

Abstract
Members of the Inhibitor of Apoptosis Protein (IAP) family are essential for cell survival in Drosophila and appear to neutralize the cell death machinery by binding to and ubiquitylating pro-apoptotic caspases. Cell death is triggered when ‘Reaper-like’ proteins bind to IAPs and liberate caspases from IAPs. We have identified the thioredoxin peroxidase Jafrac2 as an IAP-interacting protein in Drosophila cells that harbours a conserved N-terminal IAP-binding motif. In healthy cells, Jafrac2 resides in the endoplasmic reticulum but is rapidly released into the cytosol following induction of apoptosis. Mature Jafrac2 interacts genetically and biochemically with DIAP1 and promotes cell death in tissue culture cells and the Drosophila developing eye. In common with Rpr, Jafrac2-mediated cell death is contingent on DIAP1 binding because mutations that abolish the Jafrac2–DIAP1 interaction suppress the eye phenotype caused by Jafrac2 expression. We show that Jafrac2 displaces Dronc from DIAP1 by competing with Dronc for the binding of DIAP1, consistent with the idea that Jafrac2 triggers cell death by liberating Dronc from DIAP1-mediated inhibition.
Keywords: apoptosis/Drosophila/IAP/IBM-protein caspase
Introduction
Programmed cell death, or apoptosis, is central to the process of animal development and tissue homeostasis (Meier et al., 2000a). Importantly, failure to regulate apoptosis is now linked to many human pathologies such as cancer, autoimmune disease, neuro-degenerative disorders and persistent viral infection (Thompson, 1995).
Induction of apoptosis triggers the activation of a set of highly specific cysteine proteases called caspases. Caspases are ubiquitously expressed and are synthesized as zymogens with little or no intrinsic enzymatic activity. Caspases become activated during apoptosis through proteolytic cleavage whereby initiator caspases cleave and activate effector caspases, resulting in an amplification of the caspase cascade (Thornberry and Lazebnik, 1998). Active caspases cleave a wide range of structural and regulatory proteins, which culminates in disassembly of the cell. Given the irreversible nature of caspase-mediated proteolysis of cellular proteins, caspase activation and activity has to be tightly regulated. Members of the Inhibitor of Apoptosis Protein (IAP) family suppress apoptosis by directly binding to and inhibiting caspases (Salvesen and Duckett, 2002). Regulation of caspases by IAPs is essential for cell survival in Drosophila, as loss-of-function mutations in the gene thread (th) that encodes the Drosophila IAP 1 (DIAP1) causes early embryonic death due to ectopic activation of caspases (Hay et al., 1995; Wang et al., 1999; Goyal et al., 2000; Lisi et al., 2000). In mammals, the IAPs c-IAP1, 2 and XIAP suppress cell death via direct binding to, and neutralizing of, active but not precursor forms of caspase-3, -7 and -9 (Shi, 2002). In contrast, DIAP1 blocks the activity of active forms of effector caspases (e.g. drICE and DCP-1) (Kaiser et al., 1998; Hawkins et al., 1999) as well as activation of initiator caspases (e.g. Dronc) (Hawkins et al., 2000; Meier et al., 2000b; Quinn et al., 2000). IAPs are defined by the presence of the BIR domain that is homologous to the Baculovirus IAP Repeat (BIR), identified in OpIAP (Crook et al., 1993), and is required for caspase inhibition. Physical interaction between the BIR2 domain of DIAP1 and the pro-domain of DRONC is essential for Dronc regulation since DIAP1 proteins with a mutation in the BIR2 domain (th4), which impairs Dronc binding, completely fail to regulate Dronc in vivo (Wilson et al., 2002).
Genetic and molecular studies of DIAP1 and the Drosophila caspase Dronc indicate that physical interaction of IAPs with caspases is necessary, but not sufficient, to regulate caspases in vivo (Wilson et al., 2002). In addition to Dronc binding, the DIAP1 RING finger is indispensable for the regulation of apoptosis, and after binding of DIAP1 to Dronc, the RING finger mediates ubiquitylation of Dronc. Hence, IAPs appear to suppress cell death by ubiquitylating and thereby inactivating caspases. This inhibition of caspases is dependent on both binding of IAP as well as the E3 ubiquitin protein ligase activity of its RING finger.
While cell survival is ensured by members of the IAP family, apoptosis in Drosophila requires the activity of Rpr (Reaper), Grim and Hid (Head Involution Defective) (White et al., 1994; Grether et al., 1995; Chen et al., 1996). Rpr, Grim and Hid share an N-terminal motif that is sufficient to bind to the BIR2 domain of DIAP1 (Vucic et al., 1998; Wang et al., 1999; Wu et al., 2001). The current model suggests that caspase activation and consequent cell death occurs when Rpr, Grim or Hid displace caspases from DIAP1. Physical interaction of Rpr and Hid with DIAP1 is critical for the induction of apoptosis by these proteins in vivo, because flies with DIAP1 mutations that impair the binding of Rpr and Hid to DIAP1 exhibit less apoptosis (Wang et al., 1999; Goyal et al., 2000; Lisi et al., 2000). Thus, cell death occurs when Rpr-like proteins bind to IAPs and liberate caspases from IAP-mediated inhibition. Interestingly, recent studies suggest that Rpr, Grim and Hid induce cell death not only by displacing caspases from DIAP1 but also by promoting DIAP1 auto-ubiquitylation and degradation (Hays et al., 2002; Holley et al., 2002; Ryoo et al., 2002; Wing et al., 2002; Yoo et al., 2002). In addition, Rpr and Grim also block global de novo protein synthesis.
In mammals, Smac/DIABLO and HtrA2/Omi counter the anti-apoptotic activity of XIAP through binding to the BIR domains of XIAP (Wolf and Green, 2002). This interaction is mediated by a motif that is related to the IAP-binding motif (IBM) of Rpr, Grim and Hid. Typically, IBMs carry an N-terminal Ala1 that is essential to anchor this motif to the BIR surface of IAPs (Wu et al., 2000, 2001). In addition to Ala1, there is a strong preference for Pro3. Smac/DIABLO and HtrA2/Omi are synthesized as precursor proteins that localize to mitochondria. Following their mitochondrial import, the signal peptide is cleaved off generating mature and IBM-bearing forms of Smac/DIABLO and HtrA2/Omi. In healthy cells, Smac/DIABLO and HtrA2/Omi are sequestered in mitochondria but are released into the cytosol during apoptosis where they bind to IAPs.
Although it is clear that direct binding of DIAP1 to Dronc is essential to regulate Dronc in vivo, little is known about how this DIAP1-mediated caspase inhibition is overcome. Here we report the identification of the thioredoxin peroxidase Jafrac2 as an IAP-binding protein. We recovered Jafrac2 as a DIAP1- and DIAP2-interacting protein using the tandem affinity purification (TAP) system. Our results demonstrate that in healthy cells Jafrac2 is localized in the endoplasmic reticulum (ER) where its signal peptide becomes cleaved off, thereby exposing a conserved N-terminal IBM. Shortly after induction of apoptosis, Jafrac2 is released from the ER into the cytosol where it promotes apoptosis by displacing Dronc from DIAP1. Rpr- and Jafrac2-mediated cell death is contingent upon binding to DIAP1 because mutations that impair their binding to DIAP1 suppress Jafrac2- and Rpr-induced cell death. Thus, Jafrac2 represents a new IAP-binding protein that promotes cell death by liberating Dronc from DIAP1-mediated inhibition.
