

Abstract
In chronic diseases such as diabetes mellitus, continuous stress stimuli trigger a persistent, self-reinforcing reprogramming of cellular function and gene expression that culminates in the pathological state. Late-onset, stable changes in gene expression hold the key to understanding the molecular basis of chronic diseases. Renal failure is a common, but poorly understood complication of diabetes. Diabetic nephropathy begins with mesangial cell hypertrophy and hyperplasia, combined with excess matrix deposition. The vasoactive peptide endothelin promotes the mesangial cell hypertophy characteristic of diabetic nephropathy. In this study, we examined the signaling pathways and changes in gene expression required for endothelin-induced mesangial cell hypertrophy. Transcriptional profiling identified seven genes induced with slow kinetics by endothelin. Of these, p8, which encodes a small basic helix–loop–helix protein, was most strongly and stably induced. p8 is also induced in diabetic kidney. Mesangial cell hypertrophy and p8 induction both require activation of the ERK, JNK/SAPK and PI-3-K pathways. Small interfering RNA (siRNA)-mediated RNA interference indicates that p8 is required for endothelin-induced hypertrophy. Thus, p8 is a novel marker for diabetic renal hypertrophy.
Keywords: diabetic renal hypertrophy/p8/RNAi/signal transduction/transcriptional profiling
Introduction
With the rising incidence of obesity in the developed world has come an epidemic of type 2 diabetes mellitus. It is currently estimated that in the USA alone, 16–17 million individuals have type 2 diabetes, and the number of adults with type 2 diabetes increased by 49% between 1991 and 2000 (Mokdad et al., 2001). Type 2 diabetes is a complex disease caused by multiple genetic and environmental factors. A substantial fraction of the morbidity and mortality due to diabetes can be attributed to the pathophysiology of diabetic complications. In the absence of a clear genetic understanding of type 2 diabetes, addressing the root causes of diabetic complications is of great significance to the development of more effective treatment approaches.
Diabetic nephropathy, a common complication of both type 1and 2 diabetes, is the major cause of end-stage renal failure in the Western world (Fine et al., 1992). In diabetes, hypertrophy and excess extracellular matrix deposition are associated with the progression to renal failure. Hypertrophy is marked by an increase in overall protein synthesis, new gene expression—notably of embryonic and immediate-early genes—and, in some cases, reorganization of the actin cytoskeleton (Bonventre and Force, 1998; Force, 1999; Molkentin, 2000). These changes typically are not accompanied by an increase in cell numbers.
The molecular basis of diabetic renal hypertrophy is still an enigma, although diabetic renal hypertrophy and matrix deposition have been linked to pathologic changes in renal mesangial cells. Accordingly, these cells have been studied extensively for clues as to the mechanisms that underlie diabetic renal disease (Fine et al., 1992; Bonventre and Force, 1998). Multiple extracellular stimuli are thought to be involved in the pathophysiology of diabetic mesangial cell hypertrophy. Prominent among these are vasoactive peptides such as endothelin-1 (ET-1) and angiotensin II. The observation that diabetic renal disease often progresses even when glucose homeostasis is well managed suggests that self-reinforcing, long-term changes in gene expression—changes that have yet to be elucidated—may lie at the heart of the molecular events that contribute to this and other chronic conditions.
Signal transduction pathways recruited in response to the persistent presence of pro-hypertrophic and other chronic stress agonists may participate in the initiation of pathological hypertrophy and other deleterious effects. Mitogen-activated protein kinases (MAPKs) have been implicated in cardiomyocyte hypertrophy (Wang et al., 1998a,b; Force et al., 1999; Bogoyevitch, 2000; Molkentin, 2000), and activation by hypertrophic stimuli of mesangial cell MAPK subgroups [extracellular signal-regulated kinase (ERK), c-Jun-N-terminal kinase/stress-activated protein kinase (JNK/SAPK) and p38 MAPK] has been documented (Bonventre and Force, 1998), but not tracked with changes in gene expression that occur during biological responses such as hypertrophy or matrix deposition. Similarly, the phosphatidylinositol-3′-OH-kinase (PI-3-K) pathway has been implicated in cardiomyocyte hypertrophy (Haq et al., 2000; Antos et al., 2002), but a role for this pathway in diabetic renal hypertrophy has not been established.
The ability of MAPKs [through the activator protein-1 (AP-1) and other transcription factors] and downstream effectors of PI-3-K [which also include AP-1, as well as nuclear factor of activated T cell (NFAT) and forkhead (FKH) family transcription factors] to trigger changes in gene expression is thought to underlie a substantial part of the mechanism by which these pathways affect cell function (Boyle et al., 1991; Cross et al., 1995; Rao et al., 1997; Molkentin et al., 1998; Haq et al., 2000; Kops and Burgering, 2000; Kyriakis, 2000; Brazil and Hemmings, 2001; Katso et al., 2001; Kyriakis and Avruch, 2001; Lawlor and Alessi, 2001; Neal and Clipstone, 2001; Scheid and Woodgett, 2001; Antos et al., 2002). The molecular basis by which these various signaling pathways reprogram cells to adapt to chronic stresses, such as those that cause hypertrophy, remains nebulous. In particular, a picture of the more long-term, stable, changes in gene expression that occur as cells progress to a pathologically deranged state is missing. This is important since chronic diseases such as diabetic nephropathy probably involve long-term, systematic changes in gene expression that, in response to chronic stress, essentially condemn cells to a diseased state.
Here we report the results of a combined pharmacological, transcriptional profiling and genetic study aimed at beginning to identify renal mesangial cell signaling pathways and transcripts induced by ET-1 under conditions that trigger hypertrophy. We sought to identify either genes induced after the prolonged periods of ET-1 treatment (up to 48 h) needed to trigger hypertrophy, or genes induced rapidly, but stably throughout the 48 h treatment period. Our aim was to implicate ET-1-induced transcripts as necessary to the progress of mesangial cell hypertrophy. One transcript encoding the small basic helix–loop–helix (bHLH) protein p8 (Mallo et al., 1997) was strikingly induced under conditions indistinguishable from those necessary to trigger mesangial cell hypertrophy. Moreover, RNA interference (RNAi) experiments indicate that p8 induction is required for ET-1-stimulated mesangial cell hypertrophy. These findings are significant inasmuch as they identify p8 as a necessary component in the transcriptional program that triggers mesangial cell hypertrophy. In addition, these results indicate that p8 is a novel marker gene for diabetic renal hypertrophy. Given that traditional protein synthesis assays for mesangial cell hypertrophy are cumbersome and inefficient, assaying induction of p8 will streamline the search for novel inhibitors of diabetic renal disease. Finally, our results reveal a striking similarity between the signaling pathways required for renal mesangial cell hypertrophy and cardiac hypertrophy (Molkentin, 2000). Accordingly, p8 may also be important for other hypertrophic pathologies, including those of the heart.
Results
ET-1 triggers renal mesangial cell hypertrophy
Figure 1A is an illustration of the treatment protocol to which we subjected rat renal mesangial cells in these studies. Mesangial cell hypertrophy is defined as a stimulus-dependent increase in cellular protein synthesis ([3H]leucine uptake) without an accompanying increase in DNA synthesis ([3H]thymidine uptake). We routinely observe that ET-1 triggers mesangial cell hypertrophy manifested as an increase in protein synthesis and a decrease in DNA synthesis (Figure 1B).

Fig. 1. Mesangial cell treatment protocol and ET-1 induction of hypertrophy. (A) ET-1 treatment protocol. The figure describes the time course of treatment for induction of hypertrophy. For certain experiments, as indicated in other figures, cells were harvested at earlier times in order to monitor regulation of signaling pathways or gene expression. (B) The 48 h treatment protocol in (A) is sufficient to trigger mesangial cell hypertrophy, an increase in [3H]leucine uptake and a parallel decrease in [3H]thymidine uptake.
Microarray analysis
In contrast to cardiomyocyte hypertrophy, few details have emerged concerning gene induction during the progression of renal mesangial cells to hypertrophy. Specifically, no known marker genes have been clearly linked to renal mesangial cell hypertrophy. In order to identify genes induced by ET-1 under conditions that trigger mesangial cell hypertrophy, we performed microarray analysis. Inasmuch as hypertrophy is a delayed process, we were interested in genes whose induction, if rapid, was sustained for a prolonged period of ET-1 treatment. Alternatively, we sought genes induced only after extended ET-1 treatment. Rat renal mesangial cells were treated with vehicle or ET-1 as described above. RNA was collected at 1, 24, 36 and 48 h after treatment; in parallel, progression of the cells to hypertrophy was confirmed as above (Figure 1). A portion of the RNA from the cells treated for 24 and 48 h was reverse transcribed, labeled with 33P and applied to filter microarrays spotted with ∼5500 cDNAs. RNA samples were also subjected simultaneously to northern analysis. Induction values on northern blots were quantitated by phosphoimaging and normalized for gapdh expression.
To confirm functional gene induction in the ET-1-treated cells, we analyzed expression of gene 33, a gene induced in response to a variety of proinflammatory and mitogenic stimuli (Messina, 1994; Makkinje et al., 2000). gene 33 expression was stimulated by ET-1, reaching an apparent maximum at 1 h, and declining to baseline by 24 h (Figure 2; Table I). We identified seven genes that obeyed our criteria for late-onset genes. Results for the northern analyses are shown in Figure 2; Table I summarizes the transcriptional profiling for genes detected on both the microarrays and northern blots as ET-1 late-onset-inducible. Induced with slow kinetics in response to ET-1 is a set of genes associated with stress and inflammation [copper/zinc-containing superoxide dismutase (Cu/Zn-sod), bag2], signaling [ADP ribosylation factor-6 (arf6), phospholipase-Cβ4 (PLCβ4), p116 rho-interacting protein (p116rip), myr5] and/or diabetes [cystatin C, platelet-derived growth factor A chain (PDGF-A)] (Silver et al., 1989; Bhandari and Abboud, 1993; Jiang et al., 1994; Hosler and Brown, 1995; Gebbink et al., 1997; Hall, 1998; Post et al., 1998; Takayama et al., 1999; Mostov et al., 2000; Song et al., 2001; Mussap et al., 2002) (Figure 2; Table I).

Fig. 2. Early- and late-onset gene induction in mesangial cells treated with ET-1. Mesangial cells were treated with ET-1 for the indicated times. RNA was subjected to microarray analysis, and genes whose induction was confirmed on the arrays were subjected to further analysis by northern blotting. Induction is quantitated in Table I.
Table I. Time course of induction of various genes by ET-1 in rat renal mesangial cells.
| Function | Protein name | Fold induction at indicated time post ET-1 treatment |
|||
|---|---|---|---|---|---|
| 1 h | 24 h | 36 h | 48 h | ||
| Signaling | PDGF A chain | 0.8 | 1.2 | ND | 2.0 |
| p116rip | 0.7 | 1.5 | ND | 2.1 | |
| Gene 33 | 6.0 | 1.1 | 1.1 | 1.1 | |
| myr5 | 1.4 | ND | ND | 2.5 | |
| Phospholipase C-β4 | 0.9 | ND | ND | 1.9 | |
| bag2 | 5.5 | ND | ND | 1.3 | |
| Nuclear | p8 | 2.4 | 3.0 | 8.0 | 5.1 |
| Enzymes | Cu/Zn superoxide dismutase (SOD) | 2.2 | 2.1 | 2.2 | 1.9 |
| ADP ribosylation factor-6 | 1.6 | 1.3 | 2.1 | 1.6 | |
| ECM | Cystatin C | 0.6 | 1.1 | ND | 2.2 |
Gene induction was detected initially by microarray and confirmed by northern blot analysis. Fold inductions shown were determined by northern blot analysis. ND, not determined.
By far, the gene most strikingly and stably induced by ET-1 is p8. p8 encodes a small bHLH nuclear protein up-regulated in pancreatitis and implicated in V12-ras-dependent tumorigenesis and regulation of paired box transcription factors of the Pax family (Mallo et al., 1997; Hoffmeister et al., 2002; Vasseur et al., 2002b). During the course of ET-1 treatment, p8 is induced significantly at 1 h (3.3-fold), with induction continuing out to 24 h (4-fold), reaching a maximum of 6- to 10-fold induction at 36 h treatment (Figure 2; Table I). Most notably, p8 induction remains elevated at least 7-fold above basal after 48 h of ET-1 treatment.
ET-1 activates the mesangial cell ERK, JNK/SAPK and p38 MAPK pathways as well as the PI-3-K pathway
To establish a clear picture of the signaling pathways recruited by ET-1, we assessed the activation of various MAPK pathways and the PI-3-K pathway during the course of ET-1-induced mesangial cell hypertrophy. MAPKs are recruited as part of three tiered MAPK kinase kinase (MAP3K)→MAPK kinase (MKK)→MAPK core pathways. MAPKs require concomitant tyrosine and threonine phosphorylation for activity (Kyriakis and Avruch, 2001). Using antibodies specific for the phosphorylated, active forms of ERK1/2, JNK/SAPK and p38, as well as their corresponding MKKs (MEK1/2, MKK4, and MKKs 3 and 6, respectively), we find that all three of these MAPK pathways are activated in response to ET-1 (Figure 3A–D). ERK activation is comparable with that incurred by epidermal growth factor (EGF), used as positive control for cell activation and as an indicator of the reliability of the phospho-antibodies. In contrast, JNK/SAPK and p38 pathway activation, while significant, is more modest than that stimulated by hyperosmotic shock (500 mM sorbitol, like EGF used as positive control) (Figure 3A and C). In all cases, MAPK pathway activation by ET-1 is maximal after 5–10 min and declines to basal by 60 min (Figure 3A and C). After 60 min, JNK/SAPK and p38 activation remain at basal levels for the remainder of the 48 h treatment protocol (Figure 3A and B). ERK pathway activity, however, rises modestly for a second time, reaching a submaximal plateau by 48 h (Figure 3B and D), a point in time when the cells are clearly hypertrophic (Figure 1). ERK activity is also elevated after 24 h of EGF stimulation (Figure 3B).

