mitochondrial ANT1/oxidative damage/redox state
Introduction
A variety of stressful and pathological conditions induce the expression of heat shock proteins (Hsps), which enhance physiological resistance against the potentially deleterious consequences of environmental challenges. The inducible synthesis of Hsps is known to be tightly controlled by heat shock transcription factors (HSFs; Morimoto, 1998). Despite several genes encoding multiple HSFs isoforms (HSF1α and β, HSF2α and β, HSF3 and HSF4) having been identified in vertebrates (Sarge et al., 1991; Nakai and Morimoto, 1993), only HSF1 is essential for the transcriptional activation of Hsps in mammalian organisms (McMillan et al., 1998; Morimoto, 1998).
In recent years, a conceptual framework for stress-induced activation and feedback repression of HSF1 has emerged (for reviews, see Wu, 1995; Morimoto, 1998; Pirkkala et al., 2000). The HSF1 molecule contains distinct regulatory modules including an N-terminal DNA-binding domain, a central trimerization domain containing heptad repeats of hydrophobic leucine zippers, and a C-terminal transactivation domain (Wu, 1995). At physiological conditions, negative regulation of transcriptional activity occurs at multiple levels in which the transactivation domains of HSF1 monomers are sequestered by both intramolecular (Jacquier-Sarlin and Polla, 1996; Manalo et al., 2002) and intermolecular interactions (Wu, 1995; Marchler and Wu, 2001). Stressful conditions including oxidative stress might act at two levels towards HSF1 activation: (i) to generate misfolded and/or denatured proteins that increase their propensity for association with chaperones instead of HSF1, thus shifting the equilibrium away from negative feedback; and (ii) to induce a conformation change that normally represses HSF1 activity (Amin et al., 1988).
Along with the insights gained about their regulation, a substantial amount of literature has documented the beneficial effects and mechanisms by which diverse classes of Hsps mitigate pathological injury, both in cultured cells and in intact organs (for reviews, see Benjamin and McMillan, 1998; Christians et al., 2002). Among the subset of Hsps that seem to be specifically involved in the attenuation of molecular oxidative damage, for instance, are the small molecular weight Hsps whose overexpression increases the resistance of L929 fibroblasts, which are particularly vulnerable to oxidative stress induced by H2O2 (Preville et al., 1999). Specifically, Hsp25 has been shown to augment glutathione (GSH) concentration and to protect mitochondrial integrity, as indicated by the transmembrane potential during oxidative stress (Preville et al., 1999).
A well-recognized characteristic of aerobic cells is that they incessantly generate reactive oxygen species (ROS), primarily by the autoxidation of ubisemiquinone, in the mitochondrial electron transport chain (Cadenas et al., 1977; Turrens et al., 1985). Although cells contain an array of antioxidative defenses, the steady-state concentrations of reaction products of free radical attack on macromolecules are still detectable, even under normal physiological conditions. This means that antioxidative defenses are not 100% effective, leading to the concept that cells normally exist under a certain level of oxidative stress.
Of interest, far less attention has been paid to either the regulation or to the functions of the broad classes of mammalian Hsps, which are expressed constitutively in all major organelles, under ambient conditions. In this context, the question arises whether the maintenance of redox state requires HSF1-dependent regulation of Hsps, which, in turn, might play a functional role in the modulation of oxidative stress under normal physiological conditions. In other words, would the physiological level of oxidative stress be higher if HSF1-dependent expression of Hsps was lacking? Accordingly, our primary goal is to elucidate the role of HSF1-dependent regulation of constitutive Hsps under normal physiological conditions, i.e. in the absence of externally or pathologically induced oxidative stress.
We have developed a mouse model in which HSF1 expression is disrupted (McMillan et al., 1998; Xiao et al., 1999), thereby the involvement of HSF1 on constitutive Hsp expression can be directly tested in specific tissues. Consistent with the high abundance of HSF1 expression found in placenta and ovaries, our recent studies have established that HSF1 deficiency causes placenta defects, which reduce embryonic survival (Xiao et al., 1999), and renders female mice infertile (Christians et al., 2000), demonstrating the essential roles of HSF1 in selective tissues in the absence of supraphysiological stress. In turn, these results also raise the possibility that HSF1 abundance in other organs such as the heart might be linked to specific requirements for constitutive HSF1 activity, albeit the nature of these functions has not been elucidated.
Thus, the specific objective of the present study is to test the hypothesis that HSF1 activity is required for the attenuation of oxidative stress/damage in the heart under normal physiological conditions. Our results reveal not only that a novel regulatory requirement exists on the constitutive level of Hsp expression by HSF1, but also that such HSF1-dependent function is linked directly to a major antioxidative pathway through effects on the activity of glucose 6-phosphate dehydrogenase (G6PD). Furthermore, we demonstrate that an increased oxidative stress caused by HSF1 deficiency has a detrimental effect on critical molecular targets such as adenine nucleotide translocator 1 (ANT1) and define such functional consequences at the level of mitochondrial permeability transition pore (MPTP) opening in vivo. Our studies, therefore, provide the first genetic evidence for a novel mechanism, termed the oxidative stress–HSF pathway, by which the chaperone-dependent requirements of the HSF1 regulator serve to complement antioxidative defense systems in mammals.
Results
HSF1 deficiency selectively reduces expression of Hsps in the unstressed adult heart
Although we have previously reported the essential requirement for HSF1 for heat stress-induced Hsp expression in multiple organs of Hsf1 knockout mice (Xiao et al., 1999), the effects of HSF1 deficiency in the heart under physiological conditions have not been thoroughly investigated. We thus surveyed the protein expression patterns of several Hsps from Hsf1 knockout mice (5–7 months) and age-matched controls of a similar genetic background (background C, 129X1) by immunoblot analysis. Our experiments were performed using cardiac homogenates with antibodies specifically directed against αB-crystallin (αBC), Hsp25, Hsp70 and Hsp90, as well as mitochondrial Hsp60 (Figure 1A). Densitometric data shown in Figure 1B revealed that in the absence of HSF1, the Hsp25 level was decreased by 72%, αBC by 42%, Hsp70 by 34% while neither cytosolic Hsp90 nor mitochondrial Hsp60 expression was affected compared with wild-type animals. These results suggest that HSF1, besides serving an essential requirement for the heat shock response, plays a key regulatory role in the constitutive expression of bona fide members of the Hsp family, at normal temperature (37°C).
