

Abstract
The histone acetyltransferases CREB binding protein (CBP) and the related p300 protein function as key transcriptional co-activators in multiple pathways. In the case of transcriptional activation by nuclear receptors, ligand promotes the recruitment of co-activators of the p160 family, such as GRIP-1. Subsequently, the p160 co-activators recruit other co-activators via two activation domains, AD1 and AD2. AD1 binds CBP or p300, whereas AD2 has been shown to activate transcription through the recruitment of the arginine methyltransferase CARM1. Recently, the KIX domain of CBP has been shown to be methylated by CARM1 in vitro. Here, we report that another domain of CBP is specifically methylated by CARM1 on conserved arginine residues in vitro and in vivo. We also provide functional evidence that arginine residues methylated by CARM1 play a critical role in GRIP-1-dependent transcriptional activation and in hormone-induced gene activation. Altogether, our data provide strong evidence that arginine methylation represents an important mechanism for modulating co-activator transcriptional activity.
Keywords: arginine methylation/CARM1/CBP/nuclear receptors/transcriptional activation
Introduction
CREB binding protein (CBP) and the highly related E1A-associated p300 protein are essential for transcriptional activation by a large number of transcription factors including nuclear receptors (Kamei et al., 1996; Smith et al., 1996), CREB (Kwok et al., 1994), bHLH factors (Eckner et al., 1996; Yuan et al., 1996), STAT1 (Horvai et al., 1997; Korzus et al., 1998) and AP-1 (Arias et al., 1994). The co-activator functions of CBP and p300 probably involve a variety of mechanisms. First, these proteins bind to many transcription factors, to other co-activators and to various components of the transcriptional machinery, and these properties have led to the proposal that CBP and p300 may serve as scaffolds or platforms for assembly of multiprotein complexes with transcriptional activity (for a recent review see Goodman and Smolik, 2000). In addition, CBP and p300 harbour an intrinsic acetyltransferase activity (HAT) that can acetylate histones (Bannister and Kouzarides, 1996; Ogryzko et al., 1996), thereby activating transcription (Martinez-Balbas et al., 1998). Acetylation of transcription factors and proteins of the basal transcriptional machinery by CBP and p300 has also been shown to be an important mode of transcriptional regulation (for a review see Kouzarides, 2000).
CBP and p300 transcriptional activity can be regulated by post-translational modifications such as phosphorylation. For example, in vivo phosphorylation of CBP by cyclin E/cdk2 at the G1/S boundary increases CBP HAT activity (Ait-Si-Ali et al., 1998). Similarly, in vivo phosphorylation of CBP GF box, but not of p300, in response to growth factors increases CBP-mediated transcriptional co-activation (Zanger et al., 2001).
Upon addition of ligand, nuclear receptors activate gene transcription by binding to specific promoters and by recruiting co-activators of the p160 family, which consists of the three related 160 kDa proteins SRC-1, GRIP-1/TIF2 and pCIP/RAC3/ACTR/AIB1/TRAM1 (for a review see Torchia et al., 1998). The p160 co-activators propagate the activating signal through at least two activation domains, AD1 and AD2, which act by recruiting a number of secondary co-activator proteins. AD1 binds CBP or p300 (Kamei et al., 1996), whereas AD2 recruits the co-activator associated arginine methyltransferase 1 (CARM1) (Chen et al., 1999). CARM1 (also called PRMT4) belongs to a family of arginine methyltransferases which includes the mammalian protein arginine methyltransferases (PRMT)-1, -2, -3, -5 and -6 (Lin et al., 1996; Pollack et al., 1999; Frankel and Clarke, 2000; Frankel et al., 2002), and the yeast hnRNP arginine methyltransferase 1 (HMT1) (Henry and Silver, 1996). These six arginine methyltransferases share a highly conserved catalytic domain (for recent reviews see Zhang and Reinberg, 2001; Kouzarides, 2002). CARM1 was originally identified as a histone methyltransferase, since it methylates histone H3 in vitro. Recently, in vivo methylation of histone H3 has been shown to be associated with gene activation upon hormone treatment (Ma et al., 2001; Bauer et al., 2002). In addition to histones, (Xu et al., 2001) have shown that CARM1 methylates the KIX domain of CBP, at least in vitro.
Here, we show that CARM1 specifically methylates CBP and the related protein p300 on conserved arginine residues outside the KIX domain in vitro. Moreover, we provide several pieces of evidence indicating that the region we have mapped within CBP is also methylated by CARM1 in vivo. Finally, a CBP protein point mutated on the methylation sites we have characterized abolishes GRIP-1- and steroid hormone-dependent transcriptional activation. Altogether, our findings strongly support the hypothesis that arginine methylation represents a functionally important post-translational modification of CBP.
Results
CARM1 specifically methylates CBP N-terminal part in vitro
In order to test whether CBP was methylated by the co-activator arginine methyltransferase CARM1, bacterially expressed GST, GST–CBP full length (CBP FL) and purified histones were incubated with GST–CARM1 in the presence of radiolabelled S-adenosyl l-[methyl-3H] methionine (SAM). Transfer of methyl groups from SAM to the substrates was analysed by fluorography after SDS–PAGE. We found that GST–CBP FL was efficiently methylated by CARM1, whereas the control (GST protein alone, GST) was not. As expected, histone H3, a known substrate for CARM1, was also methylated by CARM1 (Figure 1A, left panel). Both CBP FL and CBP N-terminal half (N-CBP) were efficiently methylated, whereas CBP C-terminal half (CBP-C) was not (Figure 1A, middle panel). These data indicate that some regions of CBP are not methylated by CARM1 in vitro. To further confirm the specificity of CBP methylation by CARM1, we tested other bacterially produced fusion proteins as potential substrates for CARM1-specific methylation. In this assay, we tested the histone acetyltransferase P/CAF and the transcription factors MyoD, p53 and E2F-1. We found that none of these substrates was efficiently methylated by CARM1, indicating that CBP is a specific substrate for CARM1 (Figure 1B, left panel). Reciproc ally, we decided to address the possibility that CBP could be a target of other arginine methyltransferases than CARM1. To this aim, we performed methyltransferase assays using PRMT1 and HMT1 in parallel with CARM1. HMT1 represents the major methyltransferase activity in yeast and has a broader substrate specificity than PRMT1 (Gary and Clarke, 1998). Both proteins belong to the same family of methyltransferases as CARM1 (Chen et al., 1999). As shown in Figure 1C, CARM1 specifically methyl ated histone H3 (left panel) but also very efficiently methylated CBP (right panel), while HMT1 and PRMT1 methylated histone H4 (left panel), as expected (Chen et al., 1999; Strahl et al., 2001; Wang et al., 2001), but not CBP (right panel). Altogether, these data indicate that CBP is specifically methylated by CARM1 in vitro.

