

Abstract
We discovered that the hepatitis C virus (HCV) envelope glycoprotein E2 binds to human hepatoma cell lines independently of the previously proposed HCV receptor CD81. Comparative binding studies using recombinant E2 from the most prevalent 1a and 1b genotypes revealed that E2 recognition by hepatoma cells is independent from the viral isolate, while E2–CD81 interaction is isolate specific. Binding of soluble E2 to human hepatoma cells was impaired by deletion of the hypervariable region 1 (HVR1), but the wild-type phenotype was recovered by introducing a compensatory mutation reported previously to rescue infectivity of an HVR1-deleted HCV infectious clone. We have identified the receptor responsible for E2 binding to human hepatic cells as the human scavenger receptor class B type I (SR-BI). E2–SR-BI interaction is very selective since neither mouse SR-BI nor the closely related human scavenger receptor CD36, were able to bind E2. Finally, E2 recognition by SR-BI was competed out in an isolate-specific manner both on the hepatoma cell line and on the human SR-BI-transfected cell line by an anti-HVR1 monoclonal antibody.
Keywords: hepatitis C virus/hypervariable region 1/scavenger receptor class B type I/second envelope glycoprotein
Introduction
It is estimated that ∼3% of the world’s population is infected by hepatitis C virus (HCV) (Wasley and Alter, 2000). Exposure to HCV results in an overt acute disease in a small percentage of cases, while in most instances the virus establishes a chronic infection, causing liver inflammation that progresses slowly to liver failure and cirrhosis (Strader and Seeff, 2001). Epidemiological surveys indicate that HCV plays an important role in the pathogenesis of hepatocellular carcinoma (Strader and Seeff, 2001). Currently available therapies are limited to administration of interferon-α (IFNα) on its own or in combination with ribavirin (Fried and Hoofnagle, 1995; McHutchison et al., 1998). Such treatments are expensive, show low response rates and carry the risk of significant side effects. Consequently, the development of an HCV vaccine remains a high priority goal. Most efforts have been directed toward the two envelope glycoproteins E1 and E2, since chimpanzees immunized with purified recombinant E1/E2 heterodimeric proteins were shown to be protected from challenge with a low dose of homologous virus (Choo et al., 1994). However, a major concern still remains as to whether the response elicited by recombinant proteins from one particular viral isolate would be effective against heterologous viral inocula. In fact, HCV displays a high rate of mutation, and has been classified into distinct genotypes (1–6) and subtypes (a–c) whose distribution varies both geographically and between risk groups (Robertson et al., 1998; Wasley and Alter, 2000). Moreover, as a consequence of the high mutation rate during replication, HCV exists in the bloodstream of infected patients as a quasi-species. The most variable region of the genome is the hypervariable region 1 (HVR1), a 27 amino acid segment located at the N-terminus of the E2 glycoprotein (Hijikata et al., 1991; Weiner et al., 1991). This region is a target of anti-HCV neutralizing antibodies and undergoes gradual diversification of its sequence during the course of infection, probably resulting from intense immunological pressure (Farci et al., 1994, 1996; Korenaga et al., 2001; Mondelli et al., 2001). Recently, it was shown that a number of highly conserved segments exist within HVR1, suggesting that the structural and immunological variability is more limited than the heterogeneity found in the primary sequence (Puntoriero et al., 1998; Sobolev et al., 2000; Penin et al., 2001).
A major stumbling block in understanding the HCV infection mechanism and in testing the neutralizing potential of anti-HCV humoral responses elicited by candidate vaccines is the lack of an efficient cell culture system to study viral attachment and entry. An alternative approach is the identification and characterization of interactions between viral and host cell components, and considerable efforts have been directed at the identification of HCV receptor(s).
The low density lipoprotein (LDL) receptor has been shown to mediate HCV internalization via binding to virus associated LDL particles, a phenomenon common to many viruses that, like HCV, belong to the Flaviviridae family (Agnello et al., 1999). At present, it is not clear whether this mechanism of viral entry would permit productive viral infection, and alternative routes probably exist as suggested by studies with LDL receptor-deficient cells (Agnello et al., 1999).
A second putative HCV receptor, the tetraspanin CD81, has been identified by using soluble recombinant E2 from HCV genotype 1a for binding studies to human cells (Pileri et al., 1998). Since CD81 displays high affinity for E2 and because the presence of high titers of anti-E2 antibodies capable of blocking E2–CD81 interaction correlates with protection of vaccinated chimpanzees, it has been suggested that CD81 plays a role in viral entry (Rosa et al., 1996; Petracca et al., 2000). However, evidence points toward this molecule as being perhaps required, but not sufficient for HCV infection. First of all, CD81 is expressed in most cell types, while HCV is a hepatotropic virus; secondly, CD81 has a poor capacity to mediate internalization (Petracca et al., 2000). Further more, E2 binding to CD81 does not correlate with species permissiveness to HCV (Allander et al., 2000; Meola et al., 2000). Finally, CD81 binds efficiently to E2 proteins from the 1a genotype, but only weakly recognizes E2 from highly prevalent 1b variants (Yagnik et al., 2000; Triyatni et al., 2002; R.M.Roccasecca, H.Ansuini, A.Vitelli, A.Meola, E.Scarselli, S.Acali, M.Pezzanera, B.B.Ercole, J.McKeating, A.Yagnik, A.Lahm, A.Tramontano, R.Cortese and A.Nicosia, submitted).
In searching for alternative cell surface molecules capable of interacting with HCV E2, we discovered and describe below that human hepatoma HepG2 cells do not express CD81 on their surface, and yet they efficiently recognize E2 proteins from different isolates. By reversible cross-linking with E2, we identified an 82 kDa glycosylated molecular species as the most likely candidate. Characterization of E2 binding after chemical or enzymic modification of the cell surface led to the identification of the scavenger receptor type B class I (SR-BI) as the E2 receptor on HepG2 cells.
Results
E2 proteins from different viral isolates bind to hepatoma cells in a CD81-independent manner
The E2 glycoprotein derived from 1a isolates was shown to interact with a variety of cells in a CD81-dependent manner (Pileri et al., 1998; Flint et al., 1999). Recombinant E2 glycoproteins from different strains of the 1b genotype (BK-E2 and N2-E2) bound to CD81 displayed on the surface of Molt-4 cells significantly less efficiently than a genotype 1a variant E2 (Figure 1A, H77-E2). In contrast, when tested for their binding to human hepatoma cells Huh7 and HepG2, the variants showed binding levels comparable to the H77-E2 prototype (Figure 1A). Both Molt-4 and Huh7 cells express CD81 on their surface, though the latter at much lower levels (Figure 1B). In contrast, HepG2 cells did not show detectable CD81 on the cell surface (Figure 1B). Therefore, E2 binding to hepatoma cells must result from engagement of a receptor different from CD81.