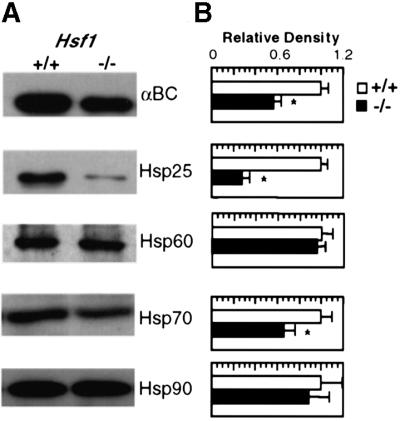
Fig. 1. HSF1 deficiency causes selective abnormalities in constitutive expression of Hsps. Immunoblots of the major classes of Hsps were determined in protein extracts isolated from the hearts of Hsf1+/+ and Hsf1–/– animals. Representative western blots for Hsp25, αBC, Hsp70/72, mitochondrial Hsp60 and chaperone Hsp90 are shown in (A). (B) shows the densitometric data from three animals determined independently in age-matched animals. * represents P < 0.05, which was considered significant using the alternate Welch’s t test.
Antioxidant mechanisms are impaired in the absence of HSF1 regulation
In cultured cells, forced overexpression of small Hsps, such as Hsp25 and αBC, has been reported to influence the cellular detoxifying/antioxidative machinery by raising the intracellular concentration of GSH and maintaining the G6PD activity, an essential requirement for reducing gluthathione disulfate (GSSG) back to GSH by the NADPH-dependent mechanism (Mehlen et al., 1996; Preville et al., 1999; Baek et al., 2000). To determine whether HSF1-dependent regulation of Hsp expression might also have similar direct biochemical effects, we first assessed the GSH:GSSG ratio in cardiac homogenates obtained from HSF1-deficient and wild-type animals. Our results show that the GSH:GSSG ratio is significantly decreased by 40% in the hearts from HSF1-deficient mice in comparison with wild-type animals (n = 3 per group; Figure 2A).
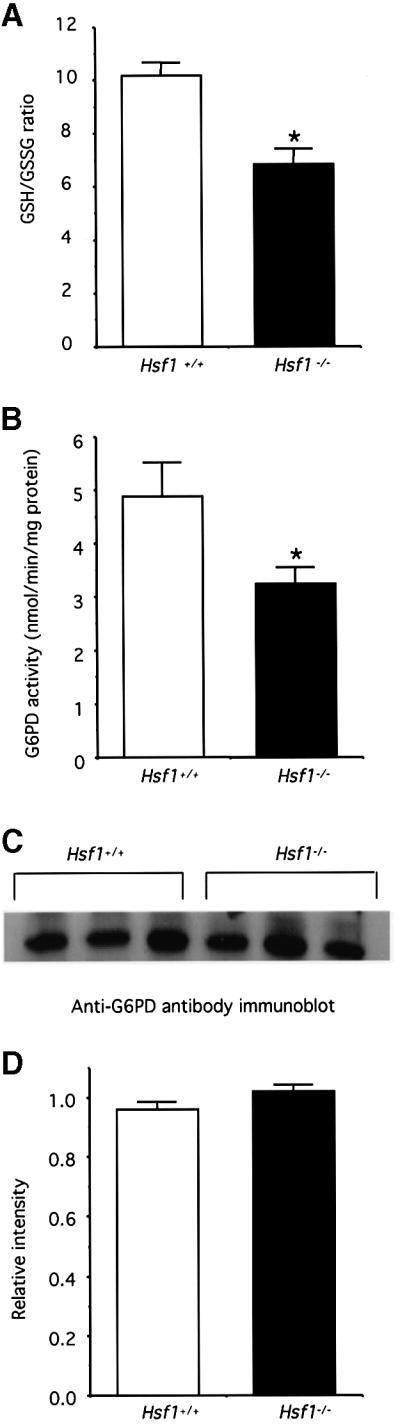
Fig. 2. (A) HSF1 disruption lowers the ratio of GSH:GSSG in heart homogenates. GSH and GSSG levels were determined by a GSH reductase-mediated recycling assay in the presence of NADPH and DTNB. (B) HSF1 deficiency decreases G6PD activity. G6PD’s ability to regenerate NADPH was used to measure its enzymatic activity, as described in detail in the text. (C) Protein expression profile of G6PD detected by anti-G6PD antibody; three independent animals were used for each group. (D) Densitometric quantitation of the immunoblot bands in (C) indicates that protein level of G6PD does not exhibit a difference between wild-type and HSF1-deficient hearts. All data are expressed as means ± SEM of three independent determinations made in separate animals (n = 3 per group). * represents P < 0.05, which was considered significant using the alternate Welch’s t test (n = 3).
To test the possibility that the G6PD function was involved in the reduction of GSH:GSSH ratio in Hsf1 knockout hearts, we next measured the G6PD activity in the supernatants obtained from cardiac mitochondria preparations of wild-type and Hsf1 knockout mice. Indeed, Figure 2B shows that G6PD activity is significantly lower by 34% in the Hsf1 knockout heart compared with that of the wild-type animals, which for the first time links the regulatory HSF1 pathway to the activity of a key metabolic/antioxidative enzyme, G6PD, in an intact organ.
The foregoing results that HSF1 deficiency was associated with the decreased activity of G6PD raised two important possibilities: either a decrease in the expression of G6PD protein lowers G6PD activity or the decreased G6PD activity is related to post-translational modifications (e.g. oxidative damage). To test the first possibility, we performed western blot analysis using anti-G6PD antibodies on heart homogenates prepared from wild-type and Hsf1 knockout mice (n = 3 per group). We observed that the level of G6PD protein expression was virtually indistinguishable between wild-type and Hsf1 null animals (Figure 2C and D), which strongly suggests that decreased activity of G6PD by HSF1 deficiency is related to post-translational mechanism(s).
Levels of oxidative stress/damage are increased in the absence of HSF1 regulation
The intracellular redox state is determined by the equilibrium between antioxidants and oxidants. Evidence for a decreased GSH:GSSG ratio in Hsf1 knockout cells implies that pro-oxidants like ROS might be generated at a higher level. Therefore, to directly evaluate this consequence of HSF1 deficiency, we prepared cardiac sub-mitochondrial particles (SMPs) and measured the rate of superoxide (O2–) generation. Our results indicate that in the Hsf1 knockout, the rate of O2– production was 43% higher (n = 3) than its wild-type counterpart (Figure 3A).

Fig. 3. Absence of HSF1 increases oxidative stress and protein oxidative damages. (A) HSF1 disruption increases the rate of O2– generation in mitochondria. SMPs were prepared from the heart of 5- to 7-month-old wild-type mice and Hsf1 knockout mice. O2– anion generation was determined as SOD-inhibitable reduction of acetylated cytochrome c. Data are expressed as means ± SEM of three independent determinations made in separate animals. *represents P < 0.05, which was considered significant using the alternate Welch’s t test (n = 3). (B) HSF1 deficiency increases protein carbonylation, a marker of protein oxidative damage, in mitochondrial extracts under unstressed conditions. Mitochondrial proteins were resolved by SDS–PAGE (10% resolving) under reducing conditions and transferred to immobilon-P membrane. Oxidized proteins were detected immunochemically using anti-DNP antibodies against the carbonyl moiety as described in the text. Several DNPH-treated proteins (labeled A–D), which are evident in extracts from both genotypes, but are more pronounced from mitochondria of Hsf1–/– mice (lane 4) compared with Hsf1+/+ animals under physiological conditions (lane 3). Lanes 1 and 2 shows Coomassie Blue staining of a parallel gel. The 32 kDa protein (labeled A) was selected for further studies described in this report.
One direct prediction of a lower redox potential and a higher rate O2– generation is the increased oxidative damage of mitochondrial proteins. Oxidative attacks by ROS on proteins have been shown to increase their carbonyl content, a post-translational modification caused by the formation of aldehydes and ketones on certain amino acid residues (Stadtman, 1992). Carbonyl detection is based on an immunochemical assay of the 2,4-dinitrophenylhydrazine (DNPH) reaction product using an anti-DNP antibody, which specifically binds the carbonyl moieties of mitochondrial proteins after DNPH treatment. To determine whether HSF1 deficiency affects the levels of carbonylation, we examined mitochondrial proteins isolated from the hearts of age-matched wild-type mice and Hsf1 knockout mice. By anti-DNPH immunodetection, we observed that several mitochondrial proteins are prominently labeled in cardiac extracts from both genotypes, albeit with a greater intensity in HSF1-deficient than in wild-type mice (Figure 3B). Among several mitochondrial proteins (arbitrarily labeled as A–D), a densitometric analysis revealed that a 32 kDa protein exhibited more carbonylation in the HSF1-deficient extracts than in the wild-type (arrow A, Figure 3B).
Identification of protein A as cardiac ANT1
To localize and determine the identity of the 32 kDa protein (designated protein A), we analyzed mitochondrial membrane SMPs and matrix subfractions for carbonylated proteins by immunochemical detection (Figure 4A). Our results indicate that both fractions contain several oxidatively modified proteins but that protein A is localized predominantly in the membrane fraction (Figure 4A, arrow). The general scheme outlined in Figure 4B was employed for the purification of the 32 kDa protein on hydroxyapatite columns using mitochondria from 3- to 5-month-old wild-type animals (n = 5). Qualitative analyses of SDS–PAGE by Coomassie Blue staining indicate that protein A elutes in the pass-through fraction, coincident with another protein of slightly larger molecular size, protein E (Figure 4C).

Fig. 4. Localization and identification of the mitochondria 32 kDa protein as cardiac ANT1 (labeled A). (A) Isolated mitochondria from wild-type animals were separated into mitochondrial matrix (lane 1) and membrane (lane 2) protein fractions. The distribution of oxidatively modified protein A was found to be in the membrane fraction. (B) A scheme for purification of protein A from heart mitochondria. (C) SDS–PAGE analysis of both purified proteins obtained in the pass-through fraction. Lane 1 shows the starting material, total mitochondrial proteins; lane 2 shows the fraction after elution through hydroxyapatite column. SDS–PAGE revealed that protein A co-purified with an additional higher molecular weight protein (E). (D) Microsequencing of one internal peptide identified the 32 kDa protein as cardiac ANT1, a mitochondrial inner membrane protein. In addition, the co-purified protein E was determined to be the VDAC protein, a mitochondrial outer membrane protein (data not shown).
After the retrieved proteins were transferred to immobilon-P membrane, we found by microsequencing that both proteins exhibited N-terminal blockage, which necessitated proteolytic digestion prior to resequencing. From computer database comparisons of the retrieved fragments, we matched the internal peptides derived from protein A to the mitochondrial ANT1 (Figure 4D). A similar approach revealed protein E to be voltage-dependent anion channel 1 (VDAC1), which, like ANT1, is also a component of the non-selective MPTP complex (Bauer et al., 1999).
HSF1 deficiency decreases functional activity of cardiac ANT1
To determine whether the effect of HSF1 deficiency on oxidative damage to ANT1 has any functional consequences, we next measured ADP/ATP exchange ability, an index for ANT1’s activity (Duan and Karmazyn, 1989). Using this assay, which relies on the uptake of tritiated ADP into intact isolated mitochondria, we observed that the ADP/ATP exchange activity was decreased by 18% in Hsf1 knockout mice in comparison with wild-type animals (Figure 5A). These results indicate that HSF1 deficiency per se has indirect functional effects on the degree of ANT1’s catalytic activity in vivo without pathological challenges.

Fig. 5. (A) HSF1 deficiency decreases the ADP/ATP exchange activity of ANT1. The inhibitor stop method was used to determine the exchange activity in Hsf+/+ and Hsf1–/– mitochondrial extracts. The values represent the mean ± SEM from three independent determinations. * represents P < 0.05, which was considered significant using the alternate Welch’s t test. (B) HSF1 deficiency increases MPTP opening. The swelling of isolated mitochondria, an index of MPTP opening, was monitored spectrophotometrically in the absence or presence of exogenous Ca2+ at 540 nm. Shown are representative tracings taken from one of four independent experiments performed on mitochondria isolated from Hsf+/+ and Hsf1–/– animals.
To determine whether increased carbonylation of ANT1 in the Hsf1 knockout mice has a direct effect on MPTP opening, we monitored the swelling of isolated mitochondria by measuring light scattering at 540 nm, in the absence or presence of exogenous calcium. Figure 5B shows that under basal conditions, the MPTP opens at a faster rate in the Hsf1 knockout than in the wild type, a finding that is independent of exogenous calcium (Figure 5B). These results demonstrate that the HSF1-dependent pathway is a causal mechanism between protein oxidative damage of cardiac ANT1 and mitochondrial functional abnormalities, and suggest a possible structure/functional mechanism to its major role in cell survival/death pathways. Since ANT1 is a major component of the MPTP, a key mediator of apoptotic and necrotic cell death (Bauer et al., 1999; Crompton, 1999), it is conceivable that alterations of MPTP might play a role in the increased vulnerability observed in Hsf1 knockout mice.