Fig. 3. Activation of mesangial cell ERK, JNK/SAPK and p38 MAPK pathways by ET-1. (A) Cells were treated with the indicated agonists for the indicated times. Crude extracts were prepared and subjected to SDS–PAGE and immunoblotting with phospho-MAPK-specific antibodies (top three panels) or total MAPK blots (bottom three panels). (B) The same as (A), except that cells were treated for longer time periods. (C) Activation of MKKs by ET-1. Cells were treated with the indicated stimuli as in (A). Extracts were prepared and subjected to SDS–PAGE and immunoblotting with the indicated phospho-MKK-specific antibodies (top three panels) or, as a loading control, with an antibody to smooth muscle actin (SMA, bottom panel). (D) The same as (C), except that blots were probed with anti-phospho-MEK only. (E) Activation of CREB by ET-1. Mesangial cells were treated with the indicated agonists for the indicated times, and crude extracts were subjected to SDS–PAGE and immunoblotting with phospho-CREB/ATF-1-specific antibody.
The transcription factor cAMP response element-binding protein (CREB) functions in diverse physiological processes, including the control of cellular metabolism and mitogen-dependent cell survival (Mayr and Montminy, 2001). In response to mitogens and stresses, CREB is phosphorylated at Ser133 and activated in a cAMP-independent, ERK- and p38 MAPK-dependent manner (Deak et al., 1998). We observe that ET-1 stimulates CREB Ser133 phosphorylation, as well as phosphorylation of the related transcription factor activating transcription factor-1 (ATF1), consistent with the idea of functionally relevant MAPK pathway activation (Figure 3E).
PI-3-K and its effectors are essential to the control of cell survival, cell size, proliferation and metabolism (Katso et al., 2001). It is of note that PI-3-K effectors have been implicated in cardiac hypertrophy (Molkentin et al., 1998; Haq et al., 2000; Antos et al., 2002). We also observe that the rat renal mesangial cell PI-3-K pathway is activated by ET-1. Recruitment of PI-3-K leads to 3′-phosphoinositide-dependent phosphorylation at Ser308 and Ser473, and consequent activation of protein kinase B (PKB) (Alessi et al., 1996, 1997a,b). ET-1 treatment triggers the rapid phosphorylation and activation of mesangial cell PKB as detected on immunoblots probed with antibodies directed towards PKB phosphorylated at Ser473 (Figure 4). Activated PKB directly phosphorylates and inhibits two key effectors: glycogen synthase kinase-3 (at Ser9 for GSK3β and Ser21 for GSK3α) and transcription factors of the FKH family (Cross et al., 1995; Kops and Burgering, 2000; Brazil and Hemmings, 2001; Scheid and Woodgett, 2001). We find that ET-1 triggers phosphorylation of GSK3β at Ser9, GSK3α at Ser21, and of the FKH transcription factors AFX (at Ser193) and FKHR (at Ser256) (Figure 4). Stimulus-induced inhibition of GSK3β has been linked to the development of cardiomyocyte hypertrophy via relief of GSK3-mediated inhibition of NFATs (Haq et al., 2000; Antos et al., 2002).

Fig. 4. Activation of the mesangial cell PI-3-K pathway by ET-1. Primary mesangial cells were treated with the indicated agonists for the indicated times. Crude extracts were subjected to SDS–PAGE and immunoblotting with phospho-PKB, phospho-GSK3, phospho-FKHR or, as a loading control, SMA as previously reported (Goruppi et al., 2001).
ET-1 induction of renal mesangial cell hypertrophy requires ERK, JNK/SAPK and PI-3-K, but not p38 MAPK
The signal transduction mechanisms that control renal mesangial cell hypertrophy are not known. Studies of cardiac hypertrophy indicate a role for MAPKs of the ERK, JNK/SAPK and p38 groups (Choukroun et al., 1998, 1999; Wang et al., 1998a,b; Force, 1999). In addition, PI-3-K and its effectors—most notably, GSK3 and NFATs—have been implicated in cardiomyocyte hypertrophy (Molkentin et al., 1998; Haq et al., 2000; Molkentin, 2000; Antos et al., 2002).
Mesangial cell ERK, JNK/SAPK, p38 and PI-3-K are activated substantially by ET-1 under conditions associated with the induction of hypertrophy (Figures 1, 3 and 4). In recent years, a suite of extremely selective, cell-permeant inhibitors of protein kinases has been developed (Davies et al., 2000). Using these specific reagents, we find that ET-1-induced mesangial cell hypertrophy requires activation of ERK, JNK/SAPK and PI-3-K. While p38 activation is observed in response to ET-1, we do not find that activation of p38α or β is necessary for hypertrophy.
U0126 is a highly specific inhibitor of the activation of MEK1/2 by the MAP3K Raf-1 (Davies et al., 2000). We find that, in parallel with inhibition of ET-1 activation of ERK, U0126 reverses the ET-1-induced increase in mesangial cell protein synthesis (Figure 5). Thus, ERK activity is necessary for optimal ET-1-induced mesangial cell hypertrophy. Similar but more striking results were obtained with LY294002, a highly specific synthetic inhibitor of PI-3-K (Davies et al., 2000), indicating a role for PI-3-K in ET-1-induced mesangial cell hypertrophy (Figure 5A). SP600125 is a high affinity anthrapyrazolone inhibitor of the JNKs/SAPKs. SP600125 does not inhibit ERK or p38 MAPK in vivo (Bennett et al., 2001; Figure 5B). SP600125 also reverses ET-1-induced mesangial cell hypertrophy (Figure 5A). Thus, as in cardiomyocytes, JNK/SAPK is an important element in ET-1-induced mesangial cell hypertrophy. In contrast, SB203580, a selective inhibitor of the α and β (but not the γ and δ) isoforms of p38 MAPK (Davies et al., 2000; Figure 5B), fails to prevent mesangial cell hypertrophy in response to ET-1, in spite of blocking the activation of p38 (Figure 5).

Fig. 5. ERK, JNK/SAPK and PI-3-K, but not p38, are required for ET-1-induced mesangial cell hypertrophy. PP2B is also required for optimal ET-1-induced hypertrophy. (A) Mesangial cells were pre- treated with the indicated inhibitor compounds for 30 min, at which time vehicle or ET-1 was added and the incubation continued for an additional 48 h. Hypertrophy was measured as in Materials and methods. (B) Efficacy of the indicated inhibitors. A portion of the cells in (A) was lysed and the lysates subjected to analysis with phospho-specific antibodies. (C) FK506 blocks mesangial cell hypertrophy. Mesangial cells were pre-treated with 100 nM FK506 and treated with ET-1 as in (A). ET-1 triggers a significant increase in leucine incorporation (indicated by *, as determined by t-test; P < 0.05, n = 3); however, FK506 pre-treatment reduces leucine incorporation to insignificant levels (indicated by **, as determined by t-test; P > 0.05, n = 3).