Fig. 1. (A) Histograms showing the E2 binding of different viral isolates to human cell lines as measured by FACS analysis. Values are expressed as a percentage of the H77-E2 isolate binding. Molt-4 cells are represented by the black histogram. The human hepatic cell line Huh7 is represented by the dark gray histogram and the HepG2 cells by the light gray histogram. (B) Cell surface expression of CD81 measured on the different cell lines by FACS analysis with the anti-CD81 (mAb1.3.3.22) antibody in a direct binding assay. Molt-4 cells are represented by the triangle, Huh7 by the circle and HepG2 by the square. On the y axis net median fluorescence intensity (MFI) values are reported.
To better characterize this interaction, HepG2 cells were incubated with increasing amounts of H77-E2 protein. E2 binding to its novel putative receptor could be saturated (half-saturation was observed with 0.5 µg E2), and from a binding saturation curve we estimated an apparent Kd of E2 for HepG2 cells of ∼10–8 M (Figure 2).

Fig. 2. Binding saturation curve of H77-E2 recombinant protein to HepG2 cells. On the y axis, net median fluorescence intensity (MFI) values were calculated by subtracting the background MFI from non-specific binding of primary and secondary antibodies to the values obtained with E2. On the x axis, the monomeric E2 content was calculated as described in Material and methods.
Deletion of the HRV1 impairs HepG2 recognition, but wild-type binding can be rescued by adaptive mutations
We recently found that deleting the HVR1 (ΔHVR1) improves binding to CD81 displayed on Molt-4 cells (Figure 3A and R.M.Roccasecca, H.Ansuini, A.Vitelli, A.Meola, E.Scarselli, S.Acali, M.Pezzanera, B.B.Ercole, J.McKeating, A.Yagnik, A.Lahm, A.Tramontano, R.Cortese and A.Nicosia, submitted). Testing the HepG2 binding efficiency of HVR1 deletion mutants produced in the context of two different E2 variants (H77 and BK) revealed an opposite phenotype. In fact, the mutants showed a reduced capability to interact with the target cells (Figure 3B).

Fig. 3. (A) A histogram showing the binding of the E2 recombinant proteins deleted of the HVR1 to Molt-4. (B) A histogram showing the binding of the E2 recombinant proteins deleted of the HVR1 to HepG2. (C) A histogram showing the binding of the E2 recombinant proteins deleted of the HVR1 with compensatory mutations to HepG2. E2 binding is measured by FACS analysis and values are expressed as a percentage of the H77-E2 isolate binding.
Deletion of the HVR1 was shown previously to affect infectivity of HCV infectious RNA in chimpanzees, and in vivo selection of adaptive mutations in the E2 ectodomain correlated with a rise in viral titers (Forns et al., 2000). We therefore introduced the same mutation (V514M or L615H) in the context of the ΔHVR1/H77-E2 construct and tested the resulting mutants for their ability to interact with HepG2 cells. As shown in Figure 3C, both ΔHVR1/H77-E2/V514M and ΔHVR1/H77-E2/L615H derivatives displayed a ‘gain of function’ phenotype, with the former mutation displaying a higher capacity to rescue the wild-type binding.
Identification and purification of a HepG2 82 kDa protein interacting with soluble E2
As a first step toward the identification of the receptor responsible for E2 binding to HepG2 cells, we enriched cells for the highest E2 binding capacity by subsequent rounds of binding and sorting with FACS. We obtained a cell population showing a 3-fold improved binding to both H77-E2 and BK-E2 proteins (data not shown).
Cell surface glycoproteins were labeled with biotin using biotin–LC-hydrazide reagent (Kahne and Ansorge, 1994). Biotinylated cells were incubated with the supernatant containing the H77-E2 protein. Binding of E2 was unaffected by the biotinylation procedure (data not shown). Bound E2 was cross-linked to the cell surface proteins by means of a thiol cleavable cross-linker. Finally, cells were lysed and the E2–receptor complexes were immunoprecipitated under non-reducing conditions with magnetic beads conjugated with an antibody specific for the His tag on E2. The immunoprecipitated samples were eluted directly in sample buffer under both reducing and non-reducing conditions, loaded on SDS–PAGE gels and analyzed by western blotting. Under reducing conditions, cell-bound E2 was detected as a diffused band at the expected molecular weight of 61 kDa, whereas under non-reducing conditions most E2 protein was detected as a higher molecular weight species (Figure 4A). By omitting the cross-linking step, HepG2-bound E2 was recovered as a monomeric species with an apparent molecular weight of 61 kDa, indicating that the higher molecular weight species was indeed a receptor–ligand complex (data not shown). In a control experiment cross-linking was performed in the absence of the E2 ligand and no E2 staining was observed in the control lanes (Figure 4A). For detection of cell surface biotinylated species immunoprecipitated with E2, western blots were developed with labeled streptavidin. By thiol cleavage of the E2–receptor complexes, we detected a predominant biotinylated band with an apparent molecular weight of 82 kDa (Figure 4B). At present we have no experimental evidence to explain the lack of streptavidin staining of the complex between E2 and the biotinylated cell surface receptor. One possibility would be that covalently bound E2 prevents labeled streptavidin from getting access to the modified glycans of the cell receptor. Streptavidin staining was not observed in the control reaction where E2 was omitted (Figure 4B).
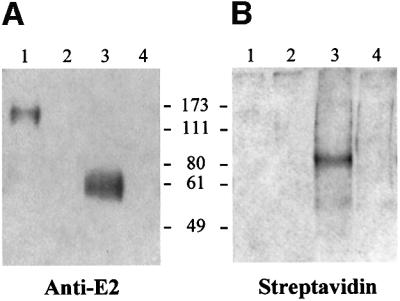
Fig. 4. (A) Western blot detection of cell surface-bound E2. Biotinylated HepG2 cells were incubated in the presence (lanes 1 and 3) or in the absence (lanes 2 and 4) of E2 recombinant protein. Bound E2 was cross-linked with DTSSP and the E2–receptor complexes were immunoprecipitated with an antibody against the His tag of the E2 recombinant protein. Samples eluted under both non-reducing (lanes 1 and 2) and reducing (lanes 3 and 4) conditions were loaded onto a 10% SDS–PAGE gel. E2 protein was detected as a monomer under reducing conditions (lane 3) and at a higher molecular weight under non- reducing conditions (lane 1), by using an anti-E2 rat mAb followed by anti-rat HRP-conjugated. (B) Immunoblot detection of biotinylated cell surface proteins interacting with E2. The reactivity with HRP conjugated streptavidin revealed a biotinylated protein band at 82 kDa only under reducing conditions (lane 3).