Discussion
Evidence has been accumulating in flies and mammals that HSF1, the major stress-inducible regulator of hsp genes, has essential functions even at normal temperatures (Jedlicka et al., 1997; Xiao et al., 1999; Christians et al., 2000). Taking into account the previous data showing that HSF is responsive to oxidative stress (Jacquier-Sarlin and Polla, 1996; Zhong et al., 1998), we reasoned that the physiological demands imposed by oxidative phosphorylation and generation of ROS will elicit HSF1-dependent requirements in specific tissues. The work presented in this paper tested the hypothesis that the transcription factor HSF1 plays a key regulatory role in maintaining cardiac redox state and decreasing protein oxidative damage, by sustaining Hsp expression. Specifically, we have demonstrated that, in the absence of HSF1, several Hsp expressions are downregulated, coincident with an increased level of oxidative stress and ROS-induced oxidative damage to mitochondrial proteins such as ANT1 under physiological conditions. To our knowledge, these results, for the first time, establish a novel requirement for HSF1 in the control of Hsps expression and define the functional consequences of deficiency in HSF1 pathway at the level of antioxidant mechanisms.
HSF1 deficiency downregulates constitutive Hsp expression
In the present study, the regulatory function of constitutive HSF1 activity is based on and deduced from the comparative levels of Hsp expression found in a highly oxidative organ (i.e. the heart) harvested from age-matched, wild-type and Hsf1 knockout animals housed under ambient conditions. Under such conditions, several representative Hsps including Hsp25, αBC and Hsp70 are significantly decreased by HSF1 deficiency, the most significant being Hsp25 (Figure 1). This is consistent with previous studies showing HSF1-dependent expression of Hsp25 (Hoang et al., 2000). In contrast, cytosolic Hsp90 and mitochondrial Hsp60 expression appears not to be similarly affected by HSF1 deficiency, suggesting that HSF1 activity, alone or in combination with other signals, exerts differential effects on the broad classes of Hsp expression. Our studies in Hsf1 knockout mice provide proof of concept for HSF1-dependent interactions with the intracellular redox state (Manalo et al., 2002), which has been recognized to exert profound effects on HSF1 DNA-binding activity (Bruce et al., 1993), hsp70 gene expression (Freeman et al., 1993), and post-translational modification of Hsp25/7 primarily using in vitro systems and cultured cells (Mehlen et al., 1997).
Small molecular weight Hsps are complementary to antioxidative systems in vivo
Evidence has been accumulating that murine Hsp25 might have direct chaperone-dependent functions on the activity of G6PD, a key antioxidative mechanism for replenishing the reducing potential. In HepG2 cells, for example, depletion of GSH has been shown to induce Hsp synthesis (Freeman et al., 1993), directly activate HSF (Hatayama and Hayakawa, 1999), increase protein oxidation of thiols, and destabilize protein structure leading to aggregation and protein denaturation (Freeman et al., 1997). Preville et al. (1999) have reported previously that Hsp25 confers cytoprotection of L929 fibroblasts against oxidative stress by stimulating the activity of G6PD under unstressed and oxidative stressed conditions. The increase in G6PD activity correlated with increased level of G6PD protein, suggesting that Hsp25 might either decrease G6PD turnover or increase its translation (Preville et al., 1999). Furthermore, a direct requirement for Hsp25 chaperone-dependent function appears unlikely for G6PD activity in L929 fibroblasts. However, our in vivo results do raise this intriguing possibility since HSF1 deficiency in the intact organism not only downregulates expression of murine Hsp25 but leaves expression of G6PD protein unaltered, coincident with a decreased G6PD activity (Figure 2). Future studies are needed to address the mechanism(s) for the discordance between G6PD content and G6PD activity.
Our findings are in agreement with studies of Pandolfi et al. (1995) that reported G6PD knockout cells exhibit increased susceptibility to oxidative stress. Large oligomers of Hsp25 have been proposed to harness chaperone activity, either to shield oxidized protein substrates from further oxidative damage or to target their proteolytic degradation via the 20S proteasome (Arrigo et al., 2002). Yet, the precise mechanism by which small molecular weight Hsps maintain reduced GSH remains to be fully elucidated. Notwithstanding, these results are significant since they provide genetic evidence for potential HSF1-dependent functions in the absence of pathological challenges: (i) the regulation of target hsp genes encoding chaperone-dependent proteins; and (ii) the maintenance of redox homeostasis.
Mitochondrial proteins are targets for increased oxidative damage in the absence of HSF1
Consistent with a lower GSH:GSSG ratio and an imbalance in redox state, O2– generation and oxidative damage of mitochondrial proteins such as ANT1 are increased in HSF1-deficient hearts (Figure 4). How might the increase of oxidative stress specifically affect cardiac ANT1? Because mitochondria are both important sources and targets of the detrimental effects of ROS (Zorov et al., 2000), the high sensitivity of ANT1 is likely to be related to the proximity, the location in inner membrane, and vulnerability of the relatively high number of lysine residues to oxidative attacks and metal-catalyzed oxidation (Yan and Sohal, 1998).
HSF1 deficiency decreases ANT1 activity and increases MPTP opening in mice
Not only does ANT1 function in the ADP/ATP exchange across the mitochondrial membrane (Kaukonen et al., 2000), but it also serves as a major component of the MPTP, a key regulatory channel for mitochondrial biogenesis and cell survival (Esposito et al., 1999). It is conceivable that increased protein oxidative modification induced by HSF1 deficiency alters the stability, conformation, activity and function of ANT1 at the level of MPTP opening (Figure 5). We provide two independent lines of evidence, a decreased ADP/ATP activity (Figure 5A) and an increased MPTP opening (Figure 5B), that suggest HSF1 deficiency is causally linked to ANT1-dependent mitochondrial dysfunction. Our results, however, do not preclude the possibility that other components of MPTP (e.g. VDAC) or other molecular targets are similarly modified by chaperone defenses in vivo.
MPTP is involved in non-selective transport of ions and small molecular weight proteins, and is a central mediator in cell death pathways (Bernardi et al., 1999). Further more, oxidative stress-induced perturbation of mitochondrial membrane potential has been recognized as a mechanism for further elevation in ROS generation, referred to as ‘ROS-induced ROS release’ (Zorov et al., 2000). It is tempting to speculate that ROS generation and MPTP opening might prevent Hsf1–/– cells from acquisition of thermotolerance (McMillan et al., 1998) and predispose knockout mice to increased mortality, coincident with exaggerated TNFα production (Xiao et al., 1999).
Nonetheless, the increased production of mitochondrial ROS and oxidative damage of ANT1 in HSF1-deficient hearts are reminiscent of similar effects reported for the heart/skeletal muscle isoform in Ant1–/– mice (Esposito et al., 1999). It is noteworthy that antioxidative enzymes such as O2– dismutase, Sod2, are not affected by ANT1 deficiency. Likewise, we observed that an increased rate of O2– production by HSF1 deficiency had no effects on either total O2– dismutase or GSH peroxidase activity (data not shown). Based on the established roles for ANT1 in mitochondrial DNA-induced diseases (Kaukonen et al., 2000), we are currently examining whether mitochondrial DNA is similarly vulnerable to oxidative damage in Hsf1–/– mice.
A proposed model for redox regulation of constitutive HSF1 functions in vivo
Figure 6 schematically illustrates a model that integrates the functions of HSF1 activity in redox homeostasis. Our study provides genetic evidence that links the hitherto unrecognized requirement between HSF1 activity and constitutive expression of selective Hsps under physiological conditions. In this scheme, regulation of the redox sensitive factor HSF1 is mediated by ROS generated under basal conditions. The downregulation of Hsp25 and αBC expression in the absence of HSF1 are the most likely mechanisms leading to the decrease in G6PD enzymatic activity and a lower GSH:GSSG ratio, although other mechanisms (e.g. mitochondrial thioredoxin 2 or Mn- SOD) might play a role, but have not been exhaustively tested (Tanaka et al., 2002). Correspondingly, a decrease in redox state and an increase in O2– generation causes an increased oxidative damage of mitochondrial proteins such as ANT1 in the intact organism. The alteration of ANT1 function might increase the permeability of MPTP, thus reinforcing ROS-induced production of excess ROS in vivo (Esposito et al., 1999; Zorov et al., 2000). Finally, our model does not preclude the possibility that other signals (e.g. misfolded proteins) may lead to stress-inducible HSF1 activation mimicking the classical heat shock response.

Fig. 6. A proposed scheme outlining a possible pathway, termed the oxidative stress–HSF pathway, by which HSF1 deficiency might potentially trigger ROS generation, at the level of protein oxidative damage of the mitochondrial ANT1 and MPTP opening, under physiological conditions. A key role for redox regulation as a proposed mechanism for activation of HSF1 is suggested by HSF1-dependent requirements for maintaining constitutive Hsp expression. Such chaperone-dependent functions appear to interact with thiol-containing molecules such as GSH and maintain the cellular redox homeostasis, a balance between pro-oxidants and antioxidants (expressed by the GSH:GSSG ratio) in order to counterbalance and detoxify the damaging effects of ROS (left panel). In the absence of HSF1, the feedback loop for regulation of selective Hsp expression is abolished that theoretically will evoke other compensatory pathways, which in the end are insufficient to mitigate protein oxidative damage, both at the source (e.g. mitochondria; this study) and other unspecified targets of ROS in the cell. ETC, electron transport chain.
Implications of these studies
Our studies challenge the current notion that metazoan HSF1 activity is transcriptionally inert under non-stressed conditions (Jedlicka et al., 1997; Xiao et al., 1999; Christians et al., 2000). However, we did not address the precise nature of the signals sensed by the transcriptional activity of HSF1 that might be responsible for constitutive Hsp expression in vivo. Beyond the importance to understand the events that govern the regulation of basal HSF1 activity, our work raises new questions to determine the signals that control HSF1-dependent functions in key biological processes (e.g. development), tissues (e.g. brain, liver and kidney) and specialized cell types (lymphocytes, endothelial cells and smooth muscle cells) in the organism. One central question, being addressed by ongoing and future studies, is whether the transcriptional requirement for HSF1 activity that maintains homeostatic balance tilts the balance and determines the susceptibility to diseases, especially ones whose onset is insidious and progressive. We suggest that the Hsf1 knockout remains a robust genetic tool for dissecting such molecular and genetic events that engage HSF1 regulation, such as protein oxidative damage and modification under basal conditions in vivo, in a complex organism.
Materials and methods
Chemicals
GSH (both reduced and oxidized), GSH reductase, SOD and antimycin A were from ICN (Costa Mesa, CA). Acrylamide/bis-acrylamide, ammonium persulfate, hydroxyapatite, rotenone and tetramethylethylenediamine were obtained from Bio-Rad (Richmond, CA). Bovine serum albumin (BSA), 2,8-[3H]adenine 5′-diphosphate ([3H]ADP) trisodium salt, glucose 6-phosphate and atractyloside, Coomassie Blue R-250, glycine, dithiothreitol, DNPH and rabbit anti-DNP antibodies were purchased from Sigma Chemical Co (St Louis, MO). Antibodies against Hsp70, Hsp90 and Hsp25 were obtained from Stressgen (Victoria BC, Canada). Anti-αBC antibody against the distal 14 AA residues to the stop codon of C-terminal was produced in our laboratory by immunizing rabbits. Anti-G6PD antibody was a kind gift from Dr Andre-Patrick Arrigo (Universite Claude Bernard Lyon-I, Villeurbanne, France). Protein standard markers were obtained from Novex (San Diego, CA). Bicinchoninic acid protein assay kit was obtained from Pierce Inc. (Rockford, IL).
Generation and breeding of Hsf1 knockout mice
The production and characterization of Hsf1 knockout mouse has been reported previously (McMillan et al., 1998) and will be described here briefly. The Hsf1–/– deletion encompasses ∼80% of the DNA-binding domain and the three leucine zippers that comprise the oligomerization domain. Hsf1 knockout mice used in this study were generated in the mixed genetic background C, 129XI (129XI/SvJ × BALB/c; McMillan et al., 1998; Xiao et al., 1999). Mice homozygous for the Hsf1 knockout allele were maintained in our breeding colony along with control littermates that were age-matched.
Preparation of mouse heart mitochondria and submitochondrial particles
Mouse heart mitochondria were isolated by differential centrifugation of heart homogenates as described previously (Arcos et al., 1968). Briefly, the heart was rinsed in cold mitochondrial isolation buffer (0.3 M sucrose, 0.03 M nicotinamide and 0.02 M EDTA pH 7.4) to remove blood and minced with scissors. A 5% (w/v) homogenate was prepared and was centrifuged at 700 g for 10 min at 4°C, and the resulting supernatant was centrifuged for 5 min under the same conditions. The supernatant was further centrifuged at 10 000 g for 5 min at 4°C. Mitochondria were stored at –80°C until analysis. To prepare SMPs, mitochondria were resuspended in 30 mM sodium phosphate (pH 7.4) and sonicated four times for 30 s with 1 min intervals in a Branson 2200 instrument (Danbury, Connecticut). The sonicated sample was centrifuged at 8250 g for 10 min to remove any unbroken mitochondria, and the supernatant was further centrifuged at 80 000 g for 40 min. The supernatant contained mitochondrial matrix, whereas the pellet contained mitochondrial SMPs (Yan et al., 1997).
Determination of GSH and GSSG
GSH and GSSG were measured according to the method described by Anderson (1985). Briefly, frozen tissues were homogenized in 5% sulfosalicylic acid and the homogenates were centrifuged at 10 000 g for 10 min. The supernatant was collected and distributed into two aliquots (100 µl) for individual measurement of GSSG and total GSH. To conjugate GSH, the first aliquot was treated with 2-vinylpyridine followed by neutralization to pH 6–7 with triethanolamine. GSSG concentrations were then determined using the enzymatic recycling assay involving the color development at 412 nm of 0.6 mM 5,5-dithiobis 2-nitro-benzoic acid in the presence of 0.2 mM NADPH and 1.8 U/ml of GSH reductase. Total GSH and GSSG concentrations were determined in the second aliquot using the same assay without adding 2-vinylpyridine. GSSG standards were used for calibration.
Measurement of the enzyme activity of G6PD
G6PD activity, determined in the supernatant fraction after mitochondrial isolation, was carried out by following the generation of NADPH at 340 nm based on the method described previously (Tian et al., 1998). Since 6-phosphategluconate dehydrogenase (6PGD) is also involved in the generation of NADPH, both 6PGD and total dehydrogenase activity (G6PD + 6PGD) were independently determined. G6PD activity was then obtained by subtracting 6PGD activity from the total dehydrogenase activity. The reaction mixture (1 ml) contained (all final concentrations) 86.3 µM triethanolamine buffer (pH 7.6), 6.7 µM MgCl2, 100 µM NADP, 200 µM glucose 6-phosphate and/or 200 µM 6-phosphategluconate. The enzyme reaction was triggered by the addition of 10 µg protein extract and monitored for up to 20 min at 340 nm.
Measurement of O2– generation
Mitochondrial SMPs, prepared as described above, were resuspended in 100 mM phosphate buffer pH 7.4. The rate of O2– generation by SMPs was measured as SOD-inhibitable reduction of acetylated ferricytochrome c (Azzi et al., 1975). The reaction mixture contained 10 µM acetylated ferricytochrome c, 6 µM rotenone, 1.2 µM antimycin A, 100 U of SOD/ml (in the reference cuvette) and 30–100 µg SMP protein in 100 mM potassium phosphate buffer pH 7.4. The reaction was initiated by addition of 7.5 mM succinate and the reduction of acetylated cytochrome c was followed at 550 nm (ε = 27 700 M–1 cm–1).
Immunochemical detection of protein carbonyls
Frozen mitochondria were solubilized in 20 mM Tris–HCl buffer (pH 6.8) containing 0.2% SDS and treated with 10 mM DNPH as described previously (Yan and Sohal, 2000). The DNPH-treated mitochondrial proteins were used for immunochemical detection, which was performed according to the methods of Keller et al. (1993) and Shacter et al. (1994). SDS–PAGE analysis of proteins was performed according to Laemmli (1970). Proteins were transferred onto immobilon-P membranes at 100 V (constant voltage, 1 h) using mini trans-blot electrophoretic transfer cell (Bio-Rad). The blots were incubated with 50 ml 5% non-fat dried milk (w/v) for at least 1 h, and then washed for 3 × 10 min with Tris-buffered saline (20 mM Tris, 500 mM NaCl pH 7.5), containing 0.1% Tween-20 (TBST). Blots were incubated for 1 h at room temperature with rabbit anti-DNP antibodies (diluted 1:1000 in TBST containing 0.2% BSA). The primary antibody was removed, and the blots were washed three times (10 min each) with TBST. The blots were then incubated in horseradish peroxidase-labeled goat anti-rabbit IgG (diluted 1:10 000 in TBST containing 0.2% BSA) for 1 h at room temperature. After the blots were washed with TBST three times (10 min each), the oxidized proteins were visualized with an enhanced chemiluminescence detection kit. Proteins on the membrane were stained with Amido Black (Sigma).
Purification and identification of the protein A
Pooled mitochondria from six hearts (wild-type animals) were resuspended in 3 ml of 150 mM NaCl, 20 mM HEPES pH 7.4 containing 2% Triton X-100. The suspension was added with 0.5 tablets of protease inhibitors and the mixture was gently stirred for 60 min at 0°C. The solubilized mitochondrial sample was centrifuged at 100 000 g for 40 min and the pellet was discarded. The supernatant was then loaded onto a hydroxyapatite column which was equilibrated with 75 ml of 50 mM NaCl, 20 mM HEPES pH 7.4 containing 0.2% Triton X-100. The target proteins, existing mainly in the pass-through fraction as determined by SDS–PAGE, were collected and concentrated using ultrafilters. To remove Triton X-100 from the concentrated protein, the sample was further treated by 10% TCA (final concentration) and the protein pellet was washed with alcohol:ethyl acetate (1:1, v/v) twice, followed by SDS–PAGE analysis. Microsequencing of the protein, blotted onto immobilon-P membranes, was determined by automated Edman degradation. Since the protein was found to be N-terminally blocked, it was further digested by proteases according to standard procedures to obtain fragments, which were subsequently identified as mitochondrial ANT1.
Assay of ANT1 exchange activity
The activity of ANT1 was measured by a slight modification of the inhibitor-stop method (Duan and Karmazyn, 1989). Briefly, mitochondria were resuspended in reaction buffer (100 mM KCl, 20 mM Tris–HCl, 1 mM EDTA pH 7.4) and 50 nmol of [3H]ADP was added to a reaction mixture consisting of the buffer and 0.5 mg/ml mitochondria proteins. The reaction was carried out at room temperature and stopped after 1 min by the addition of 100 µM atractyloside. The reaction mixture was then passed through a PD-10 column to remove free [3H]ADP and the protein concentration in the eluate was determined. For the measurement of ANT activity, 0.2 mg mitochondrial protein was mixed with 4 ml Scintisafe Plus (50%) and radioactivity was determined in a liquid scintillation counter.
Assay of mitochondrial permeability transition
Mitochondria used for the permeability transition assay was prepared by a differential centrifugation protocol (Petronilli et al., 2001). Frozen mouse hearts were homogenized by teflon homogenizer in a medium containing 180 mM KCl, 10 mM EDTA, 0.5% BSA and 10 mM HEPES pH 7.4. To remove EDTA and BSA, mitochondrial pellets were washed twice with 180 mM KCl and 10 mM HEPES pH 7.4. Mitochondrial permeability transition (swelling) was monitored as the changes at 540 nm with 250 µg of mitochondrial protein in a medium containing 250 mM sucrose, 10 mM Tris–Mops, 0.05 mM EGTA pH 7.4, 5 mM pyruvate, 5 mM malate and 1 mM phosphate. The mitochondria permeability transition was measured either in the presence or absence of 250 µM Ca2+.
Other methods
A ChemiImager 4000 (Alpha Innotech, San Leandro, CA) was used for all densitometric quantitations. Statistical analysis was performed with Welch’s t test using Instat software (Graphad Software, San Diego, CA). All protein concentrations were determined by bicinchoninic acid assay using BSA as the standard (Smith et al., 1985).
Acknowledgments
Acknowledgements
We are grateful for the many helpful comments and suggestions from colleagues during the presentation of this work, in part, at the 2002 Cold Spring Harbor meetings on Molecular Chaperones and the Heat Shock Response. This research was supported by an Established Investigator Award from the AHA and from National Institutes of Health grant RO1HL60667 (I.J.B.) of the NHLBI and grant RO1AG13563 (R.S.S.).
References
- Amin J., Ananthan,J. and Voellmy,R. (1988) Key features of heat shock regulatory elements. Mol. Cell. Biol., 8, 3761–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M.E. (1985) Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol., 113, 548–555. [DOI] [PubMed] [Google Scholar]
- Arcos J.C., Sohal,R.S., Sun,S.C., Argus,M.F. and Burch,G.E. (1968) Changes in ultrastructure and respiratory control in mitochondria of rat heart hypertrophied by exercise. Exp. Mol. Pathol., 8, 49–65. [DOI] [PubMed] [Google Scholar]
- Arrigo A.P., Paul,C., Ducasse,C., Sauvageot,O. and Kretz-Remy,C. (2002) Small stress proteins: modulation of intracellular redox state and protection against oxidative stress. Prog. Mol. Subcell. Biol., 28, 171–184. [DOI] [PubMed] [Google Scholar]
- Azzi A., Montecucco,C. and Richter,C. (1975) The use of acetylated ferricytochrome c for the detection of superoxide radicals produced in biological membranes. Biochem. Biophys. Res. Commun., 65, 597–603. [DOI] [PubMed] [Google Scholar]
- Baek S.H., Min,J.N., Park,E.M., Han,M.Y., Lee,Y.S., Lee,Y.J. and Park,Y.M. (2000) Role of small heat shock protein HSP25 in radioresistance and glutathione-redox cycle. J. Cell Physiol., 183, 100–107. [DOI] [PubMed] [Google Scholar]
- Bauer M.K., Schubert,A., Rocks,O. and Grimm,S. (1999) Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J. Cell Biol., 147, 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin I.J. and McMillan,D.R. (1998) Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ. Res., 83, 117–132. [DOI] [PubMed] [Google Scholar]
- Bernardi P., Scorrano,L., Colonna,R., Petronilli,V. and Di Lisa,F. (1999) Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur. J. Biochem., 264, 687–701. [DOI] [PubMed] [Google Scholar]
- Bruce J.L., Price,B.D., Coleman,C.N. and Calderwood,S.K. (1993) Oxidative injury rapidly activates the heat shock transcription factor but fails to increase levels of heat shock proteins. Cancer Res., 53, 12–15. [PubMed] [Google Scholar]
- Cadenas E., Boveris,A., Ragan,C.I. and Stoppani,A.O. (1977) Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys., 180, 248–257. [DOI] [PubMed] [Google Scholar]
- Christians E., Davis,A.A., Thomas,S.T. and Benjamin,I.J. (2000) Maternal effect of Hsf1 on reproductive success. Nature, 407, 693–694. [DOI] [PubMed] [Google Scholar]
- Christians E.S., Yan,L.J. and Benjamin,I.J. (2002) Heat shock factor 1 and heat shock proteins: critical partners in protection against acute cell injury. Crit. Care Med., 30, S43–S50. [PubMed] [Google Scholar]
- Crompton M. (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem. J., 341, 233–249. [PMC free article] [PubMed] [Google Scholar]
- Duan J. and Karmazyn,M. (1989) Relationship between oxidative phosphorylation and adenine nucleotide translocase activity of two populations of cardiac mitochondria and mechanical recovery of ischemic hearts following reperfusion. Can. J. Physiol. Pharmacol., 67, 704–709. [DOI] [PubMed] [Google Scholar]
- Esposito L.A., Melov,S., Panov,A., Cottrell,B.A. and Wallace,D.C. (1999) Mitochondrial disease in mouse results in increased oxidative stress. Proc. Natl Acad. Sci. USA, 96, 4820–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M.L., Sierra-Rivera,E., Voorhees,G.J., Eisert,D.R. and Meredith,M.J. (1993) Synthesis of hsp-70 is enhanced in glutathione-depleted Hep G2 cells. Radiat. Res., 135, 387–393. [DOI] [PubMed] [Google Scholar]
- Freeman M.L., Huntley,S.A., Meredith,M.J., Senisterra,G.A. and Lepock,J. (1997) Destabilization and denaturation of cellular protein by glutathione depletion. Cell Stress Chaperones, 2, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatayama T. and Hayakawa,M. (1999) Differential temperature dependency of chemical stressors in HSF1-mediated stress response in mammalian cells. Biochem. Biophys. Res. Commun., 265, 763–769. [DOI] [PubMed] [Google Scholar]