Results
Jafrac2 is an IAP-binding protein
To study the mechanisms underlying caspase activation, we sought to identify molecules that interact with IAPs and, hence, might be involved in antagonizing the DIAP1-mediated inhibition of caspases. To this end, dual epitope-tagged forms of DIAP1 and DIAP2 were stably expressed in Drosophila Schneider cells (S2), and DIAP1 and DIAP2 protein complexes were purified using the TAP system (Rigaut et al., 1999). A protein with an apparent molecular weight of 26 kDa efficiently co-purified with DIAP1 and DIAP2 (Figure 1A). Under the same conditions, no such protein was co-purified with TAP alone (data not shown) or an unrelated control protein fused to TAP. Large-scale affinity purification followed by mass spectrometry identified the 26 kDa protein as the peroxidase Jafrac2. N-terminal sequencing of the 26 kDa protein confirmed the mass spectrometric peptide mass finger-print prediction and revealed that Jafrac2 exhibits the N-terminal sequence AKP (data not shown). The isolated Jafrac2 protein lacked the N-terminal 17 amino acids (Aa) indicating that Jafrac2 is cleaved to generate AKP-Jafrac2 (Figure 1B), consistent with a predicted cleavage site at this position. Cleavage at residue 17 generates mature Jafrac2 with an N-terminal motif that is related to the IBMs of Rpr, Grim, Hid, Sickle, Smac/DIABLO and Omi/HtrA2 (Wolf and Green, 2002). In common with other IBMs (Wu et al., 2000, 2001), the putative IBM of mature Jafrac2 bears Ala at the first and Pro at the third position (Figure 1B).
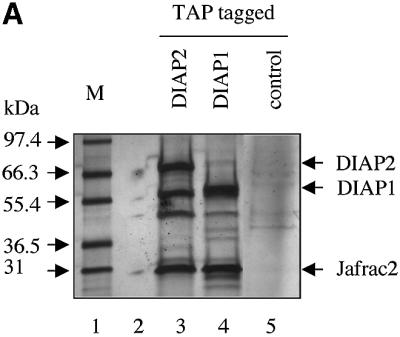



Fig. 1. Jafrac2 is an IAP-binding protein. (A) DIAP2 (3) and DIAP1 (4) but not an unrelated control protein (5) specifically co-purified a protein with an apparent molecular weight of 26 kDa (Jafrac2). Purified material was resolved by SDS–PAGE and visualized by silver staining. Molecular mass markers in kDa are shown (1). (2) is an empty lane. (B) Bar diagram representing the structure of Jafrac2 and the location of the AKP motif. ERS designates the ER targeting sequence. Cysteine residues important for the thioredoxin peroxidase activity of Jafrac2 are indicated. Lower panel: alignment of the N-terminal sequences of Rpr, Grim and Hid (left) and Jafrac2, Smac/DIABLO and HtrA2/Omi (right). Proteins were grouped based on the maturation by a methionine amino peptidase (left) and by proteolytic cleavage (right). Identical residues are shown in black, residues identical in two out of the three sequences are indicated in light grey, while residues with ≥60% similarity are shown in dark grey. (C) Co-immunoprecipitation of endogenous Jafrac2 with endogenous DIAP1 from S2 cells. Jafrac2 was co-immunoprecipitated by anti-full-length (3) and anti-RING DIAP1 (5) antibodies, but not the pre-immune serum (2 and 4). (D) The BIR2 of DIAP1 is required for Jafrac2 binding. S2 cells were transiently transfected with the indicated combinations of constructs encoding Jafrac2-V5 and wild-type or mutant DIAP1–GST. DIAP1–GST was purified from cell lysates using gluthathione beads and associated Jafrac2 was detected by immunoblot analysis with anti-V5 antibodies (top panel). Effective DIAP1 purification was determined by immunoblotting the eluate with an anti-DIAP1 RING antibody (second panel). Expression of the indicated constructs was confirmed by immunoblotting with the indicated antibodies (third and fourth panels). An asterisk denotes endogenous DIAP1.
To confirm the interaction between Jafrac2 and DIAP1, we examined whether endogenous Jafrac2 interacts with endogenous DIAP1. Antibodies raised against the full length and the RING finger of DIAP1, but not their pre-immune serum, co-immunoprecipitated Jafrac2 from S2 cells (Figure 1C).
The BIR2 domain of DIAP1 is required for Jafrac2 binding
To identify the region of DIAP1 that is required for Jafrac2 binding, we tested several DIAP1 mutants for their ability to bind to Jafrac2. To this end, we co-expressed V5-tagged Jafrac2 with either wild-type or mutant DIAP1 fused to glutathione S-transferease (GST). GST-affinity purification was used to precipitate Jafrac2 from S2 cells (Figure 1D). The full-length precursor form of Jafrac2 was efficiently processed to its mature form in S2 cells. Jafrac2 specifically co-purified with wild-type DIAP1 but did not interact with GST alone or the DIAP1 RING finger. Jafrac2 also bound to DIAP1 mutants that carry a single Aa mutation in either the BIR1 (DIAP1 6–3s, G88S) or BIR2 domain (DIAP1 23–4s, G269S), or lack the entire BIR1 region (Δ-BIR1). In contrast, the th4 DIAP1 BIR2 mutant (H283Y) displayed a greatly diminished binding to Jafrac2. Interestingly, the same th4 mutation similarly impairs the binding of DIAP1 to Dronc (Wilson et al., 2002), while the binding to Rpr and Hid remains unaffected (Goyal et al., 2000). However, the th23–4 BIR2 mutant shows reduced binding to Rpr and Hid (Goyal et al., 2000), but this mutation did not affect the binding of DIAP1 to Jafrac2 (Figure 1D). Consistent with the observation that the BIR2 domain of DIAP1 is sufficient for Jafrac2 binding, Jafrac2 co-purified with a DIAP1 BIR2 fragment but not with BIR1 and linker regions (T.Tenev, A.Zachariou and P.Meier, unpublished data). Collectively, these data indicate that the DIAP1 BIR2 domain is required for the binding of Jafrac2 to DIAP1. Furthermore, Jafrac2 and Dronc share a common binding site in the BIR2 domain that is distinct from the site of interaction between the DIAP1 BIR2 domain and Rpr and Hid.
Jafrac2 localizes to the ER and is released during apoptosis
To investigate the localization of Jafrac2, we performed immunofluorescence confocal microscopy on Drosophila SR2 and mammalian NIH 3T3 cells. Staining with Jafrac2-specific antibodies revealed that endogenous Jafrac2 localizes to the ER. This was confirmed using the ER marker GFP–Spitz (Lee et al., 2001; Urban et al., 2001), whose expression closely overlaps with that of Jafrac2 (Figure 2A). In Drosophila cells, GFP–Spitz localizes to the ER and Golgi and hence only partial overlap was obtained in merged images of Jafrac2 and GFP–Spitz. Ectopically expressed Jafrac2 completely co-localized with the ER marker ECFP-ER in mammalian NIH 3T3 cells (Figure 2C). Expression of ΔN1–17Jafrac2 lacking the N-terminal signal sequence showed a diffuse staining throughout S2 cells that closely overlapped with that of endogenous DIAP1 (Figure 2B).

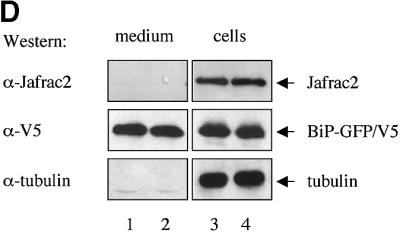


Fig. 2. Subcellular localization of Jafrac2. (A) Endogenous Jafrac2 resides in the ER. Immunofluorescence confocal microscopy of SR2 cells stained with an anti-Jafrac2 antibody (red) and GFP–Spitz (green). Right panels represent merged micrographs. (B) The N-terminal 17 Aa of Jafrac2 are required for targeting Jafrac2 to the ER. Confocal micrographs of SR2 cells transfected with ΔN1–17-Jafrac2 lacking the N-terminal signal sequence and stained with anti-V5 (red) and anti-DIAP1 antibodies (green). (C) Confocal micrographs of NIH 3T3 cells transfected with Jafrac2 (red) and the ER-marker ECFP-ER (green). (D) Jafrac2 is an intracellular protein. Endogenous Jafrac2 is present exclusively in the cellular but absent from the medium fraction. (E) Jafrac2 is released from the ER in UV-mediated apoptosis. Normal and UV-treated S2 cells were lysed in a buffer containing 0.025% digitonin and subfractionated into membrane and cytosolic fractions. The cytosolic fractions were examined by immunoblot analysis with the indicated antibodies. (F) Jafrac2 is released from the ER by the ER stress-inducing agents brefeldin A or tunicamycin. Normal and treated NIH 3T3 cells stably expressing Jafrac2-V5 were analysed as in (E). At 3–4 h post-UV (G) or -brefeldin A treatment (H) of NIH 3T3, the majority of Jafrac2 (red) is no longer co-localized with ECFP-ER (green), as seen by overlaying the green and red images (yellow).
Almost all proteins to be secreted are initially delivered to the ER lumen. However, endogenous Jafrac2 is not secreted but is present exclusively retained in the cell (Figure 2D). In parallel experiments, the secreted protein BiPSS–GFP was readily detectable in the medium and cellular fractions. α-tubulin, like Jafrac2, was also only present in the cellular fraction. Taken together, these data indicate that the N-terminal 17 Aa of Jafrac2 are required for ER localization. Upon ER import, the signal peptide is cleaved off generating mature Jafrac2 that is retained in the ER.
To determine whether Jafrac2 is released from the ER during apoptosis, we stimulated cell death with UV light and examined the intracellular localization of endogenous Jafrac2 by biochemical fractionation and immunofluorescence confocal microscopy. For the biochemical fractionation experiments, cell extracts from normal or UV-irradiated S2 cells were subfractionated into membrane and cytosolic fractions using a digitonin-based fractionation procedure (Kanuka et al., 1999; Verhagen et al., 2000, 2002; Ekert et al., 2001) and their cytosolic fractions were examined by immunoblot analysis (Figure 2E). In non-irradiated cells, Jafrac2 and cytochrome c were exclusively present in the membrane fraction (data not shown) and absent from the cytosolic fraction. Following UV irradiation, Jafrac2 was present in the cytosolic fraction indicating that Jafrac2 is released from the ER during UV-induced apoptosis. To test whether the release of Jafrac2 is also susceptible to ER stress-inducing agents, cells were treated with either brefeldin A or tunicamycin. Brefeldin A, an inhibitor of ER Golgi transport, and tunicamycin, a specific inhibitor of N-glycosylation in ER, are effective inducers of ER stress and cell death (Pahl, 1999). In non-treated cells, Jafrac2 was absent from the cytosolic fraction, but following brefeldin A or tunicamycin treatment, Jafrac2 was present in the cytosolic fraction (Figure 2F). These results indicate that Jafrac2, like Smac/DIABLO and Omi/HtrA2, is retained within specific organelles in healthy cells but is released following the initiation of apoptosis. Consistent with our biochemical fractionation assays, immunofluorescence confocal microscopy showed that Jafrac2 is released from the ER shortly after UV irradiation (Figure 2G) or brefeldin A treatment (Figure 2H), but remained localized to the ER in untreated control cells (Figure 2C). The release of Jafrac2 from the ER occurred 3–4 h after UV exposure or brefeldin A treatment, before any apoptotic morphological changes were detectable. Taken together, these results demonstrate that in normal cells the signal peptide of Jafrac2 targets it to the ER, where it becomes cleaved thereby exposing its IBM. Shortly after induction of apoptosis, Jafrac2 is released from the ER into the cytosol.
Ala1 of AKP-Jafrac2 and AVA-Rpr is essential for DIAP1 binding
To examine whether Jafrac2 binds to DIAP1 via its putative IBM, we tested full-length Jafrac2, AKP-Jafrac2 and a mutant of mature Jafrac2 lacking Ala1 (KP-Jafrac2-V5) for their DIAP1 binding ability. As expected, expression of the precursor form of Jafrac2 resulted in N-terminal cleavage and DIAP1 binding (Figure 3B). AKP- and KP-Jafrac2 were expressed in S2 cells through the ubiquitin (Ub) fusion technique (Varshavsky, 2000) in which a Ub reporter is fused to the test protein. Ubiquitin-specific proteases (UBP) cleave these fusion proteins at the C-terminus of the Ub moiety, yielding the test proteins AKP- or KP-Jafrac2. AKP- but not KP-Jafrac2 was efficiently co-purified by DIAP1 (Figure 3A). Likewise, AVA-Rpr but not VA-Rpr bound to DIAP1 (see Supplementary figure SI1 available at The EMBO Journal Online). Interestingly, a chimeric Rpr mutant (AKP-Rpr), in which the IBM of Rpr was replaced with the IBM of Jafrac2, displayed strong and unaffected binding to DIAP1. In contrast, KP-Rpr lacking Ala1 did not interact with DIAP1. In parallel experiments, neither AVA- nor AKP-Rpr bound to GST alone (Supplementary figure SI1). Disruption of the peroxidase activity of Jafrac2 had no effect upon the ability of Jafrac2 to bind to DIAP1. Jafrac2 mutants with either a single or double point mutation of catalytically active cysteines in the peroxidase domain of Jafrac2, readily co-purified with DIAP1 (Figure 3B). Intriguingly, mature Jafrac2 with an N-terminal Met (MAKP-Jafrac2) failed to interact with DIAP1, highlighting the importance of Ala1 for IAP binding. To determine whether Jafrac2 directly interacts with DIAP1, we performed in vitro co-purification assays with purified recombinant DIAP1 and Jafrac2 proteins (Figure 3C). Recombinant AKP- but not KP-Jafrac2 co-purified with DIAP1 showing that the interaction between DIAP1 and Jafrac2 is indeed direct. Taken together, these results indicate that Ala1 of mature Jafrac2 and Rpr are essential for DIAP1 binding. In common with Rpr, Grim, Hid, Smac/DIABLO and HtrA2/Omi, which bind to BIR domains of IAPs, Jafrac2 possesses a bona fide N-terminal IBM.