We concluded that the HepG2 surface molecule involved in E2 binding was an 82 kDa glycoprotein, since only glycoproteins could have been biotinylated with the biotin–hydrazide reagent. To further purify this molecule we again cross-linked E2 to the putative receptor, and recovered the E2–receptor complexes from the cell lysate with the anti-His-coated beads as before. A second affinity purification step was then performed using concanavalin A lectin (Con-A), specific for Asn-linked glycans. After Con-A purification an aliquot of the eluted material was enzymically deglycosylated using PNGase F. Control experiments were performed in the absence of E2. Specific bands migrating with apparent molecular weights of 82 and 54 kDa were detected by silver staining of a SDS–PAGE gel, before or after enzymic deglycosylation, respectively (Figure 5A). The difference in molecular weight between the two forms is compatible with the presence of about 10 potential Asn glycosylation sites.
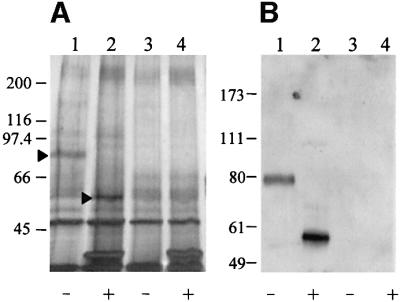
Fig. 5. (A) Silver staining of an SDS–PAGE gel loaded with samples obtained after the purification step with Con-A–Sepharose and deglycosylation with the PNGase F. The arrows show the purified receptor migrating at 82 kDa (lane 1) before PNGase F treatment (–), and migrating at 54 kDa (lane 2) after deglycosylation (+). In control samples cross-linking was performed in the absence of E2 (lanes 3 and 4). (B) Western blot detection of the glycosylated (lane 1) and deglycosylated (lane 2) receptor protein by incubation with rabbit anti-SR-BI polyclonal antibodies followed by HRP-conjugated anti-rabbit. Lanes 3 and 4 represent the control experiments performed in the absence of E2.
Identification of the HepG2 receptor responsible for E2 binding
Among known human membrane proteins a large number could theoretically correspond to the E2 binding species purified from HepG2 cells. Therefore, we decided to explore the effect on E2 binding activity of chemical and enzymatic modifications of the HepG2 cell surface. Treatment of HepG2 cells with GAG lyases (heparinase, heparitinase and hyaluronidase) did not affect the binding, while we observed a 40% reduction by incubating cells with 10 mM β-methyl cyclodextrin (data not shown). β-methyl cyclodextrin selectively removes cholesterol from the membrane and has been shown to affect the function of membrane proteins located in the lipid rafts compartment, suggesting that the E2 receptor is located in this cellular compartment (Yancey et al., 1996). The number of known proteins located in the lipid rafts is quite limited and by reviewing data from the literature we found ∼20 plasma membrane proteins from rat liver that are enriched in the lipid rafts fraction (Calvo and Enrich, 2000). Among these, the SR-BI was the only one matching the molecular pattern we had observed. Antibodies specific for SR-BI recognized both the 82 and the 54 kDa purified molecular species (Figure 5B). Moreover, HepG2 cells enriched for E2 binding activity displayed expression levels of SR-BI that were significantly higher than in the parental HepG2 cells by western blotting assays on cell extracts (data not shown).
Human SR-BI expression in CHO cells confers the ability to bind soluble E2
To prove that SR-BI is indeed able to bind soluble E2 in a specific manner, we cloned the human (hSR-BI) and mouse (mSR-BI) SR-BI coding sequences into the same eukaryotic expression vector, transfected them into CHO cells and performed E2 binding assays. Both human and mouse receptors were expressed at comparable level, as detected by staining transfected cells with an anti-SR-BI antibody reactive against an epitope highly conserved across species (Figure 6A). Only cells transfected with the human SR-BI acquired the ability to bind E2 (Figure 6B). Transfected CHO cells positive for E2 binding were sorted by FACS and kept under selection. In this way, a CHO cell population stably expressing the human SR-BI (CHO-SR-BI) was obtained. This cell line bound H77-E2 and BK-E2 with comparable efficiency (Figure 7).

Fig. 6. (A) FACS analysis of anti-SR-BI binding to CHO transfected cells. Transfection was performed with pcDNA3, pcDNA3-hSR-BI or pcDNA3-mSR-BI. (B) FACS analysis of E2-H77 binding to CHO transfected cells.

Fig. 7. FACS analysis showing the binding of E2 recombinant proteins derived from H77 and BK isolates to CHO wild type (unshaded continuous curve), CHO-CD36 (unshaded dashed curve) and CHO-SR-BI (shaded curve).
To further address the selectivity of interaction between E2 and SR-BI, we measured E2 binding to CHO cells stably transfected with human CD36. CD36 belongs to the family of scavenger receptors and shares many ligands with the SR-BI (Febbraio et al., 2001). The results of this analysis showed that the binding of the HCV E2 proteins is exclusive to the hSR-BI (Figure 7).
HVR1 is essential for E2–SR-BI interaction
HVR1 deletion mutants from the H77 and BK variants were tested for their binding to hSR-BI-transfected CHO cells and were both shown to be unable to bind SR-BI-expressing cells (Figure 8). Interestingly, neither of the two compensatory mutations V514M and L615H were able to restore E2 recognition by CHO-SR-BI transfected cells (data not shown).

Fig. 8. A histogram showing the binding of the E2 recombinant proteins deleted of the HVR1 to CHO-SR-BI transfected cells. Binding is measured by FACS analysis and values are expressed as a percentage of the H77-E2 isolate binding.
A biologically relevant question concerns the ability of anti-HVR1 antibodies to neutralize the binding of HCV E2 to cell surface-displayed SR-BI. We successfully competed out the binding of H77-E2 by using a monoclonal antibody (9/27) obtained by immunization with H77-E2 recombinant protein and reactive with the C-terminal part of the H77 HVR1 sequence (Flint et al., 2000). The antibody showed dose-dependent inhibitory activity for the binding of the H77-E2 protein to the SR-BI comparable on both HepG2 and CHO-hSR-BI transfected cells with an apparent IC50 of ∼500 nM. The antibody was not effective on the binding of the BK-E2 glycoprotein, consistent with its lack of reactivity with this and other E2 variants from 1b isolates (Figure 9).

Fig. 9. Competition of E2 binding to HepG2 cells and CHO-SR-BI by the anti-HVR1 mAb 9/27 reactive with H77-E2. Binding was detected by FACS analysis and is expressed as a percentage of the MFI values obtained in the absence of competitor. H77-E2 binding to HepG2 (open triangle) and CHO-SR-BI (open square). BK-E2 binding to HepG2 (filled triangle) and to CHO–SR-BI (filled square).
Discussion
Studies on the HCV life cycle have been limited due to the lack of a robust and reliable cell culture system for propagating the virus (Bartenschlager and Lohmann, 2001). Similarly, assessment of protective antibody responses to HCV has been hampered by the absence of an efficient in vitro neutralization assay. Several laboratories have undertaken a different approach based on the development of binding assays to cultured cell lines using different expression systems to mimic the viral envelope. The recombinant E2 protein has been used extensively for this purpose, and many efforts have also been devoted to the production of virus-like particles (VLPs) in insect cells and pseudotype viruses as alternative systems (Rosa et al., 1996; Matsuura et al., 2001; Lagging et al., 2002). At present there is no evidence that one of these viral surrogates is superior to another. However, it should be noted that exposure of the E2 HVR1 on VLPs is still a controversial issue, while the HVR1 has been demonstrated as exposed on HCV virions by capturing HCV from human sera with anti-HVR1 antibodies (Zhou et al., 2000; Cerino et al., 2001; Clayton et al., 2002).
Two putative HCV receptors have been described to date: the LDL receptor and the tetraspanin CD81 (Pileri et al., 1998; Agnello et al., 1999). However, there is no experimental evidence that these candidates are required for viral infection, and a number of observations suggest that neither would be sufficient to allow HCV infection of susceptible cells.
To identify novel HCV ligands we looked for cultured cell lines capable of binding to soluble recombinant E2 in a CD81-independent manner. Human hepatoma HepG2 cells fulfilled these requirements and were chosen as a source for purifying the molecule responsible for E2 binding. By a combination of biochemical and functional cloning approaches, we identified the human SR-BI as the HepG2 factor responsible for this interaction.
SR-BI is a 509 amino acid polypeptide belonging to the CD36 superfamily, which includes cell surface membrane proteins that bind chemically modified lipoproteins and often many other types of ligand (Acton et al., 1996; Krieger, 2001). SR-BI has been proposed to have a horseshoe-like membrane topology with short N- and C-terminal cytoplasmic domains, adjacent N- and C-terminal transmembrane domains, and the bulk of the protein in a heavily N-glycosylated, disulfide-containing extracellular loop (Krieger, 2001). SR-BI was initially cloned for its similarity to CD36, and the two receptors, which display 60% amino acid sequence identity, share many ligands (Calvo and Vega, 1993; Febbraio et al., 2001). Interestingly, E2 showed highly selective binding for SR-BI. The two scavenger receptors have quite different tissue distribution, SR-BI is a receptor highly expressed in the liver hepatocytes and steroidogenic tissues, while CD36 is expressed mainly in macrophages, platelets and endothelial cells (Calvo et al., 1997; Febbraio et al., 2001; Krieger, 2001). Thus, the selectivity of the E2 binding for SR-BI could account for the liver tropism of HCV. Moreover, in spite of its high level of homology (80% amino acid identity) mouse SR-BI was not able to bind E2, mirroring the species specificity of HCV infection.
SR-BI binds native high density lipoprotein (HDL) with high affinity and plays a functional role in lipid metabolism (Babitt et al., 1997). It was recently demonstrated that SR-BI internalizes its natural ligand HDL through a non-clathrin-dependent endocytosis mediating cholesterol uptake and recycling of HDL apoprotein (Silver et al., 2001). This observation opens up the possibility, at least theoretically, that the SR-BI is also involved in triggering internalization of ligands other than HDL, such as, for example, HCV. In line with this hypothesis, SR-BI is a fatty acylated protein that has been located in the cholesterol-rich ‘lipid rafts’ membrane compartment (Babitt et al., 1997). Rafts domains are thought to be a preferential entry site for pathogens, providing them with a way to escape from the classical degradative pathway (van der Goot and Harder, 2001). Among viruses, SV40, filoviruses and echoviruses use receptors located in the lipid rafts to enter cells (Acton et al., 1994; Bergelson et al., 1994; Empig and Goldsmith, 2002; Norkin et al., 2002). Moreover, envelope-mediated fusion of HIV has been shown to require integrity of lipid rafts (Manes et al., 2000).
To gain further insight into the biological relevance of the E2–SR-BI interaction for HCV infection, we evaluated the role of HVR1 in E2 recognition by SR-BI. The reason for focusing on HVR1 stems from the observation that this protein element, which exhibits the highest degree of genetic heterogeneity of the entire HCV polyprotein, evolves rapidly in infected individuals as a result of a strong immunological pressure (Hijikata et al., 1991; Weiner et al., 1991). In fact, the HVR1 is the only viral determinant shown to contain neutralizing epitopes (Farci et al., 1994, 1996). Furthermore, sequence analysis of the several HVR1s from different viral isolates suggested that a number of highly conserved segments exist within HVR1 (Puntoriero et al., 1998; Sobolev et al., 2000; Penin et al., 2001). Thus, it would seem that the HVR1, rather than being a truly variable segment, might actually adopt one of a range of closely related conformations compatible with recognition of a cellular receptor. Consistent with this hypothesis, we have demonstrated that different E2 variants deleted of the HVR1 are unable to recognize SR-BI, but can recognize CD81. Involvement of a variable region in virus–receptor interaction is not unprecedented. The most paradigmatic case is HIV, where the V3 loop of gp120 plays a role in the process of viral infection, determining viral tropism by selective interaction with different members of the chemokine receptors superfamily (Berger et al., 1999).
Forns et al. (2000) recently reported that deletion of the HVR1 from an HCV infectious RNA clone resulted in attenuated viral infection in a chimpanzee. On the basis of these results it was proposed that the HVR1 is not essential for the HCV life cycle. However, it must be emphasized that only the HVR1-deleted virus with a point mutation in the E2 ectodomain (V514M) was rescued in a second chimpanzee inoculated with large amounts of plasma from the first animal infected with the RNA (Forns et al., 2000). We thus believe that there is still no evidence that HVR1-deleted virus is infectious without compensatory mutations. In line with this observation, among all the HCV isolates described to date none was found to lack the HVR1, or to carry the E2 mutations selected in vivo upon injection of the HVR1-deleted mutant virus. We therefore favor the hypothesis that the HVR1 region is essential for the main route of HCV infection, but there may be an alternative mechanism of viral entry in the absence of this protein fragment. This alternative HVR1-independent infection process would be significantly less efficient, and viral infection would be rescued only by selecting compensatory mutations in the E2 ectodomain. In agreement with this model, our data showed that deleting the HVR1 impairs binding to HepG2 cells, and that introducing adaptive mutations selected in vivo restored binding. The behavior of the E2 deletion mutants on the SR-BI transfected cells only partially overlapped with that observed on the hepatoma cell line. In fact, the ΔHVR1 mutants showed complete loss of binding to CHO-SR-BI cells that was not restored by the compensatory mutations. At present we have no data to explain this discrepancy. This finding is unlikely to be due to the amount of SR-BI expression, since CHO-SR-BI cells display comparable levels of this molecule to HepG2 cells (data not shown). Folding or post-translational modification of SR-BI between these two cell lines could account for the observed difference. However, an attractive alternative would be the existence of a co-receptor molecule or an alternative receptor present on human hepatoma cells capable of binding HCV via E2 through a protein surface distinct from the HVR1. A model for HCV entry via alternative receptors would also fit with the infectivity observed in HVR1-deleted viral mutants (Forns et al., 2000).