- Hoang A.T., Huang,J., Rudra-Ganguly,N., Zheng,J., Powell,W.C., Rabindran,S.K., Wu,C. and Roy-Burman,P. (2000) A novel association between the human heat shock transcription factor 1 (HSF1) and prostate adenocarcinoma. Am. J. Pathol., 156, 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier-Sarlin M.R. and Polla,B.S. (1996) Dual regulation of heat-shock transcription factor (HSF) activation and DNA-binding activity by H2O2: role of thioredoxin. Biochem. J., 318, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedlicka P., Mortin,M.A. and Wu,C. (1997) Multiple functions of Drosophila heat shock transcription factor in vivo. EMBO J., 16, 2452–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukonen J., Juselius,J.K., Tiranti,V., Kyttala,A., Zeviani,M., Comi,G.P., Keranen,S., Peltonen,L. and Suomalainen,A. (2000) Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science, 289, 782–785. [DOI] [PubMed] [Google Scholar]
- Keller R.J., Halmes,N.C., Hinson,J.A. and Pumford,N.R. (1993) Immunochemical detection of oxidized proteins. Chem. Res. Toxicol., 6, 430–433. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Manalo D.J., Lin,Z. and Liu,A.Y. (2002) Redox-dependent regulation of the conformation and function of human heat shock factor 1. Biochemistry, 41, 2580–2588. [DOI] [PubMed] [Google Scholar]
- Marchler G. and Wu,C. (2001) Modulation of Drosophila heat shock transcription factor activity by the molecular chaperone DROJ1. EMBO J., 20, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan D.R., Xiao,X., Shao,L., Graves,K. and Benjamin,I.J. (1998) Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem., 273, 7523–7528. [DOI] [PubMed] [Google Scholar]
- Mehlen P., Kretz-Remy,C., Preville,X. and Arrigo,A.P. (1996) Human hsp27, Drosophila hsp27 and human αB-crystallin expression-mediated increase in glutathione is essential for the protective activity of these proteins against TNFα-induced cell death. EMBO J., 15, 2695–2706. [PMC free article] [PubMed] [Google Scholar]
- Mehlen P., Hickey,E., Weber,L.A. and Arrigo,A.P. (1997) Large unphosphorylated aggregates as the active form of hsp27 which controls intracellular reactive oxygen species and glutathione levels and generates a protection against TNFα in NIH-3T3-ras cells. Biochem. Biophys. Res. Commun., 241, 187–192. [DOI] [PubMed] [Google Scholar]
- Morimoto R.I. (1998) Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones and negative regulators. Genes Dev., 12, 3788–3796. [DOI] [PubMed] [Google Scholar]
- Nakai A. and Morimoto,R.I. (1993) Characterization of a novel chicken heat shock transcription factor, heat shock factor 3, suggests a new regulatory pathway. Mol. Cell. Biol., 13, 1983–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfi P.P., Sonati,F., Rivi,R., Mason,P., Grosveld,F. and Luzzatto,L. (1995) Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J., 14, 5209–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V., Penzo,D., Scorrano,L., Bernardi,P. and Di Lisa,F. (2001) The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem., 276, 12030–12034. [DOI] [PubMed] [Google Scholar]
- Pirkkala L., Alastalo,T.P., Zuo,X., Benjamin,I.J. and Sistonen,L. (2000) Disruption of heat shock factor 1 reveals an essential role in the ubiquitin proteolytic pathway. Mol. Cell. Biol., 20, 2670–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preville X., Salvemini,F., Giraud,S., Chaufour,S., Paul,C., Stepien,G., Ursini,M.V. and Arrigo,A.P. (1999) Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp. Cell Res., 247, 61–78. [DOI] [PubMed] [Google Scholar]
- Sarge K.D., Zimarino,V., Holm,K., Wu,C. and Morimoto,R.I. (1991) Cloning and characterization of two mouse heat shock factors with distinct inducible and constitutive DNA-binding ability. Genes Dev., 5, 1902–1911. [DOI] [PubMed] [Google Scholar]
- Shacter E., Williams,J.A., Lim,M. and Levine,R.L. (1994) Differential susceptibility of plasma proteins to oxidative modification: examination by western blot immunoassay. Free Radic. Biol. Med., 17, 429–437. [DOI] [PubMed] [Google Scholar]
- Smith P.K. et al. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem., 150, 76–85. [DOI] [PubMed] [Google Scholar]
- Stadtman E.R. (1992) Protein oxidation and aging. Science, 257, 1220–1224. [DOI] [PubMed] [Google Scholar]
- Tanaka T. et al. (2002) Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J., 21, 1695–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W.N., Braunstein,L.D., Pang,J., Stuhlmeier,K.M., Xi,Q.C., Tian,X. and Stanton,R.C. (1998) Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J. Biol. Chem., 273, 10609–10617. [DOI] [PubMed] [Google Scholar]
- Turrens J.F., Alexandre,A. and Lehninger,A.L. (1985) Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys., 237, 408–414. [DOI] [PubMed] [Google Scholar]
- Wu C. (1995) Heat shock transcription factors: structure and regulation. Annu. Rev. Cell. Dev. Biol., 11, 441–469. [DOI] [PubMed] [Google Scholar]
- Xiao X., Zuo,X., Davis,A.A., McMillan,D.R., Curry,B.B., Richardson,J.A. and Benjamin,I.J. (1999) HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J., 18, 5943–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L.J. and Sohal,R.S. (1998) Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc. Natl Acad. Sci. USA, 95, 12896–12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L.J. and Sohal,R.S. (2000) Analysis of oxidative modification of proteins. In Coligan,J.E., Dunn,B.M., Ploegh,H.L., Speicher,D.W. and Wingfield,P.T. (eds), Current Protocols in Protein Sciences. John Wiley & Sons Inc., New York, NY, pp. 14.4.1–14.4.24.
- Yan L.J., Levine,R.L. and Sohal,R.S. (1997) Oxidative damage during aging targets mitochondrial aconitase. Proc. Natl Acad. Sci. USA, 94, 11168–11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M., Orosz,A. and Wu,C. (1998) Direct sensing of heat and oxidation by Drosophila heat shock transcription factor. Mol. Cell, 2, 101–108. [DOI] [PubMed] [Google Scholar]
- Zorov D.B., Filburn,C.R., Klotz,L.O., Zweier,J.L. and Sollott,S.J. (2000) Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med., 192, 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]