

Abstract
Collagen XVII, a type II transmembrane protein and epithelial adhesion molecule, can be proteolytically shed from the cell surface to generate a soluble collagen. Here we investigated the release of the ectodomain and identified the enzymes involved. After surface biotinylation of keratinocytes, the ectodomain was detectable in the medium within minutes and remained stable for >48 h. Shedding was enhanced by phorbol esters and inhibited by metalloprotease inhibitors, including hydroxamates and TIMP-3, but not by inhibitors of other protease classes or by TIMP-2. This profile implicated MMPs or ADAMs as candidate sheddases. MMP-2, MMP-9 and MT1-MMP were excluded, but TACE, ADAM-10 and ADAM-9 were shown to be expressed in keratinocytes and to be actively involved. Transfection with cDNAs for the three ADAMs resulted in increased shedding and, vice versa, in TACE-deficient cells shedding was significantly reduced, indicating that transmembrane collagen XVII represents a novel class of substrates for ADAMs. Functionally, release of the ectodomain of collagen XVII from the cell surface was associated with altered keratinocyte motility in vitro.
Keywords: basement membrane/hemidesmosome/metalloprotease/secretase/skin
Introduction
The family of type II transmembrane collagens includes collagens XIII, XVII and ectodysplasin A as adhesion molecules and/or ligand binding receptors on epithelial cell surfaces, and the types I and II macrophage scavenger receptors, and the MARCO protein, which have been proposed to function as receptors in host defense (Krieger and Herz, 1994; Schäcke et al., 1998; Snellman et al., 2000; Elomaa et al., 2001; Sankala et al., 2002). Collagens XIII, XVII and ectodysplasin A are epidermal proteins with a collagenous ectodomain that colocalizes with integrins on basal keratinocyte surfaces. Collagen XIII and ectodysplasin A are associated with α3β1 integrin and the actin cytoskeleton in focal contacts (Ezer et al., 1999; Snellman et al., 2000), and collagen XVII with α6β4 integrin and the keratin intermediate filaments in hemidesmosomes (Borradori and Sonnenberg, 1999). These features imply specific functions in epithelial cell adhesion and suggest putative ligand interactions between the transmembrane collagens and the integrins in the basal keratinocytes.
Collagen XVII is a component of hemidesmosomes which mediate adhesion of keratinocytes and certain other epithelial cells to the underlying basement membrane. Its intracellular domain interacts with the β4-integrin subunit and is essential for the incorporation of the molecule into the hemidesmosome, and the extracellular juxtamembranous NC16A domain binds to the α6-integrin subunit (Borradori and Sonnenberg, 1999). Each collagen XVII molecule is a trimer of three 180 kDa α1(XVII) chains, with a globular intracellular N-terminal domain of 466 amino acids, a short transmembrane domain of 23 amino acids and an extracellular C-terminal domain of 1008 amino acids in length. The ectodomain consists of 15 collagenous subdomains characterized by typical collagenous -G-X-Y- repeat sequences, intervened by 16 short noncollagenous sequences. The overall structure is flexible, rod-like and triple helical, with a significant thermal stability (Schäcke et al., 1998; Tasanen et al., 2000a; Areida et al., 2001). The critical role of collagen XVII in maintaining epithelial adhesion is best demonstrated by human skin diseases. Mutations in the collagen XVII gene, COL17A, lead to junctional epidermolysis bullosa, a blistering disorder with epidermal detachment from the basement membrane as a consequence of minor friction (McGrath et al., 1995; Tasanen et al., 2000b). In autoimmune diseases of the pemphigoid group, autoantibodies target both full-length collagen XVII and the shed ectodomain and lead to diminished epidermal adhesion and skin blistering (Schumann et al., 2000).
Collagen XVII was the first collagen shown to be consitutively shed from the cell surface, yielding a soluble basement membrane collagen (Hirako et al., 1998; Schäcke et al., 1998). Subsequently, shedding of collagen XIII (Snellman et al., 2000) and ectodysplasin A (Chen et al., 2001; Elomaa et al., 2001) was described. Although furin has been predicted to be involved in the shedding cascade (Schäcke et al., 1998; Snellman et al., 2000; Chen et al., 2001), it has remained unclear whether it cleaves directly the ectodomains of all of these proteins or is perhaps involved in the activation of other, physiological, sheddases. In the present study we demonstrate that three members of the ADAM family act as physiological sheddases of collagen XVII, and that furin is likely to be involved in the activation of these enzymes.
Results
Domain-specific antibodies define the shed ectodomain of collagen XVII
For characterization of the two forms of collagen XVII, two novel antibodies, Endo-2, recognizing the intracellular domain, and Ecto-5, against the distal-most 50 amino acids of the extracellular C-terminus of collagen XVII (Figure 1), were produced. The antibody NC16A to the juxtamembranous extracellular domain (Schumann et al., 2000) was also used. Since both NC16A and Ecto-5 recognized full-length collagen XVII and the soluble ectodomain, the authentic shedding product clearly spans almost the entire extracellular domain of collagen XVII. Correspondingly, Endo-2 recognized only the full-length molecule (Figure 1). Thus, the sheddase cleavage site is predicted to localize close to the cell membrane within the extracellular NC16A domain.

Fig. 1. Schematic representation of collagen XVII and the epitope regions recognized by antibodies. The full-length collagen XVII is a type II integral transmembrane protein, which is cleaved to yield a soluble ectodomain. White bars, collagenous sequences; light gray bars, non-collagenous sequences; black line, the cell plasma membrane. Dark gray bars, epitope regions of the domain-specific antibodies. Endo-2 recognizes the intracellular domain, NC-16A the juxtamembranous extracellular NC16a-domain, and Ecto-1 (Schäcke et al., 1998) and Ecto-5 the distal C-terminus of the molecule. The lower panel shows immunoblots of keratinocyte cell layer (C) and media (M) with the above antibodies. Ecto-1, Ecto-5, and NC16A recognized both the 180 kDa full-length collagen XVII and the 120 kDa soluble ectodomain. In contrast, Endo-2 recognized only full-length collagen XVII.
Release of collagen XVII ectodomain from the cell surface
Human keratinocytes were surface biotinylated, washed carefully, and the release of the labeled ectodomain into fresh medium was followed over time. The biotinylated ectodomain could be immunoprecipitated from the medium after 9 min, and after 2 h significant amounts were recovered (Figure 2). Full-length collagen XVII was progressively cleaved, and after 48 h no intact molecules were found in the cell extracts. Interestingly, the intact ectodomain was still recovered from the medium after 72–96 h, indicating a high stability of the soluble form of collagen XVII. The ectodomain was purified from the medium by immunoaffinity chromatography and subjected to N-terminal sequencing. However, despite repeated trials with sufficient amounts of protein the sequence remained elusive, because the newly generated N-terminus was blocked.

Fig. 2. Shedding of collagen XVII ectodomain. (A) Time-chase experiments with normal human keratinocytes. The cells were surface biotinylated, chased with biotin-free medium and cultured for 96 h. Aliquots of culture medium and cell extract were immunoprecipitated with antibody Ecto-1 or Endo-2 and analyzed by protein blotting with streptavidin–alkaline phosphatase. The lower panel shows the densitometric analysis of the signals. Note that the intact ectodomain was stable in the medium for at least 72 h. (B) Shedding was stimulated by phorbol esters and IL-1β. Keratinocytes were cultured for 24 h in the presence of 100 nM PMA or IL-1β (5 ng/ml). Aliquots were drawn at regular intervals and analyzed by immunoblotting with Endo-2 and NC16A antibodies. The signals were analyzed densitometrically with TotalLab1D (Phoretics) software.