One of the goals of our study was to set up a surrogate assay to test the neutralizing potential of anti-HCV antibodies. In the present work we showed that a monoclonal antibody specific to the H77 strain competes for binding of the H77-E2 recombinant protein to SR-BI. It is interesting to note that the slope of the antibody competition curve is very similar for the hepatoma cells and the SR-BI-transfected cells, suggesting that the binding of E2 protein containing the HVR1 to SR-BI plays a major role in hepatoma and in transfected cells. As reported previously, the anti-HVR1 humoral response induced by infection with one viral inoculum is ineffective against a different viral inoculum (Farci et al., 1994, 1996). Consistently, the anti-HVR1 mAb used in our experiments did not affect SR-BI recognition by the heterologous BK-E2 protein. We believe that the SR-BI binding assay will be very helpful in unravelling the neutralizing potential of antibodies cross-reactive with several HVR1 sequences induced either by infection or by vaccination (Scarselli et al., 1995; Puntoriero et al., 1998).
In conclusion, we have identified the human SR-BI as a novel candidate receptor for HCV. E2 binding to hSR-BI is common to viral isolates derived from genotypes 1a and 1b, is species specific and selective because it is not shared with the hCD36 scavenger receptor. Finally, the involvement of HVR1 in this interaction strongly supports the hypothesis that SR-BI plays a role in HCV infection.
Materials and methods
Cells
Molt-4 (human T-cell leukemia) was obtained from the MRC ADP Repository. HepG2 (human hepatoma), Huh7 (human hepatoma), HEK-293 (human embryonic kidney), CHO (chinese hamster ovary) and CHO cells transfected with human CD36 were obtained from the ATCC Repository. Cells were grown under standard conditions.
Cloning of E2 glycoproteins, production and characterization of the recombinant proteins
Cloning strategy for E2 proteins (amino acids 384–661 of the HCV polyprotein) representative of genotype 1a (isolate H77) and genotype 1b (isolates BK and N2), and their mutants lacking the N-terminal first 27 amino acids (HVR1) have been described previously (Meola et al., 2000). The vector pVIjnsTPA plasmid was used for cloning to favor secretion of the recombinant proteins into the medium. To obtain HVR1 mutants with compensatory mutations, HVR1-H77-E2 plasmid was used as template for the PCRs. To obtain the mutant HVR1-H77-E2/V514M the primers Vj-up (5′-CTAACAGACTGTTCCTTTCCAT-3′) and OL251 (5′-TAGGCGCGCCCGACCTGTCGGTCGTTCCCACCACCATGGGGCTGGGAGTGA-3′) were used for amplification of the mutated insert. The PCR product was digested by PstI and AscI (the AscI restriction site is italics in OL251) and cloned into the PstI/AscI-digested pVIjnsTPA HVR1-H77-E2. The OL251 primer introduces the point mutation indicated in bold. To obtain the mutant HVR1-H77-E2/L615H the primers Vj-down (5′-CAGGGTCAAGGAAGGCACGG-3′) and OL252 (5′-TGCATGGTCGACTACCCGTATAGGCATTGGCACTATCCTTGTACCA-3′) were used for amplification of the insert, which was cloned into the SalI/BglII-digested pVIjnsTPA HVR1-H77-E2 (the BglII restriction site is in italics in OL252). OL252 introduces the point mutation indicated in bold.
All constructs carried a region coding for a His6 tag at the C-terminus of the proteins. Constructs were transfected into HEK-293 cells, recombinant proteins were harvested from culture supernatant and concentrated 40 times as described previously (Meola et al., 2000). The content of E2 in the concentrated supernatant was quantified as described (R.M.Roccasecca, H.Ansuini, A.Vitelli, A.Meola, E.Scarselli, S.Acali, M.Pezzanera, B.B.Ercole, J.McKeating, A.Yagnik, A.Lahm, A.Tramontano, R.Cortese and A.Nicosia, submitted). Briefly, concentrated supernatant preparations were loaded onto an SDS–PAGE gel under non-reducing conditions and blotted on nitrocellulose for detection of E2 by western blot. The amount of E2 in each preparation was evaluated by acquiring the images of exposed film (Kodak Biomax ML) from the Bio-Rad densitometer and analyzing them with Molecular Analyst Software. E2 concentrated supernatants were compared with each other and with a reference purified E2 preparation (Austral Biologicals). Concentration of E2 protein in the concentrated supernatants was in the range 10–50 µg/ml with ∼50% of the protein detectable as monomeric species.
FACS analysis of anti-CD81 antibody, anti-SRI and E2 glycoproteins binding to cell lines
Cells were washed twice in phosphate-buffered saline (PBS), 1% FCS, 10 mM HEPES (washing buffer). Then 2 × 105 cells were allowed to bind at 37°C for 1 h with E2 concentrated supernatants. The E2 molecular species responsible for binding to both Molt-4 and hepatoma cells is the monomeric form, while the E2 aggregated forms always present in purified preparations of recombinant E2 were shown to be functionally inactive (Flint et al., 2000; R.M.Roccasecca, H.Ansuini, A.Vitelli, A.Meola, E.Scarselli, S.Acali, M.Pezzanera, B.B.Ercole, J.McKeating, A.Yagnik, A.Lahm, A.Tramontano, R.Cortese and A.Nicosia, submitted). To determine the relative binding efficacy of each E2 protein a dose– response curve was performed and data were normalized for the content of monomeric species in each preparation. The values, expressed as percentage of binding relative to H77-E2 protein, were derived from a best fit analysis in the linear range of each curve, and represent the ratio between the different E2 protein slopes and that of H77-E2.
Cells were treated with serial dilutions of GAG lyases, heparinase (Sigma), heparitinase (Sigma) and hyaluronidase (Sigma) for 30 min prior to incubation with E2. To monitor the effect of cholesterol sequestration, cells were incubated in the presence of 10 mM methyl-β-cyclodextrin (Sigma) for 30 min prior to incubation with E2 supernatant. After incubation with the supernatants containing E2, and one wash in washing buffer, the anti-His mouse monoclonal antibody (Qiagen) was added at a concentration of 2 µg/ml for 1 h at room temperature. Finally, cell-bound mAbs were visualized with anti-mouse IgG1–phycoerythrin (PE) conjugate (Serotec). Binding competition by the 9/27 rat monoclonal antibody was performed by incubating the antibody with E2 for 30 min prior to incubation with cells. For competition by the 9/27 antibody, the anti-His monoclonal antibody was labeled directly with Alexa fluor 488 (monoclonal antibody labeling kit; Molecular Probes). Binding to anti-CD81 was also performed at room temperature for 1 h using the mouse monoclonal antibody (1.3.3.22; Santa Cruz Biotechnology), FITC conjugated. Binding of rabbit anti-SR-BI purified polyclonal antibodies (Novus Biologicals NB 400-104) diluted 1/500 was performed on cells fixed and permeabilized by using Citofix/Citoperm Kit (Pharmingen), the binding was visualized by adding anti-rabbit IgG1–PE conjugate. Flow cytometry data were acquired by using a Becton-Dickinson FACS Vantage flow cytometer. Dead cells were detected as sytox green dye or propidium iodide (Molecular Probes) stained positive and excluded from the analyses.
FACS sorting of cells with highest E2 binding capacity
HepG2 cells or CHO transfected with SR-BI were incubated with a saturating concentration of 1a-derived E2 recombinant protein for 2 h at room temperature. The E2-bound protein was revealed as described already and analyzed in a FACS Vantage flow cytometer. The cells that showed the highest fluorescence intensity were sorted and expanded.
Cell surface biotinylation and E2–receptor cross-linking
HepG2 cells (1 × 108) enriched for E2 binding were harvested by trypsinization and washed once with cold PBS. After mild oxidation with 2 mM sodium periodate in the dark for 15 min, cells were washed twice and 5 × 107 cells/ml incubated with 5 mM biotin–LC-hydrazide for 10 min at room temperature. After biotinylation cells were washed twice and incubated with H77-E2. Cells (5 × 107 cells/ml) were washed once after binding to E2 and incubated with dithiobis-sulfosuccinimidylpropionate (DTSSP; Pierce) at 2 mM concentration in PBS, for 20 min at room temperature. The reaction was stopped by incubation with 50 mM Tris–HCl pH 7.5. DTSSP is a water-soluble, homobifunctional N-hydroxysuccinimide ester cleavable by thiol. Cells were lysed in PBS 1%, Triton X-100 in the presence of protease inhibitor cocktail (Boehringer Mannheim) for 20 min at 37°C.
Immunoprecipitation of E2–receptor complexes and western blot detection
E2–receptor complexes were immunoprecipitated with anti-His Dynabeads by incubation overnight at 4°C. Dynabeads M-450 rat anti-mouse IgG1 (Dynal, Oxoid) were previously cross-linked to the anti-His monoclonal antibody (Quiagen) following Dynal handbook instructions. After five washes in PBS, 1% Triton X-100, samples were eluted either by boiling the beads directly in SDS sample buffer or by incubating the beads for 30 min at 37°C in 50 mM dithiothreitol, 50 mM NaCl, 50 mM Tris–HCl pH 8. Samples obtained by boiling the beads were loaded on to an SDS–PAGE gel and analyzed by western blot for detection of biotinylated proteins using horseradish peroxidase (HRP)-conjugated streptavidin (Pierce) diluted 1:25 000 in Tris-buffered saline 0.05% Tween 20 (TBST), 2% bovine serum albumin (BSA). Recombinant E2 protein was detected using the rat 6/1a Mab, followed by anti-rat HRP (Serotec). The chemiluminescent substrate Super Signal West Pico (Pierce) was used for detection.
Con-A–Sepharose purification and enzymic deglycosylation of the E2 receptor
Eluates obtained from the immunoprecipitation with anti-His Dynabeads were diluted 1:2 with 1 M NaCl, 0.2% Triton X-100 and incubated with Con-A beads for 2 h at room temperature. After three washes in the incubation buffer, elution was performed under denaturing conditions in 1% SDS, 1% β-mercaptoethanol and 100 mM phosphate pH 7.5 at 95°C for 10 min. The eluates were diluted 1:10 in PNGase F incubation buffer containing 0.1% SDS, 0.5% NP-40, 10 mM EDTA and 100 mM Na phosphate pH 7.5. Eluates were divided into aliquots, one of which was treated with the enzyme PNGase F (Bio-Rad) for 3 h at 37°C. Samples were loaded onto a 7.5% SDS pre-cast gel (Bio-Rad) and silver stained. Immunoreactivity of the purified samples was probed in western blot using rabbit anti-SR-BI purified polyclonal antibodies (Novus Biologicals NB 400-104) diluted 1:1500 in TBST, 2% BSA followed by anti-rabbit HRP (Pierce) diluted 1:20 000.
Cloning of SR-BI coding sequences and transfection in the CHO cell line
RNA was extracted from HepG2 cells with TRIzol reagent (Gibco BRL). First-strand cDNA was produced by using Superscript II reverse transcriptase (Gibco BRL), mixing total RNA (2 µg) with 10 pmol of the antisense primer hSR-BI 5′-CCAGTCTAGACTACAGTTTTGCTTCCTGCAGCACAGAGCCC-3′ (the XbaI restriction site is in italics). An aliquot of the cDNA was amplified by PCR using platinum Pfx DNA polymerase (Gibco BRL), the primers used were the antisense SR-BI primer and the sense hSR-BI 5′-AGGCAAGCTTGCCGCCATGGGCTGCTCCGCCAAAGCGCGCTGGG-3′, where the HindIII restriction site is in italics. PCR was performed in a Perkin-Elmer 2400 thermocycler, denaturing samples for 4 min at 94°C and then running 35 cycles of incubation at 94°C for 30 s, at 50°C for 30 s and 68°C for 2 min. The PCR product was digested with HindIII and XbaI for directional cloning into the vector pcDNA3. The mouse SR-BI sequence was amplified from IMAGE clone BC004656 using the sense primer mSR-BI 5′-AGGCAAGCTTGCCGCCATGGGCGGCAGCTCCAGGGCGCGCTGGG-3′ where the HindIII restriction site is in italics and the antisense primer mSR-BI 5′-CCAGTCTAGACTATAGCTTGGCTTCTTGCAGCACCGTGCCC-3′ where the XbaI restriction site is in italics. The PCR was performed as before for 20 cycles and the PCR product was digested with HindIII and XbaI for directional cloning in the vector pcDNA3. Clones obtained were sequenced with the Big Dye Terminator Cycle sequencing kit with AmpliTaq (Applied Biosystems) and run with an Applied Biosystem Model 373A Sequencer. The CHO cell line was transfected by mixing each plasmid DNA with the Lipofectamine 2000 reagent (Gibco BRL). Cells were harvested 24 h after transfection and analyzed by FACS, cells positive for E2 binding were sorted and put back into culture under geneticin (Gibco BRL) selection at a concentration of 0.5 mg/ml.
Acknowledgments
Acknowledgements
We gratefully acknowledge Jane McKeating for providing the hybridoma producing the 6/1a and 9/27 monoclonal antibodies and Bruno Bruni Ercole for production and purification of the monoclonal antibodies. We also thank Raffaele De Francesco for critical reviewing the manuscript and J.Clench for linguistic revision of the manuscript. Part of this work was supported by a grant from the European Community to the ENHCV Consortium.
References
- Acton S.L., Scherer,P.E., Lodish,H.F. and Krieger,M. (1994) Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J. Biol. Chem., 269, 21003–21009. [PubMed] [Google Scholar]
- Acton S., Rigotti,A., Landschulz,K.T., Xu,S., Hobbs,H.H. and Krieger,M. (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science, 271, 518–520. [DOI] [PubMed] [Google Scholar]
- Agnello V., Abel,G., Elfahal,M., Knight,G.B. and Zhang,Q.X. (1999) Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl Acad. Sci. USA, 96, 12766–12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander T., Forns,X., Emerson,S.U., Purcell,R.H. and Bukh,J. (2000) Hepatitis C virus envelope protein E2 binds to CD81 of tamarins. Virology, 277, 358–367. [DOI] [PubMed] [Google Scholar]
- Babitt J., Trigatti,B., Rigotti,A., Smart,E.J., Anderson,R.G., Xu,S. and Krieger,M. (1997) Murine SR-BI, a high density lipoprotein receptor that mediates selective lipid uptake, is N-glycosylated and fatty acylated and colocalizes with plasma membrane caveolae. J. Biol. Chem., 272, 13242–13249. [DOI] [PubMed] [Google Scholar]
- Bartenschlager R. and Lohmann,V. (2001) Novel cell culture systems for the hepatitis C virus. Antiviral Res., 52, 1–17. [DOI] [PubMed] [Google Scholar]
- Bergelson J.M., Chan,M., Solomon,K.R., St John,N.F., Lin,H. and Finberg,R.W. (1994) Decay-accelerating factor (CD55), a glycosyl phosphatidylinositol-anchored complement regulatory protein, is a receptor for several echoviruses. Proc. Natl Acad. Sci. USA, 91, 6245–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger E.A., Murphy,P.M. and Farber,J.M. (1999) Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism and disease. Annu. Rev. Immunol., 17, 657–700. [DOI] [PubMed] [Google Scholar]
- Calvo D. and Vega,M.A. (1993) Identification, primary structure and distribution of CLA-1, a novel member of the CD36/LIMPII gene family. J. Biol. Chem., 268, 18929–18935. [PubMed] [Google Scholar]
- Calvo D., Gomez-Coronado,D., Lasuncion,M.A. and Vega,M.A. (1997) CLA-1 is an 85-kD plasma membrane glycoprotein that acts as a high-affinity receptor for both native (HDL, LDL and VLDL) and modified (OxLDL and AcLDL) lipoproteins. Arterioscler. Thromb. Vasc. Biol., 17, 2341–2349. [DOI] [PubMed] [Google Scholar]
- Calvo M. and Enrich,C. (2000) Biochemical analysis of a caveolae-enriched plasma membrane fraction from rat liver. Electrophoresis, 21, 3386–3395. [DOI] [PubMed] [Google Scholar]
- Cerino A., Meola,A., Segagni,L., Furione,M., Marciano,S., Triyatni,M., Liang,T.J., Nicosia,A. and Mondelli,M.U. (2001) Monoclonal antibodies with broad specificity for hepatitis C virus hypervariable region 1 variants can recognize viral particles. J. Immunol., 167, 3878–3886. [DOI] [PubMed] [Google Scholar]
- Choo Q.L., Kuo,G., Ralston,R., Weiner,A., Chien,D., Van Nest,G., Han,J., Berger,K., Thudium,K., Kuo,C. and et al. (1994) Vaccination of chimpanzees against infection by the hepatitis C virus. Proc. Natl Acad. Sci. USA, 91, 1294–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton R.F., Owsianka,A., Aitken,J., Graham,S., Bhella,D. and Patel,A.H. (2002) Analysis of antigenicity and topology of e2 glycoprotein present on recombinant hepatitis C virus-like particles. J. Virol., 76, 7672–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empig C.J. and Goldsmith,M.A. (2002) Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol., 76, 5266–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farci P., Alter,H.J., Wong,D.C., Miller,R.H., Govindarajan,S., Engle,R., Shapiro,M. and Purcell,R.H. (1994) Prevention of hepatitis C virus infection in chimpanzees after antibody-mediated in vitro neutralization. Proc. Natl Acad. Sci. USA, 91, 7792–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farci P., Shimoda,A., Wong,D., Cabezon,T., De Gioannis,D., Strazzera,A., Shimizu,Y., Shapiro,M., Alter,H.J. and Purcell,R.H. (1996) Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl Acad. Sci. USA, 93, 15394–15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M., Hajjar,D.P. and Silverstein,R.L. (2001) CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation and lipid metabolism. J. Clin. Invest., 108, 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint M., Maidens,C., Loomis-Price,L.D., Shotton,C., Dubuisson,J., Monk,P., Higginbottom,A., Levy,S. and McKeating,J.A. (1999) Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol., 73, 6235–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint M., Dubuisson,J., Maidens,C., Harrop,R., Guile,G.R., Borrow,P. and McKeating,J.A. (2000) Functional characterization of intracellular and secreted forms of a truncated hepatitis C virus E2 glycoprotein. J. Virol., 74, 702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forns X., Thimme,R., Govindarajan,S., Emerson,S.U., Purcell,R.H., Chisari,F.V. and Bukh,J. (2000) Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc. Natl Acad. Sci. USA, 97, 13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried M.W. and Hoofnagle,J.H. (1995) Therapy of hepatitis C. Semin. Liver Dis., 15, 82–91. [DOI] [PubMed] [Google Scholar]
- Hijikata M., Kato,N., Ootsuyama,Y., Nakagawa,M., Ohkoshi,S. and Shimotohno,K. (1991) Hypervariable regions in the putative glycoprotein of hepatitis C virus. Biochem. Biophys. Res. Commun., 175, 220–228. [DOI] [PubMed] [Google Scholar]
- Kahne T. and Ansorge,S. (1994) Non-radioactive labelling and immunoprecipitation analysis of leukocyte surface proteins using different methods of protein biotinylation. J. Immunol. Methods, 168, 209–218. [DOI] [PubMed] [Google Scholar]
- Korenaga M., Hino,K., Katoh,Y., Yamaguchi,Y., Okuda,M., Yoshioka,K. and Okita,K. (2001) A possible role of hypervariable region 1 quasispecies in escape of hepatitis C virus particles from neutralization. J. Viral Hepat., 8, 331–340. [DOI] [PubMed] [Google Scholar]
- Krieger M. (2001) Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J. Clin. Invest., 108, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagging L.M., Meyer,K., Westin,J., Wejstal,R., Norkrans,G., Lindh,M. and Ray,R. (2002) Neutralization of pseudotyped vesicular stomatitis virus expressing hepatitis C virus envelope glycoprotein 1 or 2 by serum from patients. J. Infect. Dis., 185, 1165–1169. [DOI] [PubMed] [Google Scholar]
- Manes S. et al. (2000) Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep., 1, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura Y. et al. (2001) Characterization of pseudotype VSV possessing HCV envelope proteins. Virology, 286, 263–275. [DOI] [PubMed] [Google Scholar]
- McHutchison J.G., Gordon,S.C., Schiff,E.R., Shiffman,M.L., Lee,W.M., Rustgi,V.K., Goodman,Z.D., Ling,M.H., Cort,S. and Albrecht,J.K. (1998) Interferon α-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N. Engl. J. Med., 339, 1485–1492. [DOI] [PubMed] [Google Scholar]
- Meola A. et al. (2000) Binding of hepatitis C virus E2 glycoprotein to CD81 does not correlate with species permissiveness to infection. J. Virol., 74, 5933–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondelli M.U., Cerino,A., Segagni,L., Meola,A., Cividini,A., Silini,E. and Nicosia,A. (2001) Hypervariable region 1 of hepatitis C virus: immunological decoy or biologically relevant domain? Antiviral Res., 52, 153–159. [DOI] [PubMed] [Google Scholar]
- Norkin L.C., Anderson,H.A., Wolfrom,S.A. and Oppenheim,A. (2002) Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J. Virol., 76, 5156–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penin F., Combet,C., Germanidis,G., Frainais,P.O., Deleage,G. and Pawlotsky,J.M. (2001) Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J. Virol., 75, 5703–5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petracca R. et al. (2000) Structure–function analysis of hepatitis C virus envelope-CD81 binding. J. Virol., 74, 4824–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pileri P. et al. (1998) Binding of hepatitis C virus to CD81. Science, 282, 938–941. [DOI] [PubMed] [Google Scholar]
- Puntoriero G. et al. (1998) Towards a solution for hepatitis C virus hypervariability: mimotopes of the hypervariable region 1 can induce antibodies cross-reacting with a large number of viral variants. EMBO J., 17, 3521–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson B. et al. (1998) Classification, nomenclature and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch. Virol., 143, 2493–2503. [DOI] [PubMed] [Google Scholar]
- Rosa D. et al. (1996) A quantitative test to estimate neutralizing antibodies to the hepatitis C virus: cytofluorimetric assessment of envelope glycoprotein 2 binding to target cells. Proc. Natl Acad. Sci. USA, 93, 1759–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli E., Cerino,A., Esposito,G., Silini,E., Mondelli,M.U. and Traboni,C. (1995) Occurrence of antibodies reactive with more than one variant of the putative envelope glycoprotein (gp70) hypervariable region 1 in viremic hepatitis C virus-infected patients. J. Virol., 69, 4407–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver D.L., Wang,N., Xiao,X. and Tall,A.R. (2001) High density lipoprotein (HDL) particle uptake mediated by scavenger receptor class B type 1 results in selective sorting of HDL cholesterol from protein and polarized cholesterol secretion. J. Biol. Chem., 276, 25287–25293. [DOI] [PubMed] [Google Scholar]
- Sobolev B.N., Poroikov,V.V., Olenina,L.V., Kolesanova,E.F. and Archakov,A.I. (2000) Comparative analysis of amino acid sequences from envelope proteins isolated from different hepatitis C virus variants: possible role of conservative and variable regions. J. Viral Hepat., 7, 368–374. [DOI] [PubMed] [Google Scholar]
- Strader D.B. and Seeff,L.B. (2001) Hepatitis C: a brief clinical overview. ILAR J., 42, 107–116. [DOI] [PubMed] [Google Scholar]
- Triyatni M., Vergalla,J., Davis,A.R., Hadlock,K.G., Foung,S.K. and Liang,T.J. (2002) Structural features of envelope proteins on hepatitis C virus-like particles as determined by anti-envelope monoclonal antibodies and CD81 binding. Virology, 298, 124–132. [DOI] [PubMed] [Google Scholar]
- van der Goot F.G. and Harder,T. (2001) Raft membrane domains: from a liquid-ordered membrane phase to a site of pathogen attack. Semin. Immunol., 13, 89–97. [DOI] [PubMed] [Google Scholar]
- Wasley A. and Alter,M.J. (2000) Epidemiology of hepatitis C: geographic differences and temporal trends. Semin. Liver Dis., 20, 1–16. [DOI] [PubMed] [Google Scholar]
- Weiner A.J. et al. (1991) Sequence variation in hepatitis C viral isolates. J. Hepatol., 13, S6–S14. [DOI] [PubMed] [Google Scholar]
- Yagnik A.T., Lahm,A., Meola,A., Roccasecca,R.M., Ercole,B.B., Nicosia,A. and Tramontano,A. (2000) A model for the hepatitis C virus envelope glycoprotein E2. Proteins, 40, 355–366. [DOI] [PubMed] [Google Scholar]
- Yancey P.G., Rodrigueza,W.V., Kilsdonk,E.P., Stoudt,G.W., Johnson,W.J., Phillips,M.C. and Rothblat,G.H. (1996) Cellular cholesterol efflux mediated by cyclodextrins. Demonstration of kinetic pools and mechanism of efflux. J. Biol. Chem., 271, 16026–16034. [DOI] [PubMed] [Google Scholar]
- Zhou Y.H., Shimizu,Y.K. and Esumi,M. (2000) Monoclonal antibodies to the hypervariable region 1 of hepatitis C virus capture virus and inhibit virus adsorption to susceptible cells in vitro. Virology, 269, 276–283. [DOI] [PubMed] [Google Scholar]