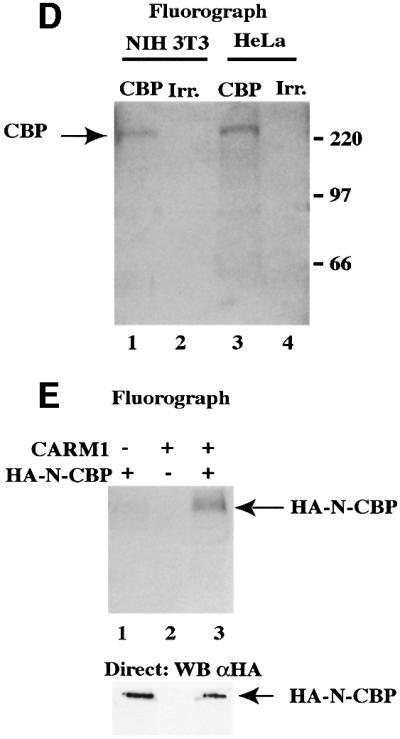
Fig. 1. Methylation of CBP by CARM1. (A) Left panels: 1 µg of purified core histones (hist.), bacterially expressed recombinant GST–CBP FL (CBP FL), control GST (GST), GST–CBP-C or GST–N-CBP, where indicated, were assayed for methylation by 500 ng of bacterially produced recombinant GST–CARM1. Reaction products were then analysed by SDS–PAGE followed by fluorography. The asterisks (*) indicate the position of the various full-length GST fusion proteins. The right panels show western blots allowing the detection of recombinant GST–CBP FL or mutants used in the left panels. The N-terminus of CBP was detected using the A22 antibody (Santa Cruz; α CBP NT), and the C-terminal half using the NM11 antibody (PharMingen; α CBP CT). Note that using the A22 antibody, many shorter bands could be detected, which probably reflects premature termination and/or protein degradation that occur during the production of large proteins in bacteria. (B) GST control, GST–CBP, GST–P/CAF, GST–MyoD, GST–p53 and GST–E2F-1 were assayed for methylation by GST–CARM1. Reaction products were then analysed by SDS–PAGE followed by fluorography. The asterisks (*) indicate the position of the various full-length GST fusion proteins. The right panel shows the corresponding Coomassie staining. Note the presence in CBP and P/CAF preparation of prominent truncated protein products, which most likely reflects premature termination and/or protein degradation that occur during the production of large proteins in bacteria. (C) One microgram of histones (left panel) or 10 ng of GST–CBP FL (right panel) were assayed for methylation by 500 ng of GST–CARM1, GST–PRMT1, GST–HMT1 or control GST protein as indicated. Reaction products were resolved on an 18% SDS–PAGE (left panel) or on an 8% SDS–PAGE (right panel). (D) HeLa cells and NIH 3T3 cells (30 × 106) were radiolabelled with SAM. Total cell extracts were then immunoprecipitated with either an anti-CBP antibody (A22; Santa Cruz) or with an irrelevant anti-HA antibody (Santa Cruz). Immunoprecipitates were then analysed by SDS–PAGE. Gels were fixed and fluorographed for 8 weeks. (E) U2OS cells (2 × 106) were transfected as indicated with 40 µg of pCMV-HA-CBP (1–1098) (N-CBP) and/or 20 µg of pSG5-CARM1, in the presence of 10 µg of pSG5-GRIP-1. Transfected cells were radiolabelled with SAM, and total cells extracts were immunoprecipitated using a monoclonal anti-HA antibody (12CA5; Roche Diagnostics). Immunoprecipitates were analysed by SDS–PAGE followed by fluorography (4 weeks) (upper panel). 1/200th of the immunoprecipitates was subjected to an anti-HA western blot (lower panel).
CBP is methylated in living cells by CARM1
We next wondered whether CBP methylation also occurs in living cells. Metabolic radiolabelling of either HeLa cells or NIH 3T3 cells with SAM followed by immunoprecipitation using either an anti-CBP antibody or an irrelevant antibody (Irr.) showed that endogenous CBP was specifically methylated in both cell lines (Figure 1D). In order to analyse whether the methylation of CBP was due, at least in part, to CARM1, we transfected U2OS cells, which could be transfected very efficiently, with N-CBP [amino acids (aa) 1–1098] in the presence or in the absence of exogenous CARM1. We used N-CBP because it is methylated in vitro by CARM1 (Figure 1A, middle panel) and better expressed than the full-length protein when transfected in eukaryotic cells (data not shown). We found that the CBP N-terminal region was much more methylated in the presence of overexpressed CARM1 than in its absence (Figure 1E, upper panel, compare lanes 1 and 3). This effect was not due to a higher expression level of N-CBP, as western blot analysis showed that an equal amount of N-CBP was immunoprecipitated in the presence or in the absence of CARM1 (Figure 1E, lower panel). Thus, these findings indicate that CARM1 can also methylate CBP in vivo.