Modulation of shedding
For characterization of the enzymes involved in the shedding of collagen XVII, keratinocytes were cultured in the presence of protease inhibitors (Table I). Release of the ectodomain was completely inhibited by phenanthroline and by the MMP- and sheddase-targeting hydroxamate derivates BB 3103, BB 3241, and IC-3. About 50% inhibition was observed with 25 µM hydroxamate FN-439 and with 500 nM TIMP-3, ∼10% with 500 nM TIMP-1, but none with the aspartate protease inhibitor pepstatin A, the cystein peptidase inhibitor E-64, or the serine protease inhibitors AEBSF, aprotinin, or benzamidine. In addition, no effect was seen with TIMP-2 or with CTTHWGFTLC, a specific cyclic peptide inhibitor of MMP-2 and MMP-9.
Table I. Effect of proteinase inhibitors on shedding of collagen XVII.
| Inhibitor | Concentration (µM) | Inhibition ± SD (%) |
|---|---|---|
| None | 0 | |
| 1,10-o-phenanthroline | 100 | 91 ± 9 |
| Pepstatin A | 100 | 3 ± 4 |
| E-64 | 100 | 8 ± 8 |
| Phosphoramidon | 10 | 4 ± 6 |
| AEBSF | 1000 | 5 ± 11 |
| Aprotinin | 25 | 11 ± 13 |
| Benzamidine | 1000 | 14 ± 9 |
| FN-439 | 20 | 20 ± 7 |
| 200 | 44 ± 10 | |
| BB 3103 | 10 | 87 ± 16 |
| BB 3241 | 10 | 92 ± 9 |
| IC-3 (TAPI) | 25 | 95 ± 4 |
| TIMP-1 | 0.5 | 11 ± 4 |
| TIMP-2 | 1 | 1 ± 4 |
| TIMP-3 | 0.15 | 15 ± 4 |
| 0.5 | 51 ± 4 | |
| Selective gelatinase inhibitora | 100 | 4 ± 3 |
aCyclic synthetic peptide: CTTHWGFTLC.
Shedding in the presence or absence of inhibitors was analyzed by immunoblotting followed by semiquantitative densitometry of the signals. The inhibition is expressed as a percentage ± SD (n = 3) of controls.
Potential stimulators of collagen XVII shedding were also added to keratinocytes. No effect on shedding of collagen XVII was observed with 5 ng/ml TNF-α or 5–10 ng/ml TGF-β2 (not shown). In contrast, 6 h after addition of 5 ng/ml IL-1β, increased ectodomain release, with a concomitant decrease of full-length collagen XVII, was seen (Figure 2B). Addition of 100 nM phorbol 12-myristate 13-acetate (PMA) induced shedding after 24 h (Figure 2B). Both agents are well-known stimulators of shedding and inducers of several ADAMs (Hooper et al., 1997; Flannery et al., 1999).
Exclusion of furin, BMP-1, MMP-2, MMP-9 and MT1-MMP as collagen XVII sheddases
The above inhibitor and stimulator profiles indicated that the protease(s) involved in shedding of collagen XVII belong(s) to the metalloproteinases, e.g. MMPs or sheddases of the ADAM family. Enzymes associated with keratinocyte surfaces, such as furin, BMP-1, MMP-2, MMP-9 and MT1-MMP (MMP-14), were tested for their capacity to cleave authentic purified collagen XVII (Figure 3A). No cleavage was observed with recombinant furin or BMP-1. All three MMPs cleaved collagen XVII into a 140 kDa fragment in vitro. Immunoblotting with the antibody Endo-2 demonstrated that in addition to the ectodomain, the fragment contained part of the endodomain and, therefore, this cleavage was not physiologically relevant (not shown). The observations were corroborated by studies on MT1-MMP- and MMP-2-deficient cells. After 24 h in culture, the shed ectodomain and full- length collagen XVII were analyzed by immunoblotting (Figure 3B). MMP-2-deficient human gastric carcinoma cells, which had only low MT1-MMP expression, and primary MT1-MMP-deficient murine keratinocytes generated the ectodomain as efficiently as control keratinocytes (Figure 3B), indicating that these MMPs were not involved in the shedding of collagen XVII.

Fig. 3. Furin, BMP-1, MMP-2, MMP-9 and MT1-MMP are not directly involved in the processing of collagen XVII. (A) Purified native collagen XVII was digested with recombinant human MT1-MMP, MMP-2, MMP-9, furin and BMP-1. The digests in the absence (–) or in the presence (+) of 5 mM EDTA were immunoblotted with antibody Ecto-1. Furin and BMP-1 did not cleave collagen XVII. MMP-2, MMP-9 and MT1-MMP generated a 140 kDa fragment which was distinct from the authentic 120 kDa fragment in culture medium (Med). (B) Ectodomain shedding in MMP-2-deficient gastric carcinoma cells (left panel) and MT1-MMP-deficient murine keratinocytes (right panel). In both cultures the 120 kDa ectodomain was generated similarly to controls. C, cell extract; M, medium. Immunoblotting was with a mixture of antibodies Endo-2 and NC16A (left panel) or Endo-2 and MO-NC16A (right panel).
Support for the involvement of TACE, instead of the above MMPs, was obtained from comparisons of the inhibitor profiles of metalloproteases and collagen XVII shedding. Recombinant human MMP-2, MMP-9 and TACE were used to cleave fluorescein-coupled gelatin or internally quenched fluorescent oligopeptides. Inhibitors of collagen XVII shedding, i.e. IC-3, BB 3241 and BB 3103 strongly inhibited MMP-2, MMP-9 and TACE, whereas the hydroxamate FN-439, a weak inhibitor of shedding, had only little effect on the activity of the MMPs and TACE (Table II). In contrast, the gelatin inhibitor, which did not inhibit collagen XVII shedding, strongly inhibited MMP-2 and MMP-9, but not TACE.
Table II. Effect of proteinase inhibitors on recombinant proteases.
| Inhibitor |
Concentration (µM) |
% activity |
|||
|---|---|---|---|---|---|
| MMP-2 | MMP-9 | TACE | Furin | ||
| BB 3103 | 25 | 0 ± 4 | 1 ± 2 | 0 ± 4 | 101 ± 12 |
| BB 3241 | 25 | 0.5 ± 3 | 0 ± 3 | 0 ± 1 | 102 ± 9 |
| FN-439 | 25 | 36 ± 3 | 58 ± 3 | 45 ± 7 | 93 ± 3 |
| Gelatinase inhibitora | 25 | 19 ± 11 | 10 ± 10 | 98 ± 12 | 98 ± 9 |
| IC-3 (TAPI) | 25 | 0 ± 4 | 0.3 ± 1 | 0 ± 1 | 102 ± 9 |
| Furin inhibitorb | 100 | 97 ± 4 | 88 ± 3 | 97 ± 8 | 6 ± 4 |
aCyclic synthetic peptide: CTTHWGFTLC.
bDecanoyl-RVKR-chloromethyl ketone.
The enzymes (5 nM MMP-2; 4 nM MMP-9; 20 nM CAT-DIS-TACE; 10 nM human furin) and inhibitors were preincubated for 30 min at room temperature and the fluorogenic substrate was added as described in Materials and methods. The proteolytic activity is expressed as a percentage ± SD (n = 3) in relation to controls.
The role of furin in collagen XVII shedding was of interest, since a furin-specific chloromethyl ketone inhib itor prevents collagen XVII processing (Schäcke et al., 1998). The fact that recombinant furin did not cleave purified collagen XVII in vitro (Figure 3A) and was not sensitive to hydroxamates (Table II) suggested that furin is indirectly involved in the activation of a convertase. To verify this, TACE expression was analyzed in keratinocytes treated with the furin inhibitor (Figure 4C). Immunoblots of control cell lysates showed two distinct TACE-positive bands, representing the proform and the mature form of the enzyme (Figure 4B and C). Addition of 50 µM furin inhibitor to the cultures inhibited the activation of TACE, as shown by nearly complete absence of the mature form (Figure 4C). In contrast, addition of BB 3103 did not block TACE activation, as shown by the presence of the proform and the mature form in both controls and hydroxamate-treated samples (Figure 4C). Importantly, 10 mM 1,10-o-phenanthroline was added to the extraction buffers, since it was recently shown that an additional form of TACE lacking the pro- and cytoplasmatic domains is detected if cell lysates are prepared without 1,10-o-phenanthroline (Schlondorff et al., 2000).

Fig. 4. Expression of ADAMs in human keratinocytes. (A) Immuno fluorescence staining of collagen XVII and ADAMs in the skin. Collagen XVII antibody NC16A produced a linear signal at the basement membrane zone (a). TACE (b) and ADAM-9 (c) immunoreactivity was stronger in the lower epidermis, in the neighborhood of collagen XVII, whereas ADAM-10 immunoreactivity (d) was distributed throughout the entire epidermis. Scale bar: 250 µm. (B) Immunoblot of keratinocyte extracts with antibodies to the ectodomains of TACE and ADAM-9, and the endodomain of ADAM-10. TACE and ADAM-9 antibodies recognized two distinct protein bands of ∼120 and 85 kDa, and 110 and 79 kDa, respectively, representing the proform and the active form of the enzymes. ADAM-10 antibody recognized one band of ∼60 kDa, corresponding to the active form. (C) Activation of TACE in keratinocytes was inhibited by a furin inhibitor, but not by hydroxamates. Keratinocytes were cultured for 24 h in serum-free medium with or without 50 µM furin inhibitor decanoyl-RVKR-chloromethyl ketone or 25 µM hydroxamate BB 3103. Keratinocyte lysates were immunoblotted with TACE antibodies. The furin inhibitor, but not BB 3103, prevented the conversion of pro-TACE to active TACE. Molecular weight markers are shown on the left.
Expression of ADAMs in human keratinocytes
Based on the above observations, the ADAM family proteinases were considered as candidate sheddases for collagen XVII. RT–PCR amplification of human keratinocyte mRNA demonstrated that TACE and the related sheddases ADAM-9 and ADAM-10 were expressed in these cells (not shown). Immunofluorescence staining of human skin with antibodies to TACE and ADAM-9 produced a positive signal in the lower epidermis in the vicinity of collagen XVII (Figure 4A), and ADAM-10 was found within the entire epidermis. Immunoblotting of keratinocyte lysates (Figure 4B) demonstrated that the proform and the active form of TACE (Schlondorff et al., 2000) and ADAM-9 (Roghani et al., 1999), and the active form of ADAM-10 (Qi et al., 1999) were present in these cells.
TACE and other ADAMs contribute to shedding of collagen XVII
To assess the role of TACE in collagen XVII shedding, HaCaT cells were transiently transfected with full-length murine TACE cDNA. Since TACE is expressed in many cell lines, including HaCaT, different cDNA concentrations (0–20 µg) were used. The transfection efficiency was estimated by analysis of TACE protein expression with immunofluorescence staining and determination of the relative number of transfected cells with a strongly positive TACE signal within a defined culture area. A transfection efficiency of ∼20% was obtained with 20 µg of TACE cDNA (Figure 5A). A dose-dependent increase in shedding, with a concomitant decrease in full-length collagen XVII, was observed with increasing cDNA concentrations. Transfection with 20 µg of TACE cDNA led to a nearly 2-fold increase of shedding (Figure 5B). Enhanced shedding in transfected cells was effectively inhibited with the hydroxamates BB 3103 or BB 3241 (Figure 6).
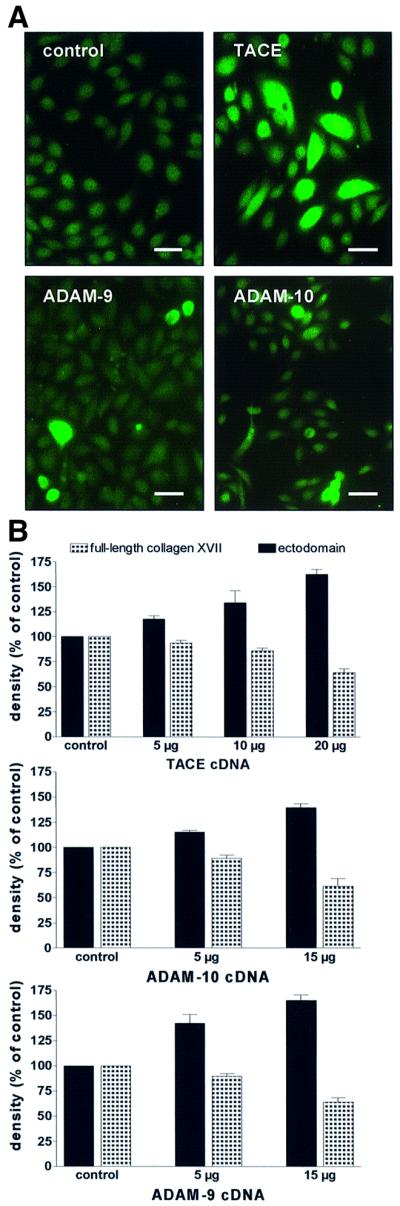
Fig. 5. Transient transfections of HaCaT cells with cDNAs for TACE, ADAM-9 and ADAM-10. (A) Immunofluorescence staining of transfected cells. Cells transfected with empty vector (control) remained negative, but ADAM-transfected cells showed strong positive signals. Scale bar: 100 µm. (B) HaCaT cells were transfected with different concentrations of cDNA for TACE (upper panel), ADAM-10 (middle panel) and ADAM-9 (lower panel) and shedding of collagen XVII was assessed with immunoblotting with a mixture of antibodies Endo-2, NC16A and Ecto-1. Densitometric analysis of the signals (expressed as a percentage of the control ± SD; n = 3) showed a dose-dependent increase of ectodomain shedding, with a concomitant decrease of full-length collagen XVII.

Fig. 6. Hydroxamates inhibit increased collagen XVII shedding in TACE-transfected cells. The cells were electroporated with empty vector (control), or with 10 or 20 µg of TACE cDNA. After 24 h the cells were washed, 10 µM BB 3103 or BB 3241 added, and after 6 h the ectodomain in the medium detected with immunoblotting using antibodies NC16A and Ecto-1. Both hydroxamates strongly inhibited shedding in this system.