Fig. 3. DIAP1 binding is mediated by the N-terminal IBMs of Jafrac2 and Rpr. (A) AKP- but not KP-Jafrac2 interacts with DIAP1. Co-purification of Jafrac2 with DIAP1 from cellular extracts. In this and subsequent figures, expression and purification of the indicated constructs was determined as in Figure 1D. (B) The thioredoxin peroxidase activity of Jafrac2 is not required for DIAP1 binding. Co-purification of wild-type and Jafrac2 mutants with DIAP1 from cellular extracts. (C) AKP-Jafrac2 directly interacts with DIAP1. In vitro co-purification of recombinant AKP- and KP-Jafrac2 with recombinant DIAP1 protein is shown. First and second panel: immunoblot analysis of Jafrac2- and DIAP1-specific antibodies. Third panel: Coomassie-stained gel of recombinant AKP- and KP-Jafrac2.
Mature Jafrac2 promotes cell death in S2 cells
Mature Jafrac2 efficiently binds to DIAP1 and thus may promote apoptosis by antagonizing IAPs in the same manner as Rpr, Grim, Hid, Smac/DIABLO or HtrA2/Omi. To address whether mature Jafrac2 promotes apoptosis by binding to DIAP1, we performed a killing assay in S2 cells. Induced expression of AKP-Jafrac2, like that of AVA-Rpr and AKP-Rpr, caused rapid and efficient killing of S2 cells (Figure 4). In contrast, Jafrac2 or Rpr mutants that lack the N-terminal Ala1 and fail to bind to DIAP1, also failed to induce apoptosis. In contrast to precursor Jafrac2, which is localized in the ER, Jafrac2 that lacks the ER-targeting sequence (ΔN1–17) is not compartmentalized and, like Rpr, can interact and antagonize IAPs. Jafrac2- and Rpr-mediated cell death is caspase dependent because treatment with z-VAD-FMK, a pan-caspase inhibitor, blocked cell death induced by expression of Jafrac2 and Rpr (data not shown). These experiments indicate that cell death induced by mature Jafrac2 and Rpr is dependent on the presence of Ala1 in their IBM, which mediates their interaction with DIAP1.

Fig. 4. Induced expression of AKP- but not KP-Jafrac2 causes induction of apoptosis in S2 cells. Similarly, AVA- and AKP-Rpr but not VA- and KP-Rpr promote cell death. The indicated Ub fusion constructs were co-transfected with a lacZ reporter plasmid, and 24 h post-transfection cells from each well were divided into two dishes to avoid variations in transfection efficiencies. Expression of the indicated constructs was induced by copper sulfate and cells were examined for β-galactosidase (β-gal) activity. (A–D) and (I–L), untreated cells; (E–H) and (M–O), cells treated with copper sulfate (induced state).
Mature Jafrac2 competes with Dronc for the binding of DIAP1
Physical interaction between the BIR2 domain of DIAP1 and the Dronc pro-domain is necessary to regulate Dronc activation in vivo. Thus, both Dronc and Jafrac2 bind to the BIR2 domain of DIAP1. We therefore tested whether Jafrac2 antagonizes DIAP1 function by preventing DIAP1 from binding to Dronc. The ability of DIAP1 to bind to a catalytically inactive mutant of Dronc (Dronc C>A) was significantly impaired in the presence of mature Jafrac2 (AKP-Jafrac2), but not a Jafrac2 mutant that lacks Ala1 (KP-Jafrac2) and fails to bind to DIAP1 (Figure 5). This result suggests that Jafrac2 and Dronc compete for the binding of DIAP1. Both Jafrac2 and Dronc bind to the BIR2 domain of DIAP1, however, Dronc had a significantly lower affinity for the binding to DIAP1 compared with the binding of Jafrac2 to DIAP1 (data not shown). These results suggest that Jafrac2, like Rpr, Grim, Hid, Smac/DIABLO and HtrA2/Omi, can disrupt the caspase–IAP interaction to promote cell death.

Fig. 5. Jafrac2 competes with Dronc for the binding of DIAP1. The ability of DIAP1 to bind to Dronc is significantly impaired in the presence of AKP- but not KP-Jafrac2. Co-purification of a catalytically inactive Dronc mutant (Dronc C>A) with DIAP1–GST in the presence of KP- or AKP-Jafrac2 from cellular extracts. Expression and purification of the indicated constructs was determined as in Figure 1D.
Ectopic expression of Jafrac2 induces a Dronc-like eye ablation phenotype in Drosophila
To determine whether ectopic expression of Jafrac2 can induce cell death in Drosophila, we expressed various forms of Jafrac2 and Rpr in the developing compound eye using the eye-specific promoter glass multimer reporter (GMR) (Hay et al., 1994). AKP- and KP-Jafrac2 as well as AVA- and VA-Rpr were expressed through the Ub fusion technique. Expression of AKP-Jafrac2 induced ectopic cell death in the developing retina causing a ‘spotted eye’ phenotype (Figure 6B). Although akp- jafrac2 flies are white+, and should therefore have red eyes, their eyes appeared white with occasional red spots. Intriguingly, the eye phenotype of akp-jafrac2 flies is highly reminiscent to the one of Dronc-expressing flies (Figure 6A) (Meier et al., 2000b). Eye-specific expression of Dronc, like AKP-Jafrac2, generates an unusual phenotype in which the eye exhibits apparently normal outer morphology with internal ablation of all photoreceptor and most pigment cells, resulting in a ‘spotted eye’. In contrast to eyes expressing AKP-Jafrac2 that have a normal external morphology and size, GMR-driven expression of AVA- and AKP-Rpr resulted in eyes that are severely reduced in size (Figure 6C and D). The phenotypes induced by mature Jafrac2 and Rpr expression are dependent upon Ala1 in their respective IBMs since expression of Ala1 deletion mutants of Jafrac2 and Rpr exerted no detectable effects on eye development (Figure 6F–H).

Fig. 6. Ectopic expression of AKP-Jafrac2, AVA- and AKP-Rpr in the developing eye causes eye ablations. (B) GMR-akp-jafrac2/+ transgenic flies display a spotted eye phenotype that is reminiscent to the eye phenotype caused by Dronc expression in the eye. (A) GMR-gal4/UAS-pro-droncW). (C) GMR-ava-rpr/+ and (D) GMR-akp-rpr/+ flies show eyes of reduced size. (E) Control flies (Canton S). (F) GMR-kp-jafrac2/+, (G) GMR-va-rpr/+ and (H) GMR-akp-rpr/+ transgenic flies show no detectable effects on eye development.
To determine whether the eye phenotype caused by ectopic expression of AKP-Jafrac2 is contingent upon IAP binding, we crossed akp-jafrac2 transgenic flies to th4 mutant flies (Figure 7). Flies carrying the th4 allele that abrogates Jafrac2 binding, strongly suppressed the AKP-Jafrac2 eye phenotype (Figure 7B), indicating that physical interaction between Jafrac2 and DIAP1 is essential for Jafrac2-mediated cell death. In contrast, th4 flies enhanced the AVA-Rpr eye phenotype (Figure 7G), consistent with previous observations (Goyal et al., 2000; Lisi et al., 2000). Importantly, the AKP-Rpr-mediated eye phenotype was also enhanced in th4 mutant flies (Figure 7L). Thus, chimeric Rpr (AKP-Rpr) carrying the Jafrac2-derived IBM appeared to retain the same DIAP1 binding specificity as wild-type Rpr. Jafrac2- and Rpr-mediated eye ablations were repressed by co-expression of DIAP1 or p35 (Figure 7). Taken together, these results indicate that Jafrac2, like Rpr, promotes caspase-dependent cell death through its ability to bind to DIAP1.

Fig. 7. The eye ablation phenotype caused by ectopic expression of Jafrac2 is contingent on DIAP1 binding and is suppressed by co-expression of DIAP1 and p35. The eye phenotypes of GMR-akp-jafrac2/+, GMR-ava-rpr/+ and GMR-akp-rpr/+ transgenic flies on their own (A, F and K), or in combination with th4 (B, G and L), GMR-diap1 (C, H and M), GMR-p35 (D, I and N) or Df(3L)AC1 [indicated as Df(Dronc); E, J and O] are shown. The th4 diap1 mutation suppresses AKP-Jafrac2 but enhances AVA- and AKP-Rpr induced eye phenotypes. Co-expression of DIAP1 and p35 rescues cell death induced by Jafrac2 and Rpr. Flies with a chromosomal deletion that removes the dronc locus (Df(3L)AC1) display a suppressed Jafrac2 and Rpr eye phenotype.
Jafrac2-induced cell death is mediated by Dronc
Rpr, Grim and Hid induce cell death by binding to DIAP1 thereby liberating caspases (Wang et al., 1999; Goyal et al., 2000). Significantly, Rpr, Grim and Hid all induce cell death via activation of Dronc (Hawkins et al., 2000; Meier et al., 2000b; Quinn et al., 2000). To test whether Jafrac2-induced cell death is also mediated by Dronc, we examined whether Jafrac2-mediated cell death is sensitive to dronc gene dosage. Heterozygosity at the dronc locus [Df(3L)AC1] significantly suppressed the AKP-Jafrac2 eye phenotype (Figure 7E). The Df(3L)AC1 similarly suppressed Rpr- (Figure 7J and O) and Hid-mediated cell killing in the eye (Meier et al., 2000b). These results are consistent with the notion that Dronc is a rate-limiting caspase in Jafrac2-mediated cell death.
Discussion
Apoptosis signalling leads to the activation of caspases. In healthy cells, members of the IAP family function to suppress the activation or activity of caspases (Shi, 2002). In order for cells to die, this IAP-mediated inhibition of caspases has to be overcome. It appears that Rpr-like proteins induce cell death by binding to IAPs thereby displacing caspases from IAPs.
We report here the identification of a new IAP-interacting protein. We recovered Jafrac2 as a DIAP1-interacting protein in the cell using the TAP system. Like Rpr, Grim, Hid, Sickle, Smac/DIABLO and HtrA2/Omi, Jafrac2 bears a conserved N-terminal IBM essential for IAP interaction. Jafrac2 is synthesized as a precursor protein with an N-terminal signal peptide that targets it to the ER. Upon import into the ER, the signal peptide of Jafrac2 is cleaved off, thereby exposing the IAP interacting domain that allows this mature Jafrac2 isoform to interact with DIAP1, DIAP2 (Figure 1) and XIAP (data not shown). Although we only tested the ability of Jafrac2 to bind to DIAP1, DIAP2 and XIAP, it is possible that Jafrac2 can also interact with other IAP family members.
In living cells Jafrac2 is compartmentalized and sequestered in the ER away from IAPs, where it exists exclusively in the processed from. This is evident because mature Jafrac2, like cytochrome c, which is compartmentalized in mitochondria, remains associated with the membrane fraction in healthy cells. Following stimulation of apoptosis by UV irradiation or ER stress-inducing agents, mature Jafrac2 is released from the membrane fraction and was present in the cytosol where it can interact with DIAP1 and DIAP2. Because the pro-apoptotic, IAP-interacting form of Jafrac2 is released only upon cell death insult, the major regulatory step for Jafrac2 appears to be its release from the ER lumen. The release of Jafrac2 from the ER of UV-irradiated cells occurs early in UV-mediated apoptosis. This is evident because Jafrac2 expression becomes diffuse in otherwise morphologically normal cells within 3–4 h following UV exposure (Figure 2F). In similar experiments, the mitochondrial release of cytochrome c, Smac/DIABLO and HtrA2/Omi that occurs, early in apoptosis, also became apparent within 3–4 h following UV treatment (Goldstein et al., 2000). Thus, Jafrac2 resembles Smac/DIABLO and HtrA2/Omi that are similarly compartmentalized in healthy cells and that promote caspase activation after their release from mitochondria following the cell death trigger (Wolf and Green, 2002). Furthermore, analogous to Smac/DIABLO and HtrA2/Omi, Jafrac2 also requires N-terminal processing to generate its pro-apoptotic form. Hence, Jafrac2, Smac/DIABLO and HtrA2/Omi all undergo a maturation process through cleaving off their signal peptide following import into their respective organelles. This organelle-specific maturation ensures that newly synthesized Jafrac2, Smac/DIABLO and HtrA2/Omi will not promote apoptosis prior to their sequestration into organelles.
In common with Rpr, Grim and Hid, Jafrac2 interacts genetically and biochemically with DIAP1 and is able to promote cell death. In the Drosophila eye and tissue culture cells, mature Jafrac2, like Rpr, efficiently induced cell death in a DIAP1-binding dependent manner. Recent studies have suggested that Rpr and Grim antagonize the anti-apoptotic activity of IAPs by two distinct mechanisms. First, by a mechanism that requires DIAP1 binding, Rpr promotes DIAP1 self ubiquitylation and proteasomal degradation (Hays et al., 2002; Holley et al., 2002; Ryoo et al., 2002; Wing et al., 2002; Yoo et al., 2002). Secondly, Rpr and Grim were also found to repress global protein translation by a mechanism that does not rely on IAP binding (Holley et al., 2002; Yoo et al., 2002). We have used the Ub fusion technique to examine whether Jafrac2 and Rpr possess apoptosis-promoting activities that are independent of IAP binding. We find that, in vivo Rpr and Jafrac2 promote cell death exclusively in an IAP-binding dependent manner because mutations that impair the binding between DIAP1 and Rpr or Jafrac2 completely abolish their ability to induce cell death in the developing eye and tissue culture cells. Thus, Rpr and Jafrac2 that fail to bind to DIAP1 also fail to induce cell death. Mutations in endogenous diap1, which greatly impair the binding of DIAP1 to Rpr or Jafrac2, suppress Rpr and Jafrac2-mediated cell killing. Together, these data argue that in common with Rpr, mature Jafrac2 promotes cell death, and this activity is contingent upon their binding to DIAP1.
The interaction between Jafrac2 and the DIAP1 BIR2 domain is indispensable for its pro-apoptotic function. Interestingly, Jafrac2 and Dronc share a common binding site in the BIR2 domain that is distinct from the site of interaction between the DIAP1 BIR2 domain and Rpr and Hid. The th4 DIAP1 BIR2 mutation greatly diminished binding to Jafrac2 and Dronc (Figure 1D) (Wilson et al., 2002), whereas the same mutation did not affect its binding to Rpr and Hid (Goyal et al., 2000). In addition, the th23–4 DIAP1 mutation that greatly impairs the binding of DIAP1 to Rpr and Hid (Goyal et al., 2000) did not affect the DIAP1–Jafrac2 interaction (Figure 1D). Consistent with our biochemical data, flies carrying the th4 mutation, which abolishes Jafrac2 binding, displayed strongly suppressed Jafrac2-induced eye ablation but enhanced Rpr-induced cell death in the eye.
Several lines of evidence show that the IBM of Jafrac2 is essential for IAP binding and induction of apoptosis. First, mutations that delete or obstruct the N-terminus of mature Jafrac2 abrogate the ability of Jafrac2 to bind to DIAP1 and trigger cell death. The view that Jafrac2 harbours a bona fide IAP-binding motif is strongly supported by crystal structure analyses that have identified Ala1 of IBMs as the critical residue to anchor this motif to the BIR surface of IAPs (Wu et al., 2000, 2001). In addition to the requirement of Ala1, there is a strong preference for Pro3 (Wu et al., 2001). In accordance with other IBMs, the putative IBM of mature Jafrac2 bears Ala1 and Pro3. Furthermore, the IBM of Rpr is functionally interchangeable with the IBM of Jafrac2. A chimeric Rpr mutant (AKP-Rpr) in which the IBM of Rpr was replaced with the IBM of Jafrac2, displayed the same phenotype and cell death promoting efficacy as wild-type Rpr (AVA-Rpr) in both the Drosophila developing eye and tissue culture cells. Together, these results reveal that whereas Jafrac2 and Rpr share a common IAP-binding motif, they also have some distinct DIAP1-binding requirements that presumably give these interactions their specificity.
Physical interaction between DIAP1 and caspases is essential to regulate apoptosis in vivo because embryos with a homozygous mutation that abolishes Dronc binding die early during embryogenesis due to widespread apoptosis (Wang et al., 1999; Goyal et al., 2000; Lisi et al., 2000; Rodriguez et al., 2002). Unrestrained cell death caused by loss of DIAP1 function requires the Drosophila Apaf-1 homologue DARK because a mutation in dark rescues DIAP1-dependent defects (Rodriguez et al., 2002). Thus, loss of DIAP1 function allows DARK-dependent caspase activation. Although activation of downstream, effector caspases is required for normal cell death, the activation of initiator caspases, such as Dronc, is rate limiting for the activation of this cascade. The observed unrestrained cell death caused by loss of DIAP1 function is likely to be triggered by the initiator caspase Dronc because DIAP1 normally suppresses Dronc activation, which in turn is mediated by DARK (Quinn et al., 2000). In line with the current model on caspase activation, we argue that the DIAP1-mediated inhibition of Dronc is the key regulatory step in controlling cell death. This view is supported by the observation that flies with diap1 mutations that either abolish binding or ubiquitylation of Dronc completely fail to suppress Dronc- mediated cell death in vivo (Wilson et al., 2002). Thus, DIAP1 suppresses Dronc activation by binding to and targeting Dronc for ubiquitylation. However, when Rpr-like molecules displace DIAP1 from Dronc, Dronc is recruited into a 700 kDa size apoptosome protein complex that results in Dronc activation (Dorstyn et al., 2002). Consequently, cell death is triggered when Dronc is liberated from DIAP1. Thus, the key event in regulating the caspase cascade appears to be inhibition of Dronc by DIAP1.
Several lines of evidence support the notion that Jafrac2 promotes cell death by interfering with the Dronc–DIAP1 interaction, thereby displacing and liberating Dronc from DIAP1. First, Jafrac2 and Dronc bind to the same site of the BIR2 domain of DIAP1, since the BIR2 th4 mutation of DIAP1 equally abolished Dronc and Jafrac2 binding. In contrast, Rpr and Hid binding to the th4 DIAP1 mutant remains unaffected (Goyal et al., 2000). Secondly, Jafrac2 competes with Dronc for the binding of DIAP1, and Jafrac2 possesses a significantly higher DIAP1-binding affinity compared with that of Dronc to DIAP1, as would be expected of a protein that displaces Dronc from DIAP1. Thirdly, ectopic expression of Jafrac2 in the developing Drosophila eye causes a phenotype that is highly reminiscent of the phenotype observed in flies ectopically expressing Dronc. Finally, heterozygosity at the dronc locus rescued the eye-ablation phenotype induced by Jafrac2, indicating that apoptotic signal transduction initiated by Jafrac2 is mediated through Dronc. Taken together, these results indicate that Jafrac2 promotes cell death by liberating Dronc from the anti-apoptotic activity of DIAP1.
The observation that Jafrac2, like the apoptotic inducers Rpr, Grim and Hid, induces apoptosis through binding to DIAP1 places Jafrac2 in a potentially pivotal position to regulate apoptosis. Our findings are consistent with a model whereby Jafrac2 promotes apoptosis by displacing DIAP1 from Dronc, so allowing activation of the caspase cascade and consequent cell death. We currently favour the idea whereby Jafrac2 function is additive to, but independent of, Rpr. The early release of Jafrac2 from the ER of UV-irradiated cells is consistent with the view that Jafrac2 is involved in the initiation of apoptosis. Thus, Jafrac2 is released from the ER at a time when other early apoptotic events occur, such as the mitochondrial release of cytochrome c, Smac/DIABLO and HtrA2/Omi in mammalian cells. Once released, Jafrac2 interacts with DIAP1 and thereby liberates Dronc, which in turn is activated by DARK. In line with the notion that Jafrac2 functions in a complementary but distinct cell death pathway to Rpr, Grim and Hid, we find that a chromosomal deletion that includes the jafrac2 locus did not suppress the eye phenotypes caused by ectopic expression of Rpr, Grim and Hid (data not shown). However, it is possible that Jafrac2 may also be part of a positive feedback mechanism, which cooperates with Rpr-like proteins to promote apoptosis in response to cellular damage. We currently cannot distinguish between these two alternatives because no jafrac2 mutant flies are available and Jafrac2 is refractory to the effect of dsRNA interference (data not shown).
Our data are consistent with the idea that Jafrac2, with its thioredoxin peroxidase activity and IAP-binding ability, contains two distinct functions. In healthy cells, Jafrac2 may fulfil a ‘housekeeping’ role through its peroxidase activity (Rodriguez et al., 2000; Radyuk et al., 2001) by protecting the cell from oxidative damage. Consistent with this view, members of the peroxiredoxin protein family play an important role in protecting cells against oxidative damage by scavenging intracellularly generated reactive oxygen species, such as H2O2. However, upon UV irradiation, mature Jafrac2 is released from the ER and competes with Dronc for the binding of DIAP1 that is independent of its peroxidase activity. Consequently, Jafrac2 liberates Dronc from DIAP1 inhibition and allows activation of the proteolytic caspase cascade, resulting in cell death.
Materials and methods
Constructs
DIAP1/2 were amplified by PCR and cloned into pIZ (Invitrogen) with a C-terminal TAP tag, or into pMT–GST (Wilson et al., 2002). Jafrac2 was cloned by RT–PCR using total RNA. Constructs encoding Jafrac2 or Rpr were generated by PCR and cloned into pMT (Invitrogen), pcDNA3.1 (Invitrogen), pExpress (Ollmann et al., 2000) and pNRTIS-33 (Tenev et al., 2000). pMT/BiP/V5-His/GFP (Invitrogen) encodes GFP with the secretion signal of BiP. Ub fusion constructs were generated by PCR and cloned into pMT. Point and deletion mutants were generated by PCR-mediated mutagenesis. All constructs were verified by DNA sequencing.
Cell culture
Cells were transfected with calcium phosphate or Effectene (Qiagen) according to manufacturer’s instructions. For the cell death assay, S2 cells were seeded in a 12-well plate and co-transfected with pAc5.1/LacZ (Invitrogen) and pMT-based Ub fusion constructs in a ratio of 1:10. Post-transfection (24 h), cells from each well were divided into two wells and expression of pMT-based constructs were induced in one of the two wells by CuSO4. Cells were then assayed for β-gal activity. Cell viability was determined based on the morphology and the reduction of β-gal positive cells of induced versus non-induced constructs.
TAP and GST protein purification
DIAP1/2-TAP expressing S2 cells were harvested and resuspended in lysis buffer [50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, Complete™ protease inhibitors (Roche)]. The lysate was centrifuged at 10 000 g in a microcentrifuge for 30 min and the supernatant was used for TAP as described previously (Rigaut et al., 1999). Purified material was resolved by SDS–PAGE, and stained with colloidal Coomassie Blue (Invitrogen) or Silver Stain Plus (Bio-Rad). GST purification was performed as described previously (Wilson et al., 2002).
Mass spectrometry and N-terminal sequencing
MALDI-TOF mass spectrum was acquired using a Voyager-DE STR (Applied Biosystems). Database searching of the mono-isotopic peptide masses was performed using the MS-Fit algorithm. Protein sequence analysis was performed using a 477A Liquid Phase Sequencer (Applied Biosystems).
Immunoprecipitations and western blot analysis
For the co-immunoprecipitation of Jafrac2 with DIAP1, Seize™X protein A immunoprecipitation system (Pierce) was used according to the manufacturer’s instructions. α-V5 (Invitrogen), α-GST (Amersham), α-cytochrome c (Pharmingen) and α-tubulin (Sigma) were used for the immunoblot analysis. α-DIAP1 RING and α-Dronc have been described previously (Wilson et al., 2002). Full-length α-DIAP1 was generated in guinea pigs using purified full-length DIAP1. α-Jafrac2 is a peptide-derived antibody generated in guinea pigs.
Confocal microscopy
Cells were fixed with 4% paraformaldyde. Jafrac2-V5 was visualized using α-V5 antibodies in conjunction with an Alexa555 conjugated secondary antibody (Molecular Probes). ECFP-ER (Clontech) staining was enhanced with an α-GFP antibody (Santa Cruz) followed by Alexa488 conjugated secondary antibody. Endogenous Jafrac2 was detected with Jafrac2-specific antibodies followed by Alexa546 conjugated secondary antibodies. Cell nuclei were detected by TO-PRO-3 DNA stain (Molecular Probes).
Purification of recombinant proteins
DIAP1, AKP- and KP-Jafrac2 were cloned into pGEX6P-1 (Pharmacia) for protein production in bacteria. The factor Xa cleavage site was inserted in front of AKP- or KP-Jafrac2. GST fusion proteins were purified and incubated over night with factor Xa (Pharmacia) according to the manufacturer’s instructions. Cleaved products were further purified by His affinity purification (Qiagen) and dialyzed in PBS.
Cell fractionation
Cell fractionation was performed as described previously (Kanuka et al., 1999; Verhagen et al., 2000, 2002; Ekert et al., 2001). In brief, S2 cells were seeded in 75 cm2 flasks and left untreated or UV irradiated. Post UV (8 h) treatment cells were harvested, resuspended in 1 ml digitonin lysis buffer (0.025% digitonin, 250 mM sucrose, 20 mM HEPES pH 7.5, 5 mM MgCl, 10 mM KCl, 1 mM EGTA, 1m M EDTA, Complete™ protease inhibitor) and homogenized by a 19 G needle. Cell extracts were centrifuged at 800 g for 10 min. The supernatant was recovered and centrifuged at 10 000 g for 10 min. The resultant pellet represents the membrane fraction and the supernatant the cytosolic fraction. For the ER stress experiment, NIH 3T3 cells stably expressing Jafrac2 were treated with 35 µM brefeldin A or 24 µM tunicamycin and harvested 24 h later.
P-element-mediated germline transformation
Transgenic flies were generated by microinjecting pExpress-based akp or kp-jafrac2, ava, va, akp or kp-rpr constructs according to standard protocols. Several independent lines were established and mapped to individual chromosomes. The following fly strains were used for the genetic analysis: GMR-diap1, GMR-p35, th4, UAS-pro-Droncw.
Supplementary data
Supplementary data are avilable at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Mathew Freeman for the GFP–Spitz construct, B.Seraphin for the TAP construct, A.Varshavsky for the ubiquitin fusion construct, David Robertson for help with the confocal microscopy, Victoria Cowling for technical assistance and the Downward laboratory for helpful discussions. We especially thank David Baker for critical reading of the manuscript.