ET-1 stimulates Ca2+ uptake (Bonventre and Force, 1998). FK506 is a potent Ca2+/calmodulin-dependent inhibitor of PP2B (Schreiber and Crabtree, 1992). ET-1 elicits cardiomyocyte hypertrophy in part through an NFATs-dependent mechanism. ET-1 inhibits GSK3, and activates PP2B, reversing GSK3’s inhibitory NFAT phosphorylation (Molkentin et al., 1998; Haq et al., 2000; Antos et al., 2002). We find that FK506 reverses ET-1-induced mesangial cell hypertrophy (Figure 5C). Taken together with the results in Figure 4 indicating that ET-1 triggers an inhibitory phosphorylation of mesangial cell GSK3, FK506 inhibition of mesangial cell hypertrophy is consistent with the idea that NFATs or other PP2B effectors are important to ET-1-induced hypertrophy.
ET-1 induction of p8 and hypertrophy requires the same signaling pathways
Despite the significant clinical importance of renal mesangial cell hypertrophy to the pathogenesis of diabetic nephropathy, little is known of the molecular mechanisms underlying this process. Accordingly, we have begun to seek transcripts induced by ET-1 in a manner indistinguishable from that of hypertrophy, and to determine if these transcripts are also induced in diabetic kidney.
To this end, we treated mesangial cells with ET-1 under conditions sufficient to trigger hypertrophy. Some of the cells were pre-treated with specific inhibitors of kinase pathways. Our findings (Figure 6) indicate that p8 induction is regulated in a manner indistinguishable from that of hypertrophy. Thus, we find that the strong ET-1 induction of p8 is reversed by the PI-3-K inhibitor LY294002, the strongest inhibitor of hypertrophy (Figures 5A and 6A). Inhibition of p8 induction is also observed with the ERK pathway inhibitor U0126 and, to a lesser degree, by the JNK/SAPK inhibitor SP600125. FK506 treatment also blocks ET-1 induction of p8, suggesting that PP2B activity is important to both ET-1-stimulated hypertrophy and p8 expression. The extent to which these inhibitors prevent p8 induction coincides well with the degree to which these drugs block mesangial cell hypertrophy (Figure 6A). SB203580, which fails to prevent ET-1-induced mesangial cell hypertrophy (Figure 5A), also fails to suppress ET-1 induction of mesangial cell p8 (Figure 6A). Induction of Cu/Zn-sod and cystatin C by ET-1 also tracks a priori with hypertrophy, insofar as expression of these genes is inhibited by LY294002, U0126 and SP600125; however, the sensitivity of these genes to the different drugs differs from that of p8 and hypertrophy. For example, SP600125 is a relatively stronger inhibitor of Cu/Zn-sod and cystatin C induction than it is an inhibitor of p8 induction and hypertrophy (Figures 5 and 6A). The rapid induction of bag2 by ET-1 after 1 h does not appear to be under the same regulation as that of p8. Thus, neither LY294002, U0126 nor SP600125 prevents bag2 induction (Figure 6A). Instead, SB203580 strongly suppresses ET-1 induction of bag2, indicating that ET-1-stimulated bag2 expression, unlike mesangial cell hypertrophy, requires p38α and/or β. The consistent robustness of p8 induction (Figures 2 and 6A; Table I) as well as the induction of p8 in diabetic nephropathy and by high glucose (Figures 6B and 7) makes p8 an excellent candidate marker for diabetic hypertrophy.

Fig. 6. Regulation of mesangial cell gene expression by ET-1 requires distinct signaling pathways. Regulation of p8 expression is indistinguishable from that of hypertrophy. (A) Suppression of gene induction by specific protein kinase pathway inhibitors. Mesangial cells were pre-treated with the indicated inhibitors and then treated with ET-1 for 24 h (upper panel) or for 1 h (lower panel). Expression was detected by northern blotting, and gapdh expression was used as a loading control. (B) High glucose induces p8 expression to a degree commensurate with ET-1. ET-1 and glucose are not synergistic. Serum-starved cells were treated with high glucose (30 mM), ET-1 or both for 24 h. p8 expression was monitored by northern blotting.

Fig. 7. Induction of diabetes in rats stimulates expression of p8. As indicated in the figure, rats were made diabetic with the injection of STZ, subjected to unilateral nephrectomy, or both. RNA was prepared from the kidneys at the indicated times. For nephrectomized animals, RNA was prepared from the remaining kidney at the indicated times. Control animals were injected with water. Northern blotting was performed and quantitated as described in Materials and methods. Ethidium bromide staining of 28S RNA served as a loading control. NP indicates nephropathy in diabetic animals as occurs 5 weeks after STZ administration.
The results shown in Figures 2, 5 and 6A are consistent with the hypothesis that p8 is a marker transcript for diabetic mesangial cell hypertrophy. To provide further support for this idea, we sought to determine if p8 was induced by high glucose (which on its own is known to trigger mesangial cell responses reminiscent of diabetes; Fine et al., 1992) and during the progression of diabetic kidney disease in rats. In Figure 6B, mesangial cells were treated with high glucose (30 mM) or with ET-1 for 24 h. High glucose alone is sufficient to elicit elevated mesangial cell p8 expression. The degree of p8 induction is comparable to that incurred by ET-1, and ET-1 does not synergize with glucose to superinduce p8 (Figure 6B).
We also subjected rats to treatment with the diabetogenic genotoxin streptozotocin (STZ), unilateral nephrectomy or both (Figure 7). Induction of p8 and Cu/Zn-sod was then determined. Unilateral nephrectomy alone does not induce p8 or Cu/Zn-sod. In contrast, at 23 h post STZ treatment, when the rats exhibit glucosuria, prominent induction of p8 is observed. Lesser induction of Cu/Zn-sod is also observed. At 5 weeks post-STZ (NP in Figure 7), the rats are clearly nephropathic, with frank proteinuria and renal hypertrophy. At this time, expression of both p8 and Cu/Zn-sod is still elevated, although the overall induction of p8 is substantially greater, having increased from that observed early in the diabetic state.
p8 is required for ET-1-stimulated mesangial cell hypertrophy
The correspondence between the regulation of ET-1 induction of p8 and hypertrophy compelled us to ask if p8 expression was required for ET-1-induced hypertrophy. To test this, we employed the recently developed method of mammalian cell RNA interference (RNAi) mediated by small interfering RNA (siRNA; Elbashir et al., 2001b). This technique is based on the observation that RNAi is initiated by 21–22 bp double-stranded RNAs (dsRNAs) generated by RNase III-like cleavage from longer dsRNAs (Elbashir et al., 2001a). In an experimental setting, this phenomenon can be exploited by introducing into cells synthetic 21 bp dsRNAs, derived from the gene of interest, that conform to the consensus sequence AA(N19)YY (Y = pyrimidine). A major advantage of this method is that it permits a facile loss-of-function analysis under conditions that do not recruit the cellular viral dsRNA response mechanism (Elbashir et al., 2001b). The rat p8 sequence contains a single suitable siRNA sequence. Introduction of the corresponding p8 dsRNA completely ablates basal p8 expression (Figure 8A). Moreover, introduction of p8 siRNA oligonucleotides abolishes basal and ET-1-induced p8 expression for up to 48 h (Figure 8B). Parallel assays of ET-1-induced hypertrophy indicate that p8 RNAi completely prevents ET-1-induced renal mesangial cell hypertrophy (Figure 8C). In contrast, serum-induced protein and DNA synthesis are unaffected by disruption of p8. (Control thymidine incorporation, 110 ± 4 c.p.m.; serum thymidine incorporation, 173 ± 1 c.p.m.; serum + p8 siRNA thymidine incorporation, 162 ± c.p.m.; P > 0.05, n = 3. Control leucine incorporation, 696 ± 41 c.p.m.; serum leucine incorporation, 1588 ± 76 c.p.m.; serum + p8 siRNA leucine incorporation, 1589 ± 324 c.p.m.; P > 0.05, n = 3.) Taken together, these results strongly indicate that p8 is a required element for ET-1-induced mesangial cell hypertrophy.
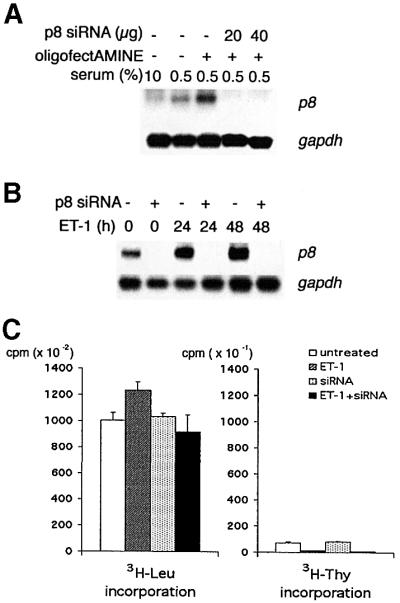
Fig. 8. Induction of p8 is required for ET-1-stimulated renal mesangial cell hypertrophy. (A) dsRNA-dependent RNAi of p8 ablates expres sion of p8. Renal mesangial cells were mock transfected with oligofectAMINE or transfected with oligofectAMINE plus p8 siRNA at the indicated concentrations before serum starvation (0.5%) for 24 h. As a control, a separate Petri dish was untreated but starved (0.5%) or left proliferating in normal serum (10%). RNA was extracted and subjected to northern analysis with a p8 probe as described above. (B) p8 RNAi blocks both basal and ET-1-dependent induction of p8 expression. Renal mesangial cells were pre-treated with p8 siRNA (10 µg/6 cm dish), serum starved for 48 h and then treated with ET-1 for the indicated times (0, 24 and 48 h). RNA was extracted and subjected to northern analysis with p8 or gapdh probes. (C) p8 RNAi completely inhibits ET-1-induced renal mesangial cell hypertrophy. p8 siRNA was introduced into the cells and, after 48 h of serum starvation, mesangial cells were subjected to the hypertrophy protocol. ET-1-stimulated [3H]leucine and [3H]thymidine incorporation were monitored as described in Figure 1.
Discussion
As the incidence of obesity and diabetes approaches epidemic proportions, improving the treatment of diabetic complications will become increasingly important. Under standing the cellular and molecular responses to chronic stresses associated with conditions such as diabetes represents a major research challenge. Often the stimuli that produce these responses act in a submaximal and cumulative manner. Moreover, most chronic conditions involve the combined, additive effects of multiple extracellular environmental inputs and genetic factors, each of which, when examined alone, may produce relatively minor effects.
Nephropathy is an important chronic and often fatal complication of diabetes. Still, in comparison with cardiac hypertrophy, the molecular details of diabetic nephropathy are poorly understood. Mesangial cell hypertrophy and excess matrix deposition play major roles in diabetic nephropathy. It has been well established that these responses can be triggered by vasoactive peptides such as ET-1, which are in excess in the diabetic kidney (Fine et al., 1992). Our results indicate that the progression of mesangial cells to ET-1-stimulated hypertrophy requires activation of PI-3-K, ERK and JNK/SAPK, but not activation of the α and β isoforms of p38. Our transcriptional profiling has revealed p8 to be a transcript strongly and stably induced in ET-1-treated mesangial cells and in diabetic kidney. The induction of p8 by ET-1 is regulated by the same pathways that trigger mesangial cell hypertrophy. Of particular importance, p8 RNAi prevents ET-1-stimulated mesangial cell hypertrophy. We conclude that p8 is required for mesangial cell hypertrophy, and can be considered a marker for this condition.
There is a striking similarity in the signaling pathways recruited in cardiomyocyte and mesangial cell hypertrophy. Several studies indicate that the JNKs/SAPKs are important to cardiomyocyte hypertrophy. Thus, inhibition of JNK/SAPK activation blocks ET-1-stimulated neonatal rat cardiomyocyte hypertrophy (Choukroun et al., 1998) and, in intact animals, pressure overload cardiac hypertrophy (Choukroun et al., 1999). Moreover, ectopic activation of the JNK/SAPK pathway triggers stimulus-independent cardiomyocyte hypertrophy (Wang et al., 1998a). Interestingly, expression of active MKK3, which in cardiomyocytes preferentially targets p38α and β, fails to induce hypertrophy. In contrast, expression of constitutively active MKK6, which targets all p38 isoforms (α–δ), leads to hypertrophy (Wang et al., 1998b). Thus, as with mesangial cells, cardiomyocyte hypertrophy apparently requires the JNK/SAPK pathway, but not p38α or β.
The role of ERK in cardiomyocyte hypertrophy is complicated and may differ from that of mesangial cell hypertrophy. Thus, it has been reported that inhibition of ERK blocks ET-1-induced cardiomyocyte hypertrophy (Yue et al., 2000). In whole animals, however, the role of ERK in cardiomyocyte hypertrophy appears to be compensatory rather than pathophysiological, inasmuch as transgenic overexpression of MEK1 protects cardiomyocytes from apoptosis and triggers a hypertrophy associated with enhanced cardiac function (Bueno et al., 2000).
The PI-3-K pathway is also thought to play a prominent role in cardiac hypertrophy (Haq et al., 2000; Antos et al., 2002), and our results suggest that, as in the myocardium, PI-3-K signaling is important to mesangial cell hypertrophy and late-onset gene expression. Recent findings indicate that stimulus-induced cardiac hypertrophy is mediated in part by PI-3-K-dependent, PKB-catalyzed phosphorylation and inhibition of GSK3β. Coincident with Ca2+/calmodulin-dependent activation of PP2B, this reverses inhibitory GSK3-catalyzed phosphorylation of NFATs (Molkentin et al., 1998; Haq et al., 2000; Antos et al., 2002). Our finding that PI-3-K and PP2B are essential to ET-1 induction of mesangial cell hypertrophy, plus the observation that GSK3 undergoes inhibitory phosphorylation in mesangial cells in response to ET-1 suggest that it will be important to determine if inhibition of GSK3 and activation of NFATs are important to mesangial cell hypertrophy.
Our results (Figures 2 and 5–8) provide compelling evidence that the p8 polypeptide plays a role in mesangial cell hypertrophy. p8 is a bHLH protein with a nuclear localization signal with a modest sequence homology to high mobility group (HMG) proteins (Mallo et al., 1997). p8 is readily phosphorylated in vitro by cAMP-dependent protein kinase (Encinar et al., 2001). In in vitro assays, phospho-p8 assumes a more ordered solution structure and binds random DNA more readily (Encinar et al., 2001). However, it is not known if this phosphorylation is regulated in vivo or if it achieves a physiologically relevant stoichiometry.
How might p8 function in eliciting hypertrophy? Recent evidence points to a role for p8 in transcriptional regulation, cell cycle control and stress responses. Induction of p8 was first observed to occur in acute pancreatitis. p8 is also induced in tumorigenesis (Mallo et al., 1997; Ree et al., 1999; Su et al., 2001a,b). The p8 polypeptide is important to cell proliferation and to the establishment of tumors; however, the function of p8 in tumorigenesis is somewhat enigmatic. Thus, disruption of p8 suppresses transformation associated with ectopic expression of activated ras and adenoviral E1A (Vasseur et al., 2002a). In contrast, p8–/– cells proliferate more rapidly in culture and are resistant to chemical stress-induced apoptosis (Vasseur et al., 2002b). Inasmuch as cultured p8–/– cells were grown on intact solid surfaces, it is possible that disruption of p8 potentiates anchorage-dependent growth, but impairs the anchorage-independent growth measured in cell transformation assays. We see strong ET-1-stimulated induction of p8 in mesangial cells under conditions in which proliferation (as measured by incorporation of [3H]thymidine) is actually reduced. Our findings that p8 is induced under conditions in which proliferation is inhibited, and in the diabetic kidney where cell proliferation is, at best, limited, suggest a more complex role for the p8 polypeptide in cellular regulation.
p8 is localized in the nucleus (Vasseur et al., 1999; S.Goruppi, unpublished data), and recent evidence points to a potential role in transcriptional regulation. Consistent with its homology to HMG proteins, p8 binds and is acetylated in vitro by the histone acetyltransferase protein p300 (in vivo acetylation has not been determined). In two-hybrid screens, p8 interacts with Pax2 trans-activation domain-interacting protein (PTIP), a strong inhibitor of Pax2 transcription factor function. In co-transfection experiments, ectopic overexpression of p8 reverses PTIP inhibition of Pax2 (Hoffmeister et al., 2002). This finding is intriguing as it suggests that, in response to stress, p8 might, by sequestering PTIP, serve to modulate transcription, perhaps stabilizing or enhancing Pax2-based transcription, thereby altering cell function. Pax2 function is critical to renal development, and disruption of pax2 is associated with renal hypoplasia and renal coloboma development (Chi et al., 2002). With regard to diabetic renal disease, Pax2 might couple p8 to renal hyper trophy. This possibility is particularly noteworthy insofar as the reappearance of embryonic gene activity frequently is associated with hypertrophy (Force et al., 1999; Molkentin, 2000).
In conclusion, our results establish the PI-3-K, ERK and JNK/SAPK pathways as critical regulators of ET-1-induced renal mesangial cell hypertrophy and late-onset gene expression. Among the late-onset genes induced by ET-1, p8 expression is required for ET-1-induced hypertrophy. p8 is also strongly and stably induced in diabetic kidney. Thus, p8 expression is an attractive marker for mesangial cell hypertrophy and diabetic nephropathy. Given the similarities between renal mesangial cell and cardiomyocyte hypertrophy, determining the role of p8 in cardiac pathology is warranted.
Materials and methods
Cell culture
Primary rat renal mesangial cells were isolated as described from the glomeruli of adult male Sprague–Dawley rats (Kreisberg and Venkatchalam, 1986). Cells were cultured in RPMI 1640/10% fetal bovine serum (FBS). The integrity of the mesangial cell phenotype was confirmed regularly by immunoblotting with anti-smooth muscle actin (SMA) and Thy1 antibodies. ET-1, wortmannin, LY294002, U0126, SB203580, FK506 and SP600125 were from Calbiochem.
Hypertrophy assays
Mesangial cells were plated in triplicate in 12-well plates and allowed to grow to 80% confluence. Cells were then starved for 48 h in RPMI 1640/0.5% FBS and treated with either 100 nM ET-1, or 20% FBS as control. After 36 h, 1 µCi of either methyl-[3H]thymidine or [3H]leucine was added to monitor S phase induction or to analyze protein synthesis, respectively. After an additional 12 h, each well was washed once with phosphate-buffered saline (PBS) and the cells were fixed by adding 1 ml of 10% trichloroacetic acid. Precipitates were dissolved in 1 ml of 0.5 M NaOH and subjected to liquid scintillation counting. As indicated, data were analyzed by the Student’s t-test. As indicated, LY294002 (20 µM), SB203580 (10 µM), SP600125 (10 µM), U0126 (10 µM) or FK506 (100 nM) were added 30 min before ET-1 addition.
DNA microarray studies
Mesangial cells were made quiescent in 0.5% FBS for 48 h and then stimulated with 100 nM ET-1 for 0, 1, 24, 36 and 48 h, either with or without the prior addition of kinase inhibitors as above. The Ambion RNA isolation kit was used as indicated by the manufacturer. A 10 µg aliquot of total RNA was reverse transcribed and labeled with 33P according to standard methods (Sambrook et al., 1989).
For microarray analysis, we used Rat GeneFilter™ (GF300, Research Genetics) arrays, each containing an average of 5500 expressed sequence tags (ESTs). Membranes were first washed in boiling 0.5% SDS for 5 min. Denatured probe was added and the membranes allowed to hybridize for 24 h at 42°C and then washed. Membranes were subjected to phosphoimager analysis. The resulting images were analyzed with the Pathways™ 2.1 software (Research Genetics). Gene expression ratios were calculated by comparing arrays from untreated and stimulated mesangial cells. Results were confirmed by stripping and reprobing the arrays with the same mRNA preparation but reversing the arrays to which RNA was applied. Genes that were regulated consistently in two independent array experiments using two separate RNA preparations were considered for further northern blot analysis.
Northern analysis
Northern blotting was performed using standard methods (Sambrook et al., 1989). The probes were prepared from the same cDNAs (Research Genetics) spotted on the GF300 array. The identity of each cDNA was confirmed by sequencing of a least one strand. Hybridization was quantitated in a phosphoimager. The fold changes in RNA levels were quantified by normalizing to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Western blots
Mesangial cells were serum starved and then stimulated as indicated in the figures with 100 nM ET-1, 100 ng/ml EGF or 0.5 M sorbitol. Cells were lysed in 500 µl of SDS–PAGE loading buffer. Equal amounts of total protein were subjected to SDS–PAGE and immunoblotting as described (Goruppi et al., 1997, 2001). Phospho-specific antibodies, as indicated in the figures, were from Cell Signaling Technology. We used rabbit anti-total ERK (Santa Cruz Biotechnology), JNK/SAPK (Cell Signaling Technology), p38 (Santa Cruz Biotechnology), PKB (Cell Signaling Technology) and anti-SMA (Sigma).
Induction of diabetes in rats
Rats were treated with STZ and monitored for diabetic complications as described (Makkinje et al., 2000). All procedures involving animals were approved by the Massachusetts General Hospital internal laboratory animal care committee.
siRNA-mediated mammalian cell RNAi
Complementary RNA oligonucleotides derived from the single rat p8 siRNA sequence (Elbashir et al., 2001b), and designed to anneal leaving 2 bp 3′ overhangs, were synthesized by Dharmacon: 5′-GGACGTACCAAGAGAGAAGCT and 5′-CTTCTCTCTTGGTACGTCCTT. Deprotection and annealing of the oligonucleotides were performed according to the manufacturer’s instructions. Procedures for RNAi were essentially as described by Elbashir et al. (2001b). The resulting dsRNAs were introduced into mesangial cells with OligofectAMINE (Invitrogen), according to the manufacturer’s instructions, at a concentration of 10–40 µg oligonucleotide per 6 cm dish. After an overnight incubation with the siRNA, mesangial cells were serum starved for 48 h in RPMI 1640/0.5% FBS before proceeding with the remainder of the experiments or before performing the hypertrophy protocol.
Acknowledgments
Acknowledgements
We thank Enzo Calautti for helpful discussions. This work was supported by USPHS grant R01-DK41513 (to J.M.K.).
References
- Alessi D.R., Andjelkovic,M., Caudwell,B., Cron,P., Morrice,N., Cohen,P. and Hemmings,B.A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J., 15, 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi D.R., James,S.R., Downes,C.P., Holmes,A.B., Gaffney,P.R.J., Reese,C.B. and Cohen,P. (1997a) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol., 7, 261–269. [DOI] [PubMed] [Google Scholar]
- Alessi D.R. et al. (1997b) 3-Phosphoinositide-dependent kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol., 7, 776–789. [DOI] [PubMed] [Google Scholar]
- Antos C.L., McKinsey,T.A., Frey,N., Kutschke,W., McAnally,J., Shelton,J.M., Richardson,J.A., Hill,J.A. and Olson,E.N. (2002) Activated glycogen synthase kinase-3β suppresses cardiac hypertrophy in vivo. Proc. Natl Acad. Sci. USA, 99, 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B.L. et al. (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl Acad. Sci. USA, 98, 13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari B. and Abboud,H.E. (1993) Platelet-derived growth factor-A chain gene expression in cultured mesangial cells: regulation by phorbol ester at the level of mRNA abundance, transcription and mRNA stability. Mol. Cell. Endocrinol., 91, 185–191. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch M.A. (2000) Signaling via stress-activated mitogen-activated protein kinases in the cardiovascular system. Cardiovasc. Res., 45, 826–842. [DOI] [PubMed] [Google Scholar]
- Bonventre J.V. and Force,T. (1998) Mitogen-activated protein kinases and transcriptional responses in renal injury and repair. Curr. Opin. Nephrol. Hypertens., 7, 425–433. [DOI] [PubMed] [Google Scholar]
- Boyle W.J., Smeal,T., Defize,L.H., Angel,P., Woodgett,J.R., Karin,M. and Hunter,T. (1991) Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA binding activity. Cell, 64, 573–584. [DOI] [PubMed] [Google Scholar]
- Brazil D.P. and Hemmings,B.A. (2001) Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem. Sci., 26, 657–664. [DOI] [PubMed] [Google Scholar]
- Bueno O.F. et al. (2000) The MEK1–ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J., 19, 6341–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi N. and Epstein,J.A. (2002) Getting your Pax straight: Pax proteins in development and disease. Trends Genet., 18, 41–47. [DOI] [PubMed] [Google Scholar]
- Choukroun G., Bonventre,J.V., Kyriakis,J.M., Rosenzweig,A. and Force,T. (1998) Endothelin stimulation of cardiomyocyte hypertrophy requires the SAPK pathway. J. Clin. Invest., 102, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choukroun G., Hajjar,R., Fry,S., del Monte,F., Haq,S., Guerrero,J.L., Picard,M. Rosenzweig,A. and Force,T. (1999) Regulation of cardiac hypertrophy in vivo by the stress-activated protein kinases/c-Jun NH2-terminal kinases. J. Clin. Invest., 104, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross D.A.E., Alessi,D.R., Cohen,P., Andjelkovich,M. and Hemmings,B.A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature, 378, 785–789. [DOI] [PubMed] [Google Scholar]
- Davies S.P., Reddy,H., Caivano,M. and Cohen,P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J., 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak M., Clifton,A.D., Lucocq,J. and Alessi,D.R. (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38 and may mediate activation of CREB. EMBO J., 17, 4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinar J.A. et al. (2001) Human p8 is a HMG-I/Y-like protein with DNA binding activity enhanced by phosphorylation. J. Biol. Chem., 276, 2742–2751. [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Lendeckel,W. and Tuschl,T. (2001a) RNA interference is mediated by 21 and 22 nt RNAs. Genes Dev., 15, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001b) Duplexes of 21 nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Fine L.G., Norman,J.T., Kujubu,D.A. and Knecht,A. (1992) Renal hypertrophy. In Seldin,D.W. and Geibisch,G. (eds), The Kidney, Physiology and Pathophysiology. Raven Press, New York, NY, pp. 3113–3134.
- Force T., Hajjar,R., Del Monte,F., Rosenzweig,A. and Choukroun,G. (1999) Signaling pathways mediating the response to hypertrophic stress in the heart. Gene Expr., 7, 337–348. [PMC free article] [PubMed] [Google Scholar]
- Gebbink M.F., Kranenburg,O., Poland,M., van Horck,F.P., Houssa,B. and Moolenaar,W.H. (1997) Identification of a novel putative Rho-specific GDP/GTP exchange factor and a RhoA-binding protein: control of neuronal morphology. J. Cell Biol., 137, 1603–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goruppi S., Ruaro,E. and Schneider,C. (1997) Requirement of PI3K-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH3T3 fibroblasts. Mol. Cell. Biol., 17, 4442–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goruppi S., Ruaro,E., Chiaruttini,C. and Schneider,C. (2001) Gas6 induces growth, β-catenin stabilization and T-cell factor transcription in contact inhibited C57 mammary cells. Mol. Cell. Biol., 21, 902–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science, 279, 509–514. [DOI] [PubMed] [Google Scholar]
- Haq S. et al. (2000) Glycogen synthase kinase-3β is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol., 151, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeister A. et al. (2002) The HMGI/Y-related protein p8 binds to p300 and Pax2 trans-activation-domain interacting protein to regulate the transactivation activity of the Pax2A and Pax2B transcription factors on the glucagon gene promoter. J. Biol. Chem., 277, 22314–22319. [DOI] [PubMed] [Google Scholar]
- Hosler B.A. and Brown,R.H.,Jr (1995) Copper/zinc superoxide dismutase mutations and free radical damage in amyotrophic lateral sclerosis. Adv. Neurol., 68, 41–46. [PubMed] [Google Scholar]
- Jiang H., Wu,D. and Simon,M.I. (1994) Activation of phospholipase Cβ4 by heterotrimeric GTP binding proteins. J. Biol. Chem., 269, 7593–7596. [PubMed] [Google Scholar]
- Katso R., Okkenhaug,K., Ahmadi,K., White,S., Timms,J. and Waterfield,M.D. (2001) Cellular function of phosphoinositide 3-kinases: implications for development homeostasis and cancer. Annu. Rev. Cell Dev. Biol., 17, 615–675. [DOI] [PubMed] [Google Scholar]
- Kops G.J. and Burgering,B.M. (2000) Forkhead transcription factors are targets of signalling by the proto-oncogene PKB (C-AKT). J. Anat., 197, 571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisberg J.I. and Venkatchalam,M.A. (1986) Vasoactive agents affect mesangial cell adhesion. Am. J. Physiol., 251, C505–C511. [DOI] [PubMed] [Google Scholar]
- Kyriakis J.M. (2000) Mammalian MAP kinase pathways. In Woodgett,J.R. (ed.), Protein Kinase Functions. Oxford University Press, Oxford, UK, pp. 40–156.
- Kyriakis J.M. and Avruch,J. (2001) Mammalian mitogen-activated protein kinase pathways activated by stress and inflammation. Physiol. Rev., 81, 807–869. [DOI] [PubMed] [Google Scholar]
- Lawlor M.A. and Alessi,D.R. (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci., 114, 2903–2910. [DOI] [PubMed] [Google Scholar]
- Makkinje A., Quinn,D.A., Chen,A., Cadilla,C.L., Force,T., Bonventre,J.V. and Kyriakis,J.M. (2000) Gene 33/Mig-6, a transcriptionally inducible adapter protein that binds GTP-Cdc42 and activates SAPK/JNK. J. Biol. Chem., 275, 17838–17847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo G.V., Fiedler,F., Calvo,E.L., Ortiz,E.M., Vasseur,S., Keim,V., Morisset,J. and Iovanna,J.L. (1997) Cloning and expression of the rat p8 cDNA, a new gene activated in pancreas during the acute phase of pancreatitis, pancreatic development and regeneration and which promotes cellular growth. J. Biol. Chem., 272, 32360–32369. [DOI] [PubMed] [Google Scholar]
- Mayr B. and Montminy,M. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol., 2, 599–609. [DOI] [PubMed] [Google Scholar]
- Messina J.L. (1994) Regulation of gene 33 expression by insulin. In Draznin,B. and LeRoith,D. (eds), Molecular Biology of Diabetes, Part II. Humana Press, Totowa, NJ, pp. 263–281.
- Mokdad A.H., Bowman,B.A., Ford,E.S., Vinicor,F., Marks,J.S. and Koplan,J.P. (2001) The continuing epidemics of obesity and diabetes in the United States. J. Am. Med. Assoc., 286, 1195–1200. [DOI] [PubMed] [Google Scholar]
- Molkentin J.D. (2000) Calcineurin and beyond: cardiac hypertrophic signaling. Circ. Res., 87, 731–738. [DOI] [PubMed] [Google Scholar]
- Molkentin J.D., Lu,J.-R., Antos,C.L., Markham,B., Richardson,J., Robbins,J., Grant,S.R. and Olson,E.N. (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell, 93, 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostov K.E., Verges,M. and Altschuler,Y. (2000) Membrane traffic in polarized epithelial cells. Curr. Opin. Cell Biol., 12, 483–490. [DOI] [PubMed] [Google Scholar]
- Mussap M., Dalla Vestra,M., Fioretto,P., Saller,A., Varagnolo,M., Nosandini,R. and Plebani,M. (2002) Cystatin C is a more sensitive marker than creatinine for the estimation of GFR in type 2 diabetic patients. Kidney Int., 61, 1453–1461. [DOI] [PubMed] [Google Scholar]
- Neal J.W. and Clipstone,N.A. (2001) Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J. Biol. Chem., 276, 3666–3673. [DOI] [PubMed] [Google Scholar]
- Post P.L., Bokoch,G.M. and Mooseker,M.S. (1998) Human myosin-Ix-b is a mechanochemically active motor and a GAP for rho. J. Cell Sci., 111, 941–950. [DOI] [PubMed] [Google Scholar]
- Rao A., Luo,C. and Hogan,P.G. (1997) Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol., 15, 707–747. [DOI] [PubMed] [Google Scholar]
- Ree A.H., Tvermyr,M., Engebraaten,O., Rooman,M., Rosok,O., Hovig,E., Meza-Zepeda,L.A., Bruland,O.S. and Fodstad,O. (1999) Expression of a novel factor in human breast cancer cells with metastatic potential. Cancer Res., 59, 4675–4680. [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Scheid M.P. and Woodgett,J.R. (2001) PKB/AKT: functional insights from genetic models. Nat. Rev. Mol. Cell Biol., 2, 760–768. [DOI] [PubMed] [Google Scholar]
- Schreiber S.L. and Crabtree,G.R. (1992) The mechanism of action of cyclosporin A and FK506. Immunol. Today, 13, 136–142. [DOI] [PubMed] [Google Scholar]
- Silver B.J., Jaffer,F.E. and Abboud,H.E. (1989) Platelet-derived growth factor synthesis in mesangial cells: induction by multiple peptide mitogens. Proc. Natl Acad. Sci. USA, 86, 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Takeda,M. and Morimoto,R.I. (2001) Bag1-Hsp70 mediates a physiological stress signalling pathway that regulates Raf-1/ERK and cell growth. Nat. Cell Biol., 3, 276–282. [DOI] [PubMed] [Google Scholar]
- Su S.B. et al. (2001a) Expression of p8 in human pancreatic cancer. Clin. Cancer Res., 7, 309–313. [PubMed] [Google Scholar]
- Su S.B., Motoo,Y., Iovanna,J.L., Bertthenzene,P., Xie,M.J., Mouri,H., Ohtsubo,K., Matsubara,F. and Sawabu,N. (2001b) Overexpression of p8 is inversely correlated with apoptosis in pancreatic cancer. Clin. Cancer Res., 7, 1320–1324. [PubMed] [Google Scholar]
- Takayama S., Xie,Z. and Reed,J.C. (1999) An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem., 274, 781–786. [DOI] [PubMed] [Google Scholar]
- Vasseur S., Vidal Mallo,G., Fiedler,F., Bodeker,H., Canepa,E., Moreno,S. and Iovanna,J.L. (1999) Cloning and expression of the human p8, a nuclear protein with mitogenic activity. Eur. J. Biochem., 259, 670–675. [DOI] [PubMed] [Google Scholar]
- Vasseur S., Hoffmeister,A., Garcia,S., Bagnis,C., Dagorn,J.C. and Iovanna,J.L. (2002a) p8 is critical for tumour development induced by ras V12 mutated protein and E1A oncogene. EMBO rep., 3, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur S., Hoffmeister,A., Garcia-Montero,A., Mallo,G.V., Fieil,R., Kühbander,S., Dagorn,J.-C. and Iovanna,J.L. (2002b) p8-deficient fibroblasts grow more rapidly and are more resistant to adriamycin-induced apoptosis. Oncogene, 21, 1685–1694. [DOI] [PubMed] [Google Scholar]
- Wang Y., Su,B., Sah,V.P., Heller Brown,J., Han,J. and Chien,K.R. (1998a) Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J. Biol. Chem., 273, 5423–5426. [DOI] [PubMed] [Google Scholar]
- Wang Y., Huang,S., Sah,V.P., Ross,J.,Jr, Brown,J.H., Han,J. and Chien,K.R. (1998b) Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J. Biol. Chem., 273, 2161–2168. [DOI] [PubMed] [Google Scholar]
- Yue T.L. et al. (2000) Extracelular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J. Biol. Chem., 275, 37895–37901. [DOI] [PubMed] [Google Scholar]