In parallel, HaCaT cells were transfected with full-length cDNA for human ADAM-9 and bovine ADAM-10. The estimated transfection efficiencies with 15 µg of cDNA for ADAM-9 and ADAM-10 were 12 and 10%, respectively (Figure 5A). Transfection with 15 µg of ADAM-10 cDNA caused an ∼1.5-fold increase, and with 15 µg of ADAM-9 cDNA a 1.75-fold increase in shedding of the ectodomain (Figure 5B).
The prediction that TACE sheds collagen XVII was confirmed with keratinocytes derived from TACE- deficient mouse embryos. Compared with age-matched normal murine keratinocytes, shedding of collagen XVII was reduced by ∼40% in TACE-deficient cells (Figure 7A). In another approach, normal and TACE-deficient murine fibroblasts were transfected with collagen XVII cDNA, and shedding was analyzed. Similarly to TACE-deficient keratinocytes, transfected TACE-deficient fibroblasts shed collagen XVII less efficiently. Also in this case the reduction was ∼40% (Figure 7B).

Fig. 7. Decreased collagen XVII shedding in TACE-deficient cells. (A) Murine keratinocytes from wild-type (WT) and TACE –/– mice were analyzed for collagen XVII shedding in vitro. Cells (C) and media (M) were sampled after 6 h and immunoblotted with a mixture of antibodies MO-NC16A and Endo-2 (left panel). The relative intensity of the signals of three individual experiments is expressed as a percentage ± SD (right panel). (B) Wild-type (WT) and TACE –/– fibroblasts were transfected with human collagen XVII cDNA. After 2 days the cells were analyzed for collagen XVII shedding. Cells (C) and media (M) were sampled after 6 h and immunoblotted with a mixture of antibodies MO-NC16A and Endo-2 (left panel). The relative intensity of the signals of two different experiments is expressed as a percentage ± SD (right panel).
Shedding of collagen XVII ectodomain is associated with altered cell motility
Since the release of collagen XVII ectodomain from the keratinocyte surface is likely to influence the interaction of the cell with its environment, we analyzed cell motility after transfection of HaCaT cells with cDNAs for TACE, ADAM-9 and -10. For this purpose, in vitro wound closure assays were performed (Cha et al., 1996). In this assay, scrape wounds were generated in confluent serum-free HaCaT cultures and, after extensive washing with PBS, the cells were allowed to migrate into the denuded area for 22 h at 37°C. Cells transfected with an empty vector migrated into the wound and ‘re-epithelialized’ it to ∼50% in 11 h and fully in ∼22 h (Figure 8A). Cells transfected with one of the ADAMs were less motile, as indicated by a lower number of cells in the denuded area after 11 h and 22 h (Figure 8A).

Fig. 8. Increased collagen XVII shedding correlates with decreased cell motility. In wound closure assays cells migrate from the edges of a ‘scrape wound’ in the monolayer (interrupted lines) to cover the denuded space (between the lines). Scrape wounds were made in serum-free cultures, and after washing the cells were allowed to re-epithelialize for 22 h at 37°C. Micrographs of non-fixed cell layers at 0, 11, 16 or 22 h after wounding are shown. Scale bars: 200 µm. (A) Transfection of HaCaT cells with ADAM cDNA negatively regulated cell motility. Transfections were with 15 µg of empty vector (control), 15 µg of TACE, 15 µg of ADAM-10, or 15 µg of ADAM-9 cDNA. (B) Addition of purified collagen XVII ectodomain to normal human keratinocytes inhibited cell motility. After wounding, 1 nM collagen XVII ectodomain was added to the culture, and the cells were permitted to migrate into the denuded space for 16 h. Ectodomain-treated cells did not cover the denuded area as efficiently as control cells.
The inhibitory activity of collagen XVII shedding on cell motility was verified with the addition of ∼1 nM purified ectodomain to the culture medium after induction of the scrape wound. Similarly to the experiment described above, untreated control cells readily migrated into the scraped area, covering it to >50% in 16 h. In contrast, keratinocyte migration from the cut edge was decreased when the purified ectodomain was added to the culture medium (Figure 8B), suggesting that the shed ectodomain negatively regulates cell motility.
Discussion
A wide variety of proteins, including cytokines, growth factors and their receptors, and cell adhesion molecules, are synthesized as integral type I or type II transmembrane proteins that can be released by selective proteolysis from the cell surface (Hooper et al., 1997). The cleavage generally occurs close to the extracellular face of the membrane, generating a physiologically active protein. Release of soluble growth factors and cytokines may change their biological activities, whereas shedding of transmembrane receptors may render cells unresponsive to particular ligands, and the resulting soluble molecules may act to modulate the activity of their ligands (Black and White, 1998). Thus, the process of ectodomain shedding can influence the interaction of cells with their environment in multiple ways.
Many of the recently characterized sheddases are metalloproteinases (Hooper et al., 1997). They are local ized at the cell surface, or are themselves integral membrane proteins which after activation can act close to the cell membrane in the extracellular space. The prototype sheddase TACE is a member of the ADAM family and was designated as ADAM-17. The substrate spectrum of TACE is broad, including TNF-α, L-selectin, IL-6 receptor and the p75 TNF receptor (Moss et al., 1997; Merlos-Suarez et al., 1998; Peschon et al., 1998). Both TACE and ADAM-10 may also participate in the processing of amyloid precursor protein (Buxbaum et al., 1998; Amour et al., 2000). Another member of the family, ADAM-9, mediates the shedding of heparin-binding epidermal growth factor-like growth factor (Izumi et al., 1998). Kuzbanian, the Drosophila homolog of mammalian ADAM-10, releases the Notch ligand Delta from cells (Qi et al., 1999). Thus, the ADAMs constitute a family of sheddases that regulate plasma membrane composition and release soluble signaling molecules and ligand binding receptors from cells. In the present study, we expand the substrate spectrum of ADAM sheddases by showing that collagen XVII, a representative of the family of transmembrane collagens, is a substrate for TACE, ADAM-9 and ADAM-10.
Genetic evidence proves collagen XVII is an essential keratinocyte adhesion molecule. Mutations in the COL17A1 gene lead to severe epidermal dysadhesion and skin blistering in humans (McGrath et al., 1995). The shed ectodomain forms a soluble collagen which remains stable for days in keratinocyte media in vitro and which can be isolated from the epidermis and amniotic fluid in vivo, suggesting that it functions as a ligand-binding matrix molecule (Zone et al., 1990; Schumann et al., 2000).
The sheddase cleavage site within the ectodomain of collagen XVII remains elusive. Epitope mapping placed the putative site within the 76 amino acid NC16A domain adjacent to the cell membrane, but determination of the cleaved peptide bond was impeded by the fact that the newly generated N-terminus was blocked and not amenable to amino acid sequencing. Since shedding of collagen XVII in keratinocytes was completely inhibited by the synthetic furin inhibitor decanoyl-RVKR-chloromethyl ketone, and since the NC16A domain contains a conserved furin cleavage motif, RIRR, we first assumed that furin releases the ectodomain from the cell surface (Schäcke et al., 1998). However, recombinant furin did not cleave immunopurified full-length collagen XVII in vitro and, more importantly, shedding was completely blocked by hydroxamate inhibitors, such as BB 3103, BB 3241 or IC-3, which do not inhibit furin.
Based on the above data and the fact that the furin inhibitor completely blocked the activation of TACE proform, it is likely that furin activates ADAMs that shed collagen XVII. TACE, ADAM-10 and -9 are expressed as an inactive proform in keratinocytes. They all contain a conserved sequence in the prodomain which is comparable to the cysteine switch sequence of MMPs (Nagase and Wossner, 1999). In addition, in the boundary between the prodomain and the metalloproteinase domain there is a four basic amino acid consensus motif for furin cleavage (RRRR in ADAM-9, RKKR in ADAM-10 and RVKR in ADAM-17), suggesting that the mechanism of activation of these ADAMs includes a cysteine switch and proteolytic removal of the prodomain by furin (Merlos-Suarez et al., 1998; Lammich et al., 1999; Roghani et al., 1999; Schlondorff et al., 2000).
Our results demonstrate that all three ADAMs can shed collagen XVII in keratinocytes, since transient transfections with TACE, ADAM-9 and ADAM-10 lead to increased shedding of the ectodomain, and since TACE-deficient keratinocytes exhibited a remaining shedding activity of ∼60%. Three observations argue that TACE may be the major sheddase. First, shedding was sensitive to TIMP-3, but not to TIMP-1 or TIMP-2. TIMP-3 is known to inhibit shedding of proTNF-α and the adhesion molecules L-selectin, syndecan-1 and -4 (Borland et al., 1999; Fitzgerald et al., 2000). Secondly, expression of TIMP-3 in keratinocytes is downregulated by IL1-β (Airola et al., 1998), a stimulator of collagen XVII shedding. Thirdly, TIMP-3, but not TIMP-1 and TIMP-2, inhibited recombinant TACE in vitro (Amour et al., 1998). In contrast, the proteolytic activity of ADAM-10 and ADAM-9 is inhibited also by TIMP-1 (Dethlefsen et al., 1998; Amour et al., 2000). Compared with TIMP-3, an equimolar concentration of TIMP-1 inhibited collagen XVII shedding by only ∼10%. The synergistic action of the three ADAMs has also been observed in another system: all three enzymes have been postulated to represent potential α-secretases that can cleave amyloid precursor protein or corresponding peptides in vitro (Buxbaum et al., 1998; Schlondorff and Blobel, 1999; Fahrenholz et al., 2000).
The biological context of collagen XVII shedding is predicted to involve keratinocyte adhesion, differentiation and migration. These processes are very complex and involve a number of receptors and ligands depending on the spatial and temporal biological context (Aumailley and Gayraud, 1998; Nguyen et al., 2000), but a scenario could be visualized in which shedding of collagen XVII ectodomain loosens or releases the cell from some of its present binding partners and allows it to embark on other functions as required. We have previously shown that recombinant Col15 domain, the largest collagenous domain of collagen XVII, promotes epithelial cell adhesion in a β1 integrin-dependent manner, and that denatured Col15 binds cells more efficiently than its native counterpart (Tasanen et al., 2000a). We concluded that the shed ectodomain, which contains Col15, may exert this function, and that shedding may generate new cell binding sites. The hypothesis is supported by the in vitro re-epithelialization assays of the present study, which showed that increased shedding of collagen XVII induced by transient transfections with ADAM cDNA, or simulation of shedding through addition of exogenous ectodomain, was associated with decreased keratinocyte motility.
Basal keratinocytes in the skin express α2, α3, α5 and αV integrin subunits in association with the β1 subunit and utilize them for cell migration on collagen and laminin e.g. during wound healing. α2β1 is used for adhesion to collagen, but this integrin does not recognize the ectodomain of collagen XVII (Nykvist et al., 2001). α6β4 and α3β1 are laminin 5-receptors and regulate anchorage and motility of epithelial cells (Ryan et al., 1999). O’Toole et al. (1997) observed an inhibitory effect of laminin 5 on keratinocyte motility and showed that this could be reversed by blocking the α3 integrin receptor. Ryan et al. (1999) demonstrated that basal keratinocytes can utilize integrin α3β1 to interact with a hitherto unknown alternative ligand. Therefore, it is feasible to postulate that the shed ectodomain of collagen XVII is an alternative ligand that can bind to α3β1 integrin and block it for other, migration-enhancing ligands.
Taken together, it is likely that an interplay between stimulatory and inhibitory regulators determines collagen XVII shedding activity in a given cell at a given time. Future studies must show how the enzyme activities, enzyme–substrate interactions and ligand binding activities are modulated. Finally, the possibility cannot be excluded that hitherto unknown constitutive sheddases, like the as yet uncharacterized shedding activities of TRANCE/OPGL (Schlondorff et al., 2001), also participate in the regulation of keratinocyte adhesion and migration.
Materials and methods
Production of bacterial fusion proteins and domain-specific collagen XVII antibodies
For production of domain-specific antibodies to collagen XVII, bacterial fusion proteins were generated as described (Tasanen et al., 2000a,b). The fusion protein GST–Ecto-5 spanned amino acids 1447–1497 and GST–Endo-2 spanned amino acids 367–466 (Figure 1). The corresponding primers for Ecto-5 were: sense primer 5′-GCGCGGATCCGGCACTCCAGGACCCAAAGGT-3′ and antisense primer 5′-GCG CGAATTCTCACGGCTTGACAGCAATATC-3′; and for Endo-2, sense primer 5′-CGGGATCCAGCGGGAAGGTCTTTACAGCC-3′ and antisense primer 5′-GGAATTCACTTCCACCAGCTGCAGCA-3′. In parallel, a fusion protein corresponding to the NC16A domain of murine collagen XVII, MO-NC16A, spanning amino acids 499–573, was produced. The corresponding cDNA was amplified by RT–PCR from murine keratinocyte mRNA with sense primer 5′-GCGCGGATCCGAGGAGGTAAGAAAGCTGAAG-3′ and antisense primer 5′-GCGCGAATTCATCTGAGATTCCCGTTCTCCTG-3′. Rabbit antibodies to the fusion proteins were produced by standard procedures (Eurogentec, Ougrée, Belgium).
Cell cultures
Normal human keratinocytes were cultured in serum-free keratinocyte growth medium (KGM; Life Technologies, Grand Island, NY) as described (Schäcke et al., 1998). The human keratinocyte cell line HaCaT was a generous gift of Dr N.Fusening (German Cancer Research Center, Heidelberg, Germany). Prior to experiments, cells were grown in the presence of 50 µg/ml l-ascorbate for 48 h in order to allow for full prolyl- and lysyl hydroxylation of newly synthesized collagens.
TACE-deficient murine keratinocytes were isolated from taceΔZn/ΔZn mouse embryos (Dotto, 1999). The mice were homozygous for a targeted mutation in the metalloproteinase domain of TACE which leads to expression of inactive TACE (Peschon et al., 1998). MT1-MMP-deficient murine keratinocytes were isolated from the skin of MT1-MMP knockout mice (Zhou et al., 2000) and cultured as described above for human keratinocytes. The MMP-2-deficient human gastric carcinoma cell line (MKN 45), which had only low MT1-MMP expression, was obtained from the Japanese Cancer Resource Bank (JCRB). Murine fibrobasts and the MKN 45 cells were maintained in DMEM supplemented with l-glutamine, 10% fetal calf serum (FCS) and penicillin/streptomycin (Life Technologies GmbH, Karlsruhe, Germany).
Surface biotinylation and time chase experiments
For time chase analysis of the release of the ectodomain, semiconfluent keratinocytes were cultured with 50 µg/ml for 48 h, washed extensively and surface biotinylated with d-biotinoyl-ε-aminocaproic acid N-hydroxysuccinimide ester according to the manufacturer’s recommendations (Roche Biochemicals, Mannheim, Germany). After washing with phosphate-buffered saline (PBS) five times, the cells were cultured in fresh medium supplemented with ascorbate, and aliquots were collected at regular intervals over 96 h. Culture medium and cell extract were immunoprecipitated with the antibodies Ecto-1 and Endo-2, respectively. The precipitates were immunoblotted with a streptavidin-coupled alkaline phosphatase (Sigma, Germany) to detect biotinylated collagen XVII. The software TotalLab1D V1.00 from Phoetix (Biostep GmbH, Jahnsdorf, Germany) was used for semiquantitative analysis of the signals.
Protein extractions
For protein extractions, the media and cells were processed separately as described (Schäcke et al., 1998). As a modification, 10 mM 1,10-o-phenanthroline was added to the extraction buffer. After centrifugation at 13 000 g for 5 min at 4°C the supernatant was used for further analysis. The medium was collected on ice, proteinase inhibitors were added immediately and cell debris removed by centrifugation. The supernatant was precipitated with ethanol and resuspended in a buffer containing 50 mM Tris–HCl, 65 mM NaCl, 5 mM EDTA, 1 mM Pefabloc pH 7.4. Total protein content was determined with a micro-Lowry assay kit (Bio-Rad), and samples containing 50 µg of protein were analyzed by immunoblotting.
Immunoblotting and immunoprecipitation of collagen XVII
SDS–PAGE and immunoblotting with collagen XVII antibodies Endo-2, Ecto-5, murine or human NC16A (Schumann et al., 2000), or Ecto-1 (Schäcke et al., 1998) were performed using standard techniques. For immunoprecipitation, cell extracts were precleared with protein A–Sepharose (Amersham Pharmacia Biotech) for 30 min and centrifuged at 2000 g for 5 min. The supernatants were incubated with antibody Endo-2/protein A–Sepharose complexes at 4°C overnight. After extensive washing, collagen XVII was eluted with 0.1 M glycine pH 2.5 for 5 min and immediately neutralized with 1 M Tris.
Digestion of collagen XVII with recombinant proteases
Immunoprecipitated collagen XVII was digested with recombinant MMP-14 (MT1-MMP) and pro-MMP-9 (Alexis Deutschland, Grünberg, Germany), recombinant pro-MMP-2, recombinant BMP-1, and recombinant furin (Calbiochem-Novabiochem, Bad Soden, Germany). The proforms of MMP-2 and MMP-9 were activated with 1 mM APMA for 1 h at 37°C. Collagen XVII was incubated with 1–5 nM recombinant enzymes in 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 5 mM CaCl2, 15 µM ZnCl2 and 0.3% Brij 35 for up to 4 h at 37°C. The reactions were stopped with 5 mM EDTA and the samples precipitated with methanol/chloroform for immunoblotting with antibodies Ecto-1 and Endo-2.
Protease inhibitors
The following protease inhibitors were used: AEBSF [4-(2-aminoethyl)-benzolsulfonylfluoride hydrochloride; Merck, Darmstadt, Germany), phosphoramidon, 1,10-o-phenolanthroline, aprotinin, benzamidine, E-64 [l-3-carboxy-2,3-trans-epoxypropionyl-leucylamido(4-guanidino)butane], pepstatin A (Sigma), the MMP-inhibitor 4-Abz-Gly-Pro-D-Leu-D-Ala-NHOH (FN-439; Calbiochem-Novabiochem) and the furin inhibitor decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone (Calbiochem-Novabiochem). The hydroxamate inhibitors BB 3103, BB 3241 were from British Biotech and IC-3 from Immunex Corp. TIMP-1, -2 and TIMP-3 were from Chemicon International (Hofheim, Germany) and the selective gelatinase inhibitor H-Cys-Thr-Thr-His-Trp-Gly-Phe-Thr-Leu-Cys-OH (Koivunen et al., 1999) from Bachem (Heidelberg, Germany).
Collagen XVII shedding assay
Subconfluent keratinocytes or HaCaT cells were washed twice with PBS, and fresh medium containing 50 µg/ml ascorbate and inhibitors or stimulators was added for 4–6 h. Media were precipitated with methanol/chloroform and aliquots containing 75 µg of total protein were immunoblotted with antibody NC16A. Semiquantitative densitometry of the signals was performed using the computer program TotalLab1D (Biostep GmbH).
Protease activity assay with fluorescent substrates
Pro-MMPs were activated with 1 mM APMA for 60 min at 37°C, and 5 nM MMP-2 and 4 nM MMP-9 in buffer containing 50 mM Tris–HCl, 200 mM NaCl, 5 mM CaCl2, 1 µM ZnCl2, 0.05% Brij 35 pH 7.5, were incubated with 15 µg/ml fluorescein-conjugated gelatin (DQ-gelatin from pig skin; Molecular Probes), and the release of soluble fluorescent fragments was followed with a spectrofluorometer with microplate reader set (FluoroMax-2; Jobin Yvon, Munich, Germany; λex = 485 nm, λem = 530 nm). The activities of recombinant furin and CAT-DIS TACE were determined with internally quenched fluorescent substrates in assay buffer. For furin, 5 µM Abz-RVKRGLA-m-nitro-YD-OH (Bachem; λex = 320 nm, λem = 425 nm; Angliker et al., 1995) and for TACE, 15 µM MCA-PLAQAV-dinitrophenyl-2,3-diaminopropianyl-RSSSR-NH2 (Sigma; λex = 325 nm, λem = 395 nm) was used.
Immunofluorescence staining
Immunofuorescence staining of human skin and keratinocytes was performed as described (Schumann et al., 2000). As first antibodies, collagen XVII antibody NC16A, TACE antibodies against the extracellular pro- and metalloproteinase domains (Santa Cruz Biotechnology) and the intracellular C-terminus (Chemicon International, Hofheim, Germany), and antibodies to ADAM-10 endodomain (Chemicon) and ADAM-9 ectodomain (R&D Systems, Wiesbaden, Germany) were used. The second antibodies were fluorescein isothiocyanate (FITC)-labeled anti-rabbit immunoglobulins (Sigma).
Transient transfections
One day before transfection with pcDNA3 vectors containing full-length cDNAs for murine TACE, human ADAM-9 cDNA and bovine ADAM-10, HaCaT cells were seeded and grown to 70% confluence. The cells were harvested and counted, washed twice with PBS and resuspended in 1.25% DMSO in KGM. The cDNA constructs were added to 0.6 × 107 cells and electroporated with Gene Pulser (Bio-Rad) with 0.25 V and 960 µFD capacitance. After electroporation the cells were transferred into pre-warmed medium, washed after 24 h, and shedding assays were performed.
TACE-deficient murine fibrobasts were transfected with pcDNA3 vector encoding the full-length human collagen XVII (kindly provided by L.Borradori, Geneva). One day before transfection the cells were seeded at a density of ∼25 000 cells/cm2 and grown in DMEM/10% FCS. The transfection was performed with Effectene. Transfection reagent (Qiagen, Hilden, Germany) was used following the manufacturer’s recommendations. Cells were used 24–48 h post-transfection to obtain maximum levels of gene expression.
Migration assays
Scratch assays were performed with normal human keratinocytes or HaCaT cells in 6-well plates (Cha et al., 1996). The cells were grown to confluence and a 2 mm broad denuded scratch was produced in the culture with a pipette tip. After washing with PBS to remove all floating cells, the cells at the cut edge were allowed to migrate into the empty zone for 11–22 h. Photographs of identical locations within each scratch were taken at different time points. Since the doubling time of keratinocytes under these conditions was ∼30 h, the assay assessed mostly cell motility, not proliferation (Woodley et al., 1988).
Acknowledgments
Acknowledgements
The authors gratefully acknowledge the generous gifts of TACE cDNA, antibodies, IC-3 and TACE-deficient fibroblasts from J.Peschon, Immunex Corp., Seattle. The hydroxamate inhibitors BB 3103 and BB 3241 were kindly provided by H.Mills, British Biotech Pharmaceuticals Ltd, Oxon, UK. The cDNA for ADAM-9 was a generous gift from C.Blobel and G.Westkamp, Sloan–Kettering Institute, Memorial Sloan–Kettering Cancer Center, NY. We are grateful to P.Dotto and Y.Aoyama, Cutaneous Biology Research Center, Harvard Medical School, Charlestown, MA, for help with culturing murine keratinocytes, to W.Stetler-Stevenson, NCI, Bethesda, MD, for a gift of recombinant MMP-2, to R.Timpl, Max-Planck-Institute for Biochemistry, Martinsried, Germany, for a gift of recombinant BMP-1, and to N.Koshikawa and V.Quaranta, Scripps Research Institute, La Jolla, CA, for help with the MMP-2-deficient cell line. This work was supported in part by grants from the German Research Council, DFG (Grant Nos. SFB 293-B3 and BR1475/6-1) and from the European Union (Contract No. QLG1-CT-2001-02007) to L.B.-T., and from the Academy of Finland and the Oulu University Hospital to K.T.
References
- Airola K., Ahonen,M., Johansson,N., Heikkila,P., Kere,J., Kahari,V.M. and Saarialho-Kere,U.K. (1998) Human TIMP-3 is expressed during fetal development, hair growth cycle, and cancer progression. J. Histochem. Cytochem., 46, 437–447. [DOI] [PubMed] [Google Scholar]
- Amour A. et al. (1998) TNF-α converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett., 435, 39–44. [DOI] [PubMed] [Google Scholar]
- Amour A., Knight,C.G., Webster,A., Slocombe,P.M., Stephens,P.E., Knauper,V., Docherty,A.J. and Murphy,G. (2000) The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett., 473, 275–279. [DOI] [PubMed] [Google Scholar]
- Angliker H., Neumann,U., Molloy,S.S. and Thomas,G. (1995) Internally quenched fluorogenic substrate for furin. Anal. Biochem., 224, 409–412. [DOI] [PubMed] [Google Scholar]
- Areida S.K., Reinhardt,D.P., Muller,P.K., Fietzek,P.P., Kowitz.J., Marinkovich,M.P. and Notbohm,H. (2001) Properties of the collagen type XVII ectodomain. Evidence for N- to C-terminal triple helix folding. J. Biol. Chem., 276, 1594–1601. [DOI] [PubMed] [Google Scholar]
- Aumailley M. and Gayraud,B. (1998) Structure and biological activity of the extracellular matrix. J. Mol. Med., 76, 253–265. [DOI] [PubMed] [Google Scholar]
- Black R.A. and White,J.M. (1998) ADAMs: focus on the protease domain. Curr. Opin. Cell Biol., 10, 654–659. [DOI] [PubMed] [Google Scholar]
- Borland G., Murphy,G. and Ager,A. (1999) Tissue inhibitor of metalloproteinases-3 inhibits shedding of L-selectin from leukocytes. J. Biol. Chem., 274, 2810–2815. [DOI] [PubMed] [Google Scholar]
- Borradori L. and Sonnenberg,A. (1999) Structure and function of hemidesmosomes: more than simple adhesion complexes. J. Invest. Dermatol., 112, 411–418. [DOI] [PubMed] [Google Scholar]
- Buxbaum J.D. et al. (1998) Evidence that tumor necrosis factor α converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem., 273, 27765–27767. [DOI] [PubMed] [Google Scholar]
- Cha D., O’Brien,P., O’Toole,E.A., Woodley,D.T. and Hudson,L.G. (1996) Enhanced modulation of keratinocyte motility by transforming growth factor-α (TGF-α) relative to epidermal growth factor (EGF). J. Invest. Dermatol., 106, 590–597. [DOI] [PubMed] [Google Scholar]
- Chen Y., Molloy,S.S., Thomas,L., Gambee,J., Bachinger,H.P., Ferguson,B., Zonana,J., Thomas,G. and Morris,N.P. (2001) Mutations within a furin consensus sequence block proteolytic release of ectodysplasin-A and cause X-linked hypohidrotic ectodermal dysplasia. Proc. Natl Acad. Sci. USA, 98, 7218–7223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen S.M., Raab,G., Moses,M.A., Adam,R.M., Klagsbrun,M. and Freeman,M.R. (1998) Extracellular calcium influx stimulates metalloproteinase cleavage and secretion of heparin-binding EGF-like growth factor independently of protein kinase C. J. Cell. Biochem., 69, 143–153. [DOI] [PubMed] [Google Scholar]
- Dotto P.G. (1999) Signal transduction pathways controlling the switch between keratinocyte growth and differentiation. Crit. Rev. Oral Biol. Med., 10, 442–457. [DOI] [PubMed] [Google Scholar]
- Elomaa O., Pulkkinen,K., Hannelius,U., Mikkola,M., Saarialho-Kere,U. and Kere,J. (2001) Ectodysplasin is released by proteolytic shedding and binds to the EDAR protein. Hum. Mol. Genet., 10, 953–962. [DOI] [PubMed] [Google Scholar]
- Ezer S., Bayes,M., Elomaa,O., Schlessinger,D. and Kere,J. (1999) Ectodysplasin is a collagenous trimeric type II membrane protein with a tumor necrosis factor-like domain and co-localizes with cytoskeletal structures at lateral and apical surfaces of cells. Hum. Mol. Genet., 8, 2079–2086. [DOI] [PubMed] [Google Scholar]
- Fahrenholz F., Gilbert,S., Kojro,E., Lammich,S. and Postina,R. (2000) Alpha-secretase activity of the disintegrin metalloprotease ADAM 10. Influences of domain structure. Ann. N.Y. Acad. Sci., 920, 215–222. [DOI] [PubMed] [Google Scholar]
- Flannery C.R., Little,C.B., Caterson,B. and Hughes,C.E. (1999) Effects of culture conditions and exposure to catabolic stimulators (IL-1 and retinoic acid) on the expression of matrix metalloproteinases (MMPs) and disintegrin metalloproteinases (ADAMs) by articular cartilage chondrocytes. Matrix Biol., 18, 225–237. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M.L., Wang,Z., Park,P.W., Murphy,G. and Bernfield,M. (2000) Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell Biol., 148, 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirako Y., Usukura,J., Uematsu,J., Hashimoto,T., Kitajima,Y. and Owaribe,K. (1998) Cleavage of BP180, a 180-kDa bullous pemphigoid antigen, yields a 120-kDa collagenous extracellular polypeptide. J. Biol. Chem., 273, 9711–9717. [DOI] [PubMed] [Google Scholar]
- Hooper N.M., Karran,E.H. and Turner,A.J. (1997) Membrane protein secretases. Biochem. J., 321, 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y. et al. (1998) A metalloprotease-disintegrin, MDC9/meltrin-γ/ADAM9 and PKCδ are involved in TPA-induced ectodomain shedding of membrane-anchored heparin-binding EGF-like growth factor. EMBO J., 17, 7260–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen E. et al. (1999) Tumor targeting with a selective gelatinase inhibitor. Nat. Biotechnol., 17, 768–774. [DOI] [PubMed] [Google Scholar]
- Krieger M. and Herz,J. (1994) Structures and functions of multiligand lipoprotein receptors; macrophage scavenger receptors and LDL receptor-related protein (LRP). Annu. Rev. Biochem., 63, 601–637. [DOI] [PubMed] [Google Scholar]
- Lammich S., Kojro,E., Postina,R., Gilbert,S., Pfeiffer,R., Jasionowski,M., Haass,C. and Fahrenholz,F. (1999) Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl Acad. Sci. USA, 96, 3922–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J.A., Gatalica,B., Christiano,A.M., Li,K., Owaribe,K., McMillan,J.R., Eady,R.A. and Uitto,J. (1995) Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat. Genet., 11, 83–86. [DOI] [PubMed] [Google Scholar]
- Merlos-Suarez A., Fernandez-Larrea,J., Reddy,P., Baselga,J. and Arribas,J. (1998) Pro-tumor necrosis factor-α processing activity is tightly controlled by a component that does not affect notch processing. J. Biol. Chem., 273, 24955–24962. [DOI] [PubMed] [Google Scholar]
- Moss M.L. et al. (1997) Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature, 385, 733–736. [DOI] [PubMed] [Google Scholar]
- Nagase H. and Woessner,J.F.,Jr (1999) Matrix metalloproteinases. J. Biol. Chem., 274, 21491–21494. [DOI] [PubMed] [Google Scholar]
- Nguyen B.P., Ryan,M.C., Gil,S.G. and Carter,W.G. (2000) Deposition of laminin 5 in epidermal wounds regulates integrin signaling and adhesion. Curr. Opin. Cell Biol., 12, 554–562. [DOI] [PubMed] [Google Scholar]
- Nykvist P., Tasanen,K., Viitasalo,T., Kapyla,J., Jokinen,J., Bruckner-Tuderman,L. and Heino,J. (2001) The cell adhesion domain of type XVII collagen promotes integrin-mediated cell spreading by a novel mechanism. J. Biol. Chem., 276, 38673–38679. [DOI] [PubMed] [Google Scholar]
- O’Toole E.A., Marinkovich,M.P., Hoeffler,W.K., Furthmayr,H., and Woodley,D.T. (1997) Laminin-5 inhibits human keratinocyte migration. Exp. Cell Res., 233, 330–339. [DOI] [PubMed] [Google Scholar]
- Peschon J.J. et al. (1998) An essential role for ectodomain shedding in mammalian development. Science, 282, 1281–1284. [DOI] [PubMed] [Google Scholar]
- Qi H., Rand,M.D., Wu,X., Sestan,N., Wang,W., Rakic,P., Xu,T. and Artavanis-Tsakonas,S. (1999) Processing of the notch ligand delta by the metalloprotease Kuzbanian. Science, 283, 91–94. [DOI] [PubMed] [Google Scholar]
- Roghani M. et al. (1999) Metalloprotease-disintegrin MDC9: intra cellular maturation and catalytic activity. J. Biol. Chem., 274, 3531–3540. [DOI] [PubMed] [Google Scholar]
- Ryan M.C., Lee,K., Miyashita,Y. and Carter,W.G. (1999) Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J. Cell Biol., 145, 1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankala M., Brannstrom,A., Schulthess,T., Bergmann,U., Morgunova,E., Engel,J., Tryggvason,K. and Pikkarainen,T. (2002) Characterization of recombinant soluble macrophage scavenger receptor marco. J. Biol. Chem., 277, in press. [DOI] [PubMed] [Google Scholar]
- Schäcke H., Schumann,H., Hammami-Hauasli,N., Raghunath,M. and Bruckner-Tuderman,L. (1998) Two forms of collagen XVII in keratinocytes. A full-length transmembrane protein and a soluble ectodomain. J. Biol. Chem., 273, 25937–25943. [DOI] [PubMed] [Google Scholar]
- Schlondorff J. and Blobel,C.P. (1999) Metalloprotease-disintegrins: modular proteins capable of promoting cell–cell interactions and triggering signals by protein-ectodomain shedding. J. Cell Sci., 112, 3603–3617. [DOI] [PubMed] [Google Scholar]
- Schlondorff J., Becherer,J.D. and Blobel,C.P. (2000) Intracellular maturation and localization of the tumour necrosis factor α convertase (TACE). Biochem. J., 347, 131–138. [PMC free article] [PubMed] [Google Scholar]
- Schlondorff J., Lum,L. and Blobel,C.P. (2001) Biochemical and pharmacological criteria define two shedding activities for TRANCE/OPGL that are distinct from the tumor necrosis factor α convertase. J. Biol. Chem., 276, 14665–14674. [DOI] [PubMed] [Google Scholar]
- Schumann H., Baetge,J., Tasanen,K., Wojnarowska,F., Schacke,H., Zillikens,D. and Bruckner-Tuderman,L. (2000) The shed ectodomain of collagen XVII/BP180 is targeted by autoantibodies in different blistering skin diseases. Am. J. Pathol., 156, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snellman A., Tu,H., Väisänen,T., Kvist,A.P., Huhtala,P. and Pihlajaniemi,T. (2000) A short sequence in the N-terminal region is required for the trimerization of type XIII collagen and is conserved in other collagenous transmembrane proteins. EMBO J., 19, 5051–5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasanen K., Eble,J.A., Aumailley,M., Schumann,H., Baetge,J., Tu,H., Bruckner,P. and Bruckner-Tuderman,L. (2000a) Collagen XVII is destabilized by a glycine substitution mutation in the cell adhesion domain Col15. J. Biol. Chem., 275, 3093–3099. [DOI] [PubMed] [Google Scholar]
- Tasanen K., Floeth,M., Schumann,H. and Bruckner-Tuderman,L. (2000b) Hemizygosity for a glycine substitution in collagen XVII: unfolding and degradation of the ectodomain. J. Invest. Dermatol., 115, 207–212. [DOI] [PubMed] [Google Scholar]
- Woodley D.T., Bachmann,P.M. and O’Keefe,E.J. (1988) Laminin inhibits human keratinocyte migration. J. Cell Physiol., 136, 140–146. [DOI] [PubMed] [Google Scholar]
- Zhou Z., Apte,S.S., Soininen,R., Cao,R., Baaklini,G.Y., Rauser,R.W., Wang,J., Cao,Y. and Tryggvason,K. (2000) Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl Acad. Sci. USA, 97, 4052–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zone J.J., Taylor,T.B., Kadunce,D.P. and Meyer,L.J. (1990) Identification of the cutaneous basement membrane zone antigen and isolation of antibody in linear immunoglobulin A bullous dermatosis. J. Clin. Invest., 85, 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]