References
- Chen P., Nordstrom,W., Gish,B. and Abrams,J.M. (1996) grim, a novel cell death gene in Drosophila. Genes Dev., 10, 1773–1782. [DOI] [PubMed] [Google Scholar]
- Crook N.E., Clem,R.J. and Miller,L.K. (1993) An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol., 67, 2168–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorstyn L., Read,S., Cakouros,D., Huh,J.R., Hay,B.A. and Kumar,S. (2002) The role of cytochrome c in caspase activation in Drosophila melanogaster cells. J. Cell Biol., 156, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekert P.G., Silke,J., Hawkins,C.J., Verhagen,A.M. and Vaux,D.L. (2001) DIABLO promotes apoptosis by removing MIHA/XIAP from processed caspase 9. J. Cell Biol., 152, 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein J.C., Nigel,N.J., Juin,P., Evan,G. and Green,D.R. (2000) The coordinated release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol., 2, 156–162. [DOI] [PubMed] [Google Scholar]
- Goyal L., McCall,K., Agapite,J., Hartwieg,E. and Steller,H. (2000) Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J., 19, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grether M.E., Abrams,J.M., Agapite,J., White,K. and Steller,H. (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev., 9, 1694–1708. [DOI] [PubMed] [Google Scholar]
- Hawkins C.J., Wang,S.L. and Hay,B.A. (1999) A cloning method to identify caspases and their regulators in yeast: identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc. Natl Acad. Sci. USA, 96, 2885–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins C.J., Yoo,S.J., Peterson,E.P., Wang,S.L., Vernooy,S.Y. and Hay,B.A. (2000) The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM. J. Biol. Chem., 275, 27084–27093. [DOI] [PubMed] [Google Scholar]
- Hay B.A., Wolff,T. and Rubin,G.M. (1994) Expression of baculovirus P35 prevents cell death in Drosophila. Development, 120, 2121–2129. [DOI] [PubMed] [Google Scholar]
- Hay B.A., Wassarman,D.A. and Rubin,G.M. (1995) Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell, 83, 1253–1262. [DOI] [PubMed] [Google Scholar]
- Hays R., Wickline,L. and Cagan,R. (2002) Morgue mediates apoptosis in the Drosophila retina by promoting degradation of DIAP1. Nat. Cell Biol., 4, 425–431. [DOI] [PubMed] [Google Scholar]
- Holley C.L., Olson,M.R., Colon-Ramos,D.A. and Kornbluth,S. (2002) Reaper eliminates IAP protein through stimulated IAP degradation and generalized translational inhibition. Nat. Cell Biol., 4, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser W.J., Vucic,D. and Miller,L.K. (1998) The Drosophila inhibitor of apoptosis D-IAP1 suppresses cell death induced by the caspase drICE. FEBS Lett., 440, 243–248. [DOI] [PubMed] [Google Scholar]
- Kanuka H., Hisahara,S., Sawamoto,K., Shoji,S., Okano,H. and Miura,M. (1999) Proapoptotic activity of Caenorhabditis elegans CED-4 protein in Drosophila: implicated mechanisms for caspase activation. Proc. Natl Acad. Sci. USA, 96, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.R., Urban,S., Garvey,C.F. and Freeman,M. (2001) Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell, 107, 161–171. [DOI] [PubMed] [Google Scholar]
- Lisi S., Mazzon,I. and White,K. (2000) Diverse domains of THREAD/DIAP1 are required to inhibit apoptosis induced by REAPER and HID in Drosophila. Genetics, 154, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier P., Finch,A. and Evan,G. (2000a) Apoptosis in development. Nature, 407, 796–801. [DOI] [PubMed] [Google Scholar]
- Meier P., Silke,J., Leevers,S.J. and Evan,G.I. (2000b) The Drosophila caspase DRONC is regulated by DIAP1. EMBO J., 19, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollmann M. et al. (2000) Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell, 101, 91–101. [DOI] [PubMed] [Google Scholar]
- Pahl H.L. (1999) Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol. Rev., 79, 683–701. [DOI] [PubMed] [Google Scholar]
- Quinn L.M., Dorstyn,L., Mills,K., Colussi,P.A., Chen,P., Coombe,M., Abrams,J., Kumar,S. and Richardson,H. (2000) An essential role for the caspase dronc in developmentally programmed cell death in Drosophila. J. Biol. Chem., 275, 40416–40424. [DOI] [PubMed] [Google Scholar]
- Radyuk S.N., Klichko,V.I., Spinola,B., Sohal,R.S. and Orr,W.C. (2001) The peroxiredoxin gene family in Drosophila melanogaster. Free Radic. Biol. Med., 31, 1090–1100. [DOI] [PubMed] [Google Scholar]
- Rigaut G., Shevchenko,A., Rutz,B., Wilm,M., Mann,M. and Seraphin,B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol., 17, 1030–1032. [DOI] [PubMed] [Google Scholar]
- Rodriguez A., Chen,P., Oliver,H. and Abrams,J.M. (2002) Unrestrained caspase-dependent cell death caused by loss of Diap1 function requires the Drosophila Apaf-1 homolog, Dark. EMBO J., 21, 2189–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J., Agudo,M., Van Damme,J., Vandekerckhove,J. and Santaren,J.F. (2000) Polypeptides differentially expressed in imaginal discs define the peroxiredoxin family of genes in Drosophila. Eur. J. Biochem., 267, 487–497. [DOI] [PubMed] [Google Scholar]
- Ryoo H.D., Bergmann,A., Gonen,H., Ciechanover,A. and Steller,H. (2002) Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nat. Cell Biol., 4, 432–438. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S. and Duckett,C.S. (2002) Apoptosis: IAP proteins: blocking the road to death’s door. Nat. Rev. Mol. Cell Biol., 3, 401–410. [DOI] [PubMed] [Google Scholar]
- Shi Y. (2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell, 9, 459–470. [DOI] [PubMed] [Google Scholar]
- Tenev T., Bohmer,S.A., Kaufmann,R., Frese,S., Bittorf,T., Beckers,T. and Bohmer,F.D. (2000) Perinuclear localization of the protein-tyrosine phosphatase SHP-1 and inhibition of epidermal growth factor-stimulated STAT1/3 activation in A431 cells. Eur. J. Cell Biol., 79, 261–271. [DOI] [PubMed] [Google Scholar]
- Thompson C.B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science, 267, 1456–1462. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A. and Lazebnik,Y. (1998) Caspases: enemies within. Science, 281, 1312–1316. [DOI] [PubMed] [Google Scholar]
- Urban S., Lee,J.R. and Freeman,M. (2001) Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell, 107, 173–182. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. (2000) Ubiquitin fusion technique and its descendants. Methods Enzymol., 327, 578–593. [DOI] [PubMed] [Google Scholar]
- Verhagen A.M., Ekert,P.G., Pakusch,M., Silke,J., Connolly,L.M., Reid,G.E., Moritz,R.L., Simpson,R.J. and Vaux,D.L. (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell, 102, 43–53. [DOI] [PubMed] [Google Scholar]
- Verhagen A.M. (2002) HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol. Chem., 277, 445–454. [DOI] [PubMed] [Google Scholar]
- Vucic D., Kaiser,W.J. and Miller,L.K. (1998) A mutational analysis of the baculovirus inhibitor of apoptosis Op-IAP. J. Biol. Chem., 273, 33915–33921. [DOI] [PubMed] [Google Scholar]
- Wang S.L., Hawkins,C.J., Yoo,S.J., Muller,H.A. and Hay,B.A. (1999) The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell, 98, 453–463. [DOI] [PubMed] [Google Scholar]
- White K., Grether,M.E., Abrams,J.M., Young,L., Farrell,K. and Steller,H. (1994) Genetic control of programmed cell death in Drosophila. Science, 264, 677–683. [DOI] [PubMed] [Google Scholar]
- Wilson R., Goyal,L., Ditzel,M., Zachariou,A., Baker,D.A., Agapite,J., Steller,H. and Meier,P. (2002) The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat. Cell Biol., 4, 445–450. [DOI] [PubMed] [Google Scholar]
- Wing J.P. et al. (2002) Drosophila morgue is a novel F box/ubiquitin conjugase domain protein important in grim-reaper mediated programmed cell death. Nat. Cell Biol., 4, 451–456. [DOI] [PubMed] [Google Scholar]
- Wolf B.B. and Green,D.R. (2002) Apoptosis: letting slip the dogs of war. Curr. Biol., 12, R177–R179. [DOI] [PubMed] [Google Scholar]
- Wu G., Chai,J., Suber,T.L., Wu,J.W., Du,C., Wang,X. and Shi,Y. (2000) Structural basis of IAP recognition by Smac/DIABLO. Nature, 408, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Wu J.W., Cocina,A.E., Chai,J., Hay,B.A. and Shi,Y. (2001) Structural analysis of a functional DIAP1 fragment bound to grim and hid peptides. Mol. Cell, 8, 95–104. [DOI] [PubMed] [Google Scholar]
- Yoo S.J. et al. (2002) Apoptosis inducers Hid, Rpr and Grim negatively regulate levels of the caspase inhibitor DIAP1 by distinct mechanisms. Nat. Cell Biol., 4, 416–424. [DOI] [PubMed] [Google Scholar]