A region of 90 amino acids is necessary and sufficient for CBP methylation by CARM1
To further narrow down the region of CBP targeted by methylation, we used a set of deletions within the N-terminal half of CBP, which was then tested for methylation by CARM1. We found that the regions of CBP corresponding to aa 1–502 (A), aa 468–685 (B) and aa 770–1098 (D) were not methylated by CARM1, while the deletion mutant spanning aa 685–774 (C) of CBP was efficiently methylated by CARM1 (Figure 2A). Thus, these data indicate that CBP is methylated between aa 685 and 774. In addition, faint bands migrating at the size of GST–CARM1 were present in all lanes, and most likely corresponded to the automethylation of CARM1 (Figure 2A, open circle) (see Supplementary data, available at The EMBO Journal Online). Finally, we tested whether this region of CBP was also methylated by CARM1 in the context of the full-length protein. In order to do so, we produced a mutated version of CBP (CBPΔ685–774) that is deleted for the region encompassing aa 685–774. As shown in Figure 2B, CBPΔ685–774 was not methylated by CARM1, whereas CBP FL was efficiently modified by CARM1. Equivalent amounts of CBP FL and CBPΔ685– 774 were used in this assay as confirmed by western blot using an antibody directed against the C-terminus of CBP (Figure 2B, lower panel). In conclusion, our results indicate that aa 685–774 of CBP are necessary and sufficient for CBP methylation by CARM1 in vitro.

Fig. 2. The region methylated by CARM1 maps onto a 90 amino acid region within the N-terminal part of CBP. (A) One microgram of control GST, 100 ng of GST–N CBP or CBP mutants A, B, C and D were assayed for methylation by GST–CARM1 and analysed by SDS–PAGE followed by fluorography. The asterisks (*) indicate the position of the various full-length GST fusion proteins. Note that fusion protein D migrates at a larger molecular weight than expected for an unknown reason. The open circle indicates a band reflecting CARM1 weak automethylation. The right panel shows the corresponding Coomassie staining. (B) Ten nanograms of GST–CBP FL and of GST–CBP Δ685–774 (which has been deleted of 90 amino acids encompassing the methylated region) were assayed for methylation by GST–CARM1 and were analysed by SDS–PAGE followed by fluorography. Lower panel: The amounts of GST–CBP FL and of GST–CBPΔ685–774 were assayed by western blot using the NM11 antibody.
CARM1 methylates CBP on arginine residues conserved in p300
CBP and the highly related protein p300 share most of their biochemical properties known so far. We thus hypothesized that p300 could also be methylated by CARM1. As shown in Figure 3A, p300 FL (FL) and p300 1–744 (N) (corresponding to aa 1–757 in CBP) were efficiently methylated by CARM1, while p300 1–595 (NN) (corresponding to aa 1–615 in CBP) was weakly methylated. These data indicate that p300 is also methylated by CARM1 on the same region as CBP. However, the CBP region methylated by CARM1 (aa 685–774) is not highly conserved in p300. It is worth noting that among the four arginine residues located within this region of CBP (R702, R714, R742 and R768), three (R714, R742 and R768) are conserved between CBP and p300 (Figure 3B). This conservation suggests that methylation could be an important post-translational modification regulating CBP and p300 function.


Fig. 3. CBP is methylated on three arginine residues conserved in p300. (A) GST–p300 FL, GST–p300 1–744 and GST–p300 1–595, as indicated, were assayed for methylation by GST–CARM1 and were analysed by SDS–PAGE followed by fluorography. The asterisks (*) indicate the position of the various full-length GST fusion proteins. Note that the major band of methylated GST–p300 FL (#) migrates below the 180 kDa marker: this band corresponds to a major truncated product of p300. In the right panel, the expression of p300 and of the deletion mutants were assayed by western blot using the NM11 antibody (α p300 CT) or by Coomassie Blue staining, as indicated. Note the presence in the p300 FL preparation of prominent truncated protein products, which most likely reflects premature termination and/or protein degradation that occur during the production of large proteins in bacteria. (B) Sequence alignment of the CBP methylated region with the corresponding sequence of p300. Conserved residues are indicated in bold. The # indicate the three conserved arginines (R). Sequences and names of the peptides used in (C) are indicated on the top of the sequence alignment. (C) Biotinylated peptides (6 µM final concentration) were assayed for methylation using either 1 µg of GST, 50 ng of GST–CARM1 or 50 ng of GST–HMT1 as indicated. Peptide methylation was measured using a scintillation counter. (D) Upper panel: GST–CBP FL and GST–CBP 3R→A (in which R714, R742 and R768 were replaced by alanines) were assayed for methylation by GST–CARM1 and were analysed on an 8% SDS–PAGE followed by fluorography. Lower panel: the amount of GST–CBP FL and GST–CBP 3R→A was assayed by western blot using the NM11 antibody.
In order to determine which arginine residue was specifically methylated by CARM1, we designed three synthetic peptides derived from CBP, each containing one or two putative CARM1 methylation sites (Figure 3B). We found that all three peptides were very efficiently methylated by CARM1 (Figure 3C), whereas a control peptide, A742, similar to the peptide R742 but in which the arginine was changed into an alanine, was not methylated at all. In addition, a peptide derived from the first 22 amino acids of CBP and containing a unique arginine was not methylated by CARM1 (data not shown). As controls, neither HMT1 nor GST alone, which did not methylate CBP FL (Figure 1C), could methylate any of the peptides (Figure 3C). These results show that at least three out of the four arginine residues present within the region of CBP we have mapped, are methylated by CARM1 in vitro.
We thus expressed a CBP protein point mutated at all three conserved arginines R714, R742 and R768 (CBP 3R→A) and we tested it for its methylation by CARM1. As shown on upper panel in Figure 3D, CBP 3R→A was not methylated by CARM1, whereas CBP FL was efficiently modified. Note that equivalent amounts of GST–CBP FL and GST–CBP 3R→A were used in this assay, as confirmed by western blotting (Figure 3D, lower panel). Altogether, these data show that in the CBP full-length protein, residues R714, R742 and R768 are the major methylation sites for CARM1 in vitro.
CBP is methylated by CARM1 on R742 in vivo
We next attempted to test whether CBP was also methylated on these sites in vivo. To this aim, an antibody specific for CBP methylated on R742 (the major site as determined in Figure 3C) was purified by affinity chromatography and was then tested for its specificity. This antibody efficiently recognized a peptide derived from CBP and dimethylated on R742, but not the same unmodified peptide or various other peptides derived from CBP (Figure 4A, left panel). In addition, we found that the purified antibody was specifically recognizing CBP methylated by CARM1 in vitro (Figure 4A, right panel, lane 3). No signal was observed in the absence of methylation (Figure 4A, right panel, lane 1) or when the non-methylatable form of CBP 3R→A was used as a substrate for CARM1 (Figure 4A, right panel, lane 2). In addition, the purified antibody did not recognize histone H3 methylated by CARM1 (Supplementary data). Thus, this antibody is highly specific for CBP methylated on R742.

Fig. 4. CBP is methylated by CARM1 on R742 in vivo. (A) Left panel: the anti-methylated CBP antibody was tested for its specificity by a slot blot experiment with the indicated amount of various peptides. Right panel: 10 ng of GST–CBP FL and of GST–CBP 3R→A were methylated by GST–CARM1 (lanes 3 and 2, respectively) or not (lane 1). Reaction products were resolved onto an 8% SDS–PAGE followed by a western blot using the purified anti-methylated CBP antibody. (B) Immunoprecipitations from HeLa nuclear extracts (100 µl) were performed using an anti-HA polyclonal antibody (Irr.), an anti-CBP antibody and the affinity purified anti-methylated CBP antibody. Immunoprecipitates and the input (1 µl of HeLa nuclear extracts) were analysed by western blot using the anti-methylated CBP antibody. (C) Lysates of HeLa cells transfected either with control siRNA duplexes (CTR) or with specific CARM1 siRNA duplexes (CARM1) were tested by western blot for the expression level of CARM1, of HDAC-1, -2 and -3 (anti-HDAC3; Transduction Laboratories), of CBP and of CBP specifically methylated on R742.
We next used this specific antibody to analyse whether R742 of CBP was methylated in vivo. This antibody recognized a band at the expected size for CBP in HeLa nuclear extracts (Figure 4B, input). To demonstrate that this band corresponded to CBP, we immunoprecipitated HeLa nuclear extracts with either an irrelevant antibody, or an anti-CBP antibody, or the purified anti-methylated CBP antibody. We found that a band migrating at the expected size was detected in CBP (CBP) and methylated CBP (Me CBP), but not in the control, immunoprecipitates (Irr.) (Figure 4B). This result indicates that endogenous CBP is methylated on R742. Finally, to strengthen the above data, we analysed the level of CBP methylation when endogenous CARM1 expression was suppressed. We designed a small interfering RNA (siRNA) duplex to specifically abolish CARM1 expression. As a control, we used a siRNA duplex, which does not anneal to any mRNA. Lysates of HeLa cells transfected with these siRNA duplexes were tested in parallel for the expression of CARM1, of histone deacetylases (HDACs)-1, -2 and -3, of CBP and of CBP methylated on R742. CARM1 expression level was very efficiently decreased in the lysate of HeLa cells transfected by CARM1-specific siRNAs compared with the control siRNA, whereas HDAC-1, -2 and -3 levels were unaffected (Figure 4C, first and second panels). This result indicates that CARM1 siRNAs silenced CARM1 specifically and did not show any toxic effect on the cells. Similarly, a western blot using an anti-CBP antibody showed that the expression level of CBP protein was not affected by CARM1-specific siRNAs (Figure 4C, third panel). These data indicate that CARM1 does not seem to play any major role in the control of CBP expression. However, a western blot of the same HeLa cell lysates using the purified anti-methylated CBP antibody showed a significant decrease of CBP methylation in cells that have been treated with CARM1 siRNAs compared with the control siRNAs (Figure 4C, fourth panel). This finding strongly supports the hypothesis that CARM1 is the actual enzyme that methylates CBP on R742 in vivo.
CBP methylation is required for GRIP-1- and steroid hormone-induced gene activation
CBP and p300 have been shown to act as co-activators for nuclear receptors, in combination with the p160 family of co-activators and CARM1 (Chen et al., 2000). A possibility is that methylation of CBP by CARM1 is involved in the process of transcriptional activation by GRIP-1. To test this hypothesis, we constructed two mammalian expression vectors encoding non-methylatable CBP proteins, HA-CBPΔ685–774 and the triple point mutant HA-CBP 3R→A. It proved impossible experimentally to test directly the function of the mutated CBP proteins in conjunction with CARM1, because in our hands, the synergy between exogenous CBP and CARM1 on GRIP-1 activity was too weak (<3-fold) (data not shown). Consequently, any result with loss-of-function mutants (such as CBPΔ685–774 or CBP 3R→A) was difficult to interpret. In addition, these experiments could not be adequately controlled because overexpression of the exogenous transcriptional regulators resulted in changes in the activity of the promoters driving the expression of co-expressed proteins (for example, in the presence of exogenous CARM1, the amount of GRIP-1 was much greater than in its absence). Consequently, we decided to use a simpler assay system. GRIP-1 is known to possess two transcriptional activation domains, one interacting with CBP and the other with CARM1. Thus, GRIP-1 transcriptional activity is probably the result of endogenous CARM1 and CBP recruitment. We reasoned that, if CBP methylation by CARM1 is involved in GRIP-1 activity, then a non-methylatable form of CBP would act as a dominant-negative mutant by replacing the endogenous protein from GRIP-1. In order to test this possibility, we transfected U2OS cells with a luciferase reporter gene containing Gal4 binding sites in its promoter, together with an expression vector for Gal4–GRIP-1 fusion protein. This experiment resulted in the artificial targeting of GRIP-1 to a heterologous promoter. Co-transfection of increasing amounts of CBP with Gal4–GRIP-1 led to a dose-dependent transcriptional activation from 2- to 6-fold (Figure 5A, upper panel, compare lane 1 with lanes 8–10). In contrast, co-transfection of CBP mutants Δ685–774 and 3R→A did not stimulate GRIP-1 activity, thereby suggesting that CBP methylation by CARM1 plays a crucial role for its co-activating activity. Strikingly, increasing amounts of CBP Δ685–774 or CBP 3R→A resulted in a strong dose-dependent repression of GRIP-1 activity (Figure 5A, upper panel, lanes 1–7). This decrease was unlikely to be due to any defect in Gal4–GRIP-1 expression level or an aberrant expression of CBP mutants compared with the wild type, as indicated by western blots (Figure 5A, middle and lower panels). This repression was specific to GRIP-1, since the two mutated CBP proteins did not show any repressive effect in similar experiments with a control vector expressing only the Gal4 DNA binding domain or when a reporter without Gal4 sites was used (Supplementary data). In addition, we found that the point mutant CBP 3R→A was as efficient as wild-type CBP in stimulating transcriptional activation by phosphorylated Gal4–CREB fusion protein, another transcription factor functioning through CBP (Figure 5B). Taken together, these experiments indicate that CBP methylation plays an important role in transcriptional activation by GRIP-1, a co-activator of steroid hormones receptors, but not by phosphorylated CREB.


Fig. 5. CBP methylation is required for GRIP1- and steroid hormone-induced transcriptional activation. (A) Upper panel: U2OS cells were transiently transfected with 2 µg of reporter vector pBS-Gal4-luciferase, 2.5 µg of pCMV-Gal4-HA-GRIP1 and 50 ng of pCMV LacZ to monitor transfection efficiency and with increasing amounts of pCMV-HA-CBPΔ685–774, of pCMV-HA-CBP 3R→A or of pCMV-HA-CBP (1, 2 or 5 µg, as indicated by the height of the triangle). Luciferase and β-galactosidase activities were measured 48 h later. The luciferase data shown here are representative of at least four independent transfection experiments (the mean and error bars from the four experiments can be seen in the Supplementary data). Only points where β-galactosidase activity was equivalent were taken into consideration. Middle and lower panels: lysates used in upper panel were tested by western blot for the expression of HA-CBPΔ685–774, HA-CBP 3R→A or HA-CBP, and for the expression of HA-Gal4-GRIP-1 (12CA5 anti-HA antibody; Roche Diagnostics). (B) U2OS cells were transfected with 2 µg of reporter vector pBS-Gal4-luciferase, 50 ng of pCMV LacZ, 2 µg of pHK-Gal4-CREB, 1 µg of PKA+ (an expression vector for a constitutively active PKA) and increasing amounts of pCMV-HA-CBP 3R→A or pCMV-HA-CBP (100 ng, 500 ng or 2 µg as indicated by the height of the triangle). Luciferase and β-galactosidase activities were measured as in (A). (C) MCF7 cells were transiently transfected with 2 µg of reporter vector p17ERE-β-globin-luciferase, 50 ng of pCMV LacZ to monitor transfection efficiency and with increasing amounts of pCMV-HA-CBP 3R→A or pCMV-HA-CBP (1, 2 or 5 µg, as indicated by the height of the triangle). 10–8 M of 17 β estradiol (E2) was added as indicated 24 h before harvesting the cells. Luciferase and β-galactosidase activities were measured as in (A). (D) HeLa cells were transiently transfected with 2 µg of reporter vector pRARE-luciferase, 50 ng of pCMV LacZ to monitor transfection efficiency and with increasing amounts of pCMV-HA-CBP 3R→A (1, 2 or 5 µg, as indicated by the height of the triangle). All-trans retinoic acid (10–7 M final concentration) (RA) was added 24 h before harvesting the cells. Luciferase and β-galactosidase activities were measured as in (A).
In order to confirm the role of CBP methylation in steroid hormone signalling, we chose to compare the co-activating activity of CBP with that of CBP 3R→A mutant in MCF7 cells upon estrogen stimulation. MCF7 cells express endogenous estrogen receptors at high levels. Thus, this assay only needed the co-transfection of a luciferase reporter gene containing estrogen responsive elements in its promoter with increasing amounts of CBP or of the non-methylatable form CBP 3R→A. Cells were treated with estrogens or with the vehicle only before gene activation was assessed. As expected, estrogen (E2) treatment led to efficient promoter stimulation (Figure 5C). Co-transfection of increasing amounts of CBP did not significantly affect hormone response (Figure 5C, compare lane 2 with lanes 10, 12 and 14), probably reflecting the fact that endogenous CBP was not limiting in MCF7 cells for transcriptional activation by endogenous estrogen receptors. In contrast, co-transfection of increasing amounts of CBP 3R→A led to a dose-dependent repression of promoter activity (Figure 5C, compare lanes 4, 6 and 8 with lane 2). Thus, altogether our results provide strong evidence that CBP methylation sites are critical for its co-activating activity and suggest that CBP methylation by CARM1 plays a crucial role in steroid hormone signalling.
To test whether it could also be important for other signalling pathways, we investigated the effect of the CBP point mutant (CBP 3R→A) on retinoic acid signalling, a process also relying on CBP function. We tranfected HeLa cells with a reporter vector in which the luciferase gene was under the control of a retinoic acid responsive element. As expected, treatment of cells with all-trans retinoic acid resulted in efficient promoter stimulation (Figure 5D). Increasing doses of the CBP 3R→A point mutant did not affect significantly retinoic acid response, indicating that CBP methylation is unlikely to play any major role in this process.
Discussion
Both CBP or p300 and CARM1 are co-activators of nuclear receptors and bind to different non-overlapping activation domains of the p160 family members of nuclear receptor co-activators. This suggests that CBP or p300 and CARM1 may work in concert to mediate transcriptional activation by nuclear receptors (Chen et al., 2000). Our results show an interesting link between these two co-activators, since we found that CARM1 methylates CBP. CARM1 was originally characterized as a methyltransferase specific to the histone H3 N-terminal tail in vitro (Chen et al., 1999). This finding has been recently confirmed in vivo on hormone-regulated promoters and suggests a link between histone methylation and transcriptional activation (Ma et al., 2001; Bauer et al., 2002). We found that CBP is also a highly specific substrate for CARM1. Furthermore, in vivo methylation of CBP was observed in various cell lines such as NIH 3T3, HeLa and the osteosarcoma cell line U2OS, and was increased upon co-expression of CARM1. By using an antibody specific for CBP methylated on R742, one of the residues methylated by CARM1 in vitro, we could show that endogenous CBP was methylated on this specific site in living cells. Given the low number of arginine methyltransferases of the PRMT family, and especially the difference of substrate specificity among them, this finding makes a strong case of CARM1 being the actual enzyme that methylates CBP in vivo. In line with this hypothesis, we observed a dramatic reduction of endogenous CBP methylation on that site when CARM1 expression was inhibited by RNA interference. Altogether, these data strongly support the hypothesis that CBP is methylated by CARM1 in vivo. Interestingly, the three arginine residues methylated by CARM1 in CBP are conserved between CBP and p300, which was also efficiently methylated within the same region. Moreover, we observed that R742 is conserved in CBP between human, mouse, Drosophila and Caenorhabditis elegans (data not shown). This finding supports the hypothesis that methylation of CBP/p300 may be an important modification that has been conserved through evolution.
Our results suggest that methylation of CBP by CARM1 can drastically regulate its activity. One of the best ways to provide information on the role of CBP methylation on specific arginines in cells consists in the use of non-methylatable forms of CBP. Indeed, we found that unlike the wild type CBP, these non-methylatable forms of CBP were acting as dominant-negative mutants on GRIP-1 as well as on steroid hormone-dependent transcriptional activation. Thus, the methylation sites within CBP are necessary for CBP co-activating functions in steroid hormone-induced gene activation. However, the possibility still remains that these CBP mutants may present other defects besides methylation. It is impossible to check mutated CBP proteins for all biochemical properties assigned to CBP, as hundreds of interacting partners have been described for CBP or p300 (Vo and Goodman, 2001). However, we would like to point out that most of our functional results have been obtained using a point mutant of CBP, which is less likely to present defects than deletion mutants. Furthermore, we tested this CBP point mutant for its intrinsic HAT activity and for its stability in living cells, and we found no difference compared with wild-type CBP (Supplementary data). These results indicate that this mutant, which showed a dramatic effect on GRIP-1- or hormone-dependent gene activation, is not impaired for all CBP properties.
Along this line, it is worth noting that we found the CBP point mutant did not act as a dominant-negative in retinoic acid-induced gene activation or in phosphorylated CREB-dependent transactivation. Thus, the point mutant can probably mediate key functions of wild-type CBP with respect to transcriptional activation. Accordingly, our results strongly indicate that CBP methylation is specifically involved in transcriptional activation by steroid hormone receptors, but that at least some promoters involving CBP or p300 are regulated independently of CBP/p300 methylation.
The molecular mechanism by which CBP methylation can affect the response to steroid hormones remains unclear. From our results, it can be anticipated that upon hormone treatment, CARM1 is recruited to the activating complex through physical interaction with GRIP-1, where it can methylate both histones and CBP (or p300). These molecular events, together with histone acetylation by the CBP/p300 acetyltransferases, will in turn lead to transcriptional activation of hormone responsive genes. Transcriptional activation by estrogen receptor is a complex process, involving waves of assembly/disassembly of the activating complex on an endogenous estrogen-regulated promoter (Shang et al., 2000). The precise kinetic of transcription (as assayed by RNA pol II presence), of CBP, p300 and Src3 (a p160 family member) recruitment and of histone acetylation has been characterized on this promoter (Shang et al., 2000). It would be very informative to assess the relative kinetic of CARM1 recruitment, CBP methylation and histone methylation on this promoter.
What then could be the biochemical consequence of CBP methylation? One obvious possibility is that CBP methylation regulates its HAT activity. However, we did not find any significant change in CBP HAT activity following CBP methylation by CARM1 (data not shown). Another interesting possibility is that methylation could change CBP conformation and/or regulate some protein– protein interactions responsible for CBP-mediated transcriptional activation on steroid hormone receptors. Interestingly, methylation of two proteins has been shown to link arginine methylation to the regulation of protein–protein interaction (Bedford et al., 2000; Mowen et al., 2001). To date, no protein has been shown to bind directly to the domain of CBP that is methylated by CARM1.
While our manuscript was in preparation, Xu et al. (2001) reported that CBP was methylated by CARM1 within the KIX domain (aa 582–672) in vitro. However, we found that the KIX domain of CBP was very weakly methylated in vitro. Also, CBP mutants in which R702, R714, R742 and R768 are deleted or mutated (but still containing the KIX domain) were not methylated at all, or very weakly methylated, by CARM1 in vitro. One possibility to explain this discrepancy could be that methylation on the sites we identified is required for methylation within the KIX domain. If this is the case, then both methylation events will be involved in the same pathway. Methylation of CBP within the KIX domain was proposed by Xu et al. (2001) to be involved in the CREB pathway. We found that a CBP protein mutated on the sites we identified functions as a co-activator for phosphorylated CREB, and this in a manner indistinguishable from wild-type CBP. The two methylation events are thus most likely unrelated and independent processes.
Consequently, the fact that a CBP mutant deleted for the sites we identified could only be methylated to very low levels by CARM1 suggests that the methylation sites described by Xu et al. (2001) are very minor methylation sites, at least in vitro. An interesting possibility would be that weak methylation sites could play major functional roles in vivo. Alternatively, due to the presence of co-factors or after some post-translational modifications of CBP or of CARM1, the sites within the KIX domain could be more efficiently methylated in vivo. Consistent with this possibility, we found by metabolic labelling that a CBP molecule deleted for the methylation sites we identified was still methylated in cells to some extend (Supplementary data), although it was not methylated by CARM1 in vitro. These results indicate that CBP can be methylated on sites other than the ones we have identified. That methylation could be mediated by CARM1 and could occur on the sites described by Xu et al. (2001). However, because SAM is the only methyl donor group, metabolic labelling cannot distinguish between arginine methylation, lysine methylation and methylation elsewhere on the protein, such as C-terminal methylation. Because of this limitation, a study of the extent and the dynamic of CBP methylation on the sites described by Xu et al. (2001) would require the use of an antibody specific for CBP methylated on one of these sites.
In summary, arginine methylation is an additional modification that regulates CBP co-activator function. It is likely that other non-histone methylation targets involved in the regulation of transcription will be identified in the near future. Whether arginine methylation will turn out to be a post-translational modification as important as phosphorylation or acetylation is an attractive, but rather provocative, idea.
Materials and methods
Plasmids and expression of recombinant proteins
CBP C (aa 685–774) and CBP D (aa 770–1098) were made by inserting the corresponding cDNAs into pGEX2tk vector by PCR. CBPΔ685–774 was created by deleting by PCR a region spanning aa 685–774 from pGEX2tk CBP FL. PGEX2tk CBP 3R→A (corresponding to R714, R742 and R768 mutated to alanines) was generated by in vitro site-directed mutagenesis with GeneEditor™ (Promega). PGEX CBP B was provided by Dr T.Kouzarides and all the other pGEX CBP constructs were a kind gift from Dr A.Harel-Bellan. PGEX-KG-p300 full length, aa 1–595 and aa 1–744 were a kind gift from Drs Y.Chang and P.S.Moore. pGEX-E2F-1 (aa 89–437) was a gift from Dr N.La Thangue, pGEX-p53 was from Dr G.Bouche, pGEX-MyoD was from Dr S.A.Leibovitch, pGEX-HMT1 was from Dr M.Ferrer and pGEX-PRMT1 was from Dr H.Herschman.
The reporter vector pBS-Gal4-luciferase, the Gal4 DNA binding domain expression vector pCMV-Gal4-HA and the transfection efficiency control vector pCMV-lacZ were as described previously (Vandel et al., 2001). The reporter vectors p17ERE-β-globin-luciferase and pRARE-luciferase were from Dr H.Richard-Foy and Dr P.Balaguer, respectively. PCMV-HA-CBP was given by Dr A.Harel-Bellan. PCMV-HA-CBPΔ685–774 and pCMV-HA-CBP 3R→A were obtained by inserting an AflII–BglII fragment from pGEX2tk CBPΔ685–774 or pGEX2tk CBP 3R→A, respectively, into pCMV-HA-CBP FL cut by AflII–BglII. PCMV-Gal4-GRIP 1 was constructed by inserting an EcoRI–EcoRI GRIP-1 cDNA fragment from pSG5-GRIP1 (a kind gift from Dr M.R.Stallcup) in-frame with Gal4 into pCMV-Gal4. PHK-Gal4, pHK-Gal4 CREB and pSV-PKA+ vectors were from Dr T.Kouzarides. Details of constructs are available upon request.
GST fusion proteins were expressed in BL21 bacteria as described previously (Vandel et al., 2001).
In vitro methyltransferase assay
GST–CARM1, GST–HMT1 or GST–PRMT1 (500 ng) were incubated in the presence of various substrates. Biotinylated peptides overlapping potential methylation sites within CBP sequence were synthesized by Millegen or by Sigma Genosys and their sequences were as follows: R702/714, biotin-PPAQSVRPPNGPLPLPVNRMQVSQG; R742, biotin-NVQLPQAPMGPRAASPMNHSVQM; A742 as for R742, except that R742 was mutated to an alanine; R768, biotin-MASVPGMAISPSRMPQPPNMMGT; R15, biotin-MAENLLDGPPNPKRAKLSSPGF. Methylation reactions were performed with 6 µM of peptides (final concentration) in the presence of 50 ng of GST–CARM1, GST–PRMT1 or GST–HMT1.
Reactions were performed at 30°C for 30 min in 30 µl (final volume) of IPH buffer (50 mM Tris, pH 8, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40) after addition of 1 µCi of radiolabelled SAM (specific activity 82 Ci/mmol; Amersham). Reactions were analysed by SDS–PAGE followed by fluorography.
When using biotinylated peptides as substrates, the reaction products were diluted in 500 µl of IPH buffer and were centrifuged through a Genelute agarose spin column to get rid off the GST proteins linked to agarose beads. Streptavidin–agarose beads were then added to the flow-through and biotin–streptavidin interaction was performed for 1 h at 4°C on a rotating wheel. Beads were washed three times in IPH buffer and were added to the scintillation liquid before monitoring their radioactivity.
In vivo labelling of CBP
For endogenous CBP radiolabelling, 3 × 107 HeLa or NIH 3T3 cells were trypsinized, washed twice in phosphate-buffered saline (PBS) and cell pellet was resuspended in 2 ml of PBS containing 400 µCi of SAM. Cell labelling was performed at 37°C for 1 h on a rocking platform and excess radioactivity was removed by four washes in PBS. Cell lysis and immunoprecipitation were performed as described previously (Ait-Si-Ali et al., 1998). Immunoprecipitated proteins were resolved on an 8% SDS–PAGE; gels were then fixed, dried and fluorographed for 8 weeks.
For metabolic radiolabelling of transfected HA tagged-CBP, 2 × 106 U2OS cells were transfected by the classical calcium/phosphate technique with 10 µg of pSG5-GRIP1, 40 µg of pCMV-HA-N-CBP or with 13 µg of empty pCMV-HA vector (completed to 40 µg with pUC vector) as a control in the presence of 20 µg of pSG5-CARM1 or of empty vector pSG5 as a control. Radiolabelling of the cells was performed as for endogenous CBP and was detected after 4 weeks of exposure. In parallel with the fluorograph, 1/200th of the immunoprecipitations were loaded on an 8% SDS–PAGE and HA-N-CBP expression was detected by western blot using an anti-HA antibody followed by ECL (Roche Diagnostics).
Antibody generation
Rabbits were immunized with a CBP peptide corresponding to aa 739–746 in which R742 was asymmetrically dimethylated. The dimethylated peptide or the corresponding unmethylated peptide was coupled to keyhole limpet haemocyanin (KLH) using m-maleimido benzoyl-N-hydroxysuccinimide ester. For affinity purification, the peptides were immobilized on sulfo-linked agarose beads (Pierce). Immuno reactive serum was applied first onto the CBP R742-methylated peptide column and the obtained eluate was dialysed against PBS and was then applied onto the corresponding CBP R742 unmethylated peptide column. The flow-through obtained after the second column was concentrated with a Centricon centrifugal filter (Millipore). Specificity of each batch of purified antibody was tested by blot on a peptide dimethylated on R742 [R742(me)2], compared with various peptides derived from CBP, which were spotted onto Hybond-C in a volume of 1 µl. Specificity of the anti methylated CBP antibody was also checked by western blot after in vitro methylation of CBP by CARM1.
siRNAs and transfections
The siRNA sequence targeting CARM1 corresponded to the coding region 129–149 relative to the first nucleotide of the start codon. CARM1 specific siRNAs or the control siRNAs (Dharmacon’s control) were purchased from Dharmacon. Transfections were performed on 2 × 106 HeLa cells with a final concentration of 200 nM siRNA duplex using oligofectamine reagent (Invitrogen) according to the manufacturer’s guidelines. Sixty hours after transfection, cells were lysed in RIPA buffer and cell lysates were tested for the suppression of endogenous CARM1.
Cell culture and transfections
U2OS cells were maintained in DMEM supplemented with 10% fetal calf serum. Cells were transfected by the calcium/phosphate co-precipitation procedure. For reporter activity assays, transfections were performed with 3 × 104 cells in 12-well plates.
HeLa cells were transfected by Fugene 6 (Roche Diagnostics). For reporter activity assays, transfections were performed with 3 × 104 cells in 12-well plates. Cells were treated with 10–7 M all-trans retinoic acid 24 h before being harvested, as indicated.
MCF-7 cells were grown in Phenol Red-free DMEM with 5% dextran-charcoal stripped fetal bovine serum for 3 days before being transfected with Fugene 6. Transfections were performed with 5 × 104 cells in 6-well plates. Cells were treated with 10–8 M 17β estradiol 24 h before being harvested, as indicated.
The amount of CMV promoter was kept constant using empty vectors, and the total amount of DNA was 15 µg/well. Cells were harvested 36 h after transfection. For luciferase assays, pCMV-lacZ was included in each experiment as a control for transfection efficiency and only transfections with similar β-galactosidase values were taken into account. Luciferase and β-galactosidase activities were measured using Promega and Tropix kits respectively, according to the manufacturer’s instructions.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Drs A.Harel-Bellan, Y.Chang, P.S.Moore, G.Bouche, T.Kouzarides, M.Ferrer, H.Richard-Foy, P.Balaguer and H.Herschman for reagents. We also thank M.Calligé for helpful advice with MCF7 cell experiments and L.Fauquier for help with antibody purification. We are grateful to Dr M.G.Poupinot for critical reading of the manuscript. This work was supported by a grant from ‘La Ligue Nationale Contre Le Cancer’ to D.T. as an ‘équipe labellisée’.
References
- Ait-Si-Ali S. et al. (1998) Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature, 396, 184–186. [DOI] [PubMed] [Google Scholar]
- Arias J., Alberts,A.S., Brindle,P., Claret,F.X., Smeal,T., Karin,M., Feramisco,J. and Montminy,M. (1994) Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature, 370, 226–229. [DOI] [PubMed] [Google Scholar]
- Bannister A.J. and Kouzarides,T. (1996) The CBP co-activator is a histone acetyltransferase. Nature, 384, 641–643. [DOI] [PubMed] [Google Scholar]
- Bauer U.M., Daujat,S., Nielsen,S.J., Nightingale,K. and Kouzarides,T. (2002) Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO rep., 3, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford M.T., Frankel,A., Yaffe,M.B., Clarke,S., Leder,P. and Richard,S. (2000) Arginine methylation inhibits the binding of proline-rich ligands to Src homology 3, but not WW, domains. J. Biol. Chem., 275, 16030–16036. [DOI] [PubMed] [Google Scholar]
- Chen D., Ma,H., Hong,H., Koh,S.S., Huang,S.M., Schurter,B.T., Aswad,D.W. and Stallcup,M.R. (1999) Regulation of transcription by a protein methyltransferase. Science, 284, 2174–2177. [DOI] [PubMed] [Google Scholar]
- Chen D., Huang,S.M. and Stallcup,M.R. (2000) Synergistic, p160 coactivator-dependent enhancement of estrogen receptor function by CARM1 and p300. J. Biol. Chem., 275, 40810–40816. [DOI] [PubMed] [Google Scholar]
- Eckner R., Yao,T.P., Oldread,E. and Livingston,D.M. (1996) Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes Dev., 10, 2478–2490. [DOI] [PubMed] [Google Scholar]
- Frankel A. and Clarke,S. (2000) PRMT3 is a distinct member of the protein arginine N-methyltransferase family. Conferral of substrate specificity by a zinc-finger domain. J. Biol. Chem., 275, 32974–32982. [DOI] [PubMed] [Google Scholar]
- Frankel A., Yadav,N., Lee,J., Branscombe,T.L., Clarke,S. and Bedford,M.T. (2002) The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J. Biol. Chem., 277, 3537–3543. [DOI] [PubMed] [Google Scholar]
- Gary J.D. and Clarke,S. (1998) RNA and protein interactions modulated by protein arginine methylation. Prog. Nucleic Acid Res. Mol. Biol., 61, 65–131. [DOI] [PubMed] [Google Scholar]
- Goodman R.H. and Smolik,S. (2000) CBP/p300 in cell growth, transformation and development. Genes Dev., 14, 1553–1577. [PubMed] [Google Scholar]
- Henry M.F. and Silver,P.A. (1996) A novel methyltransferase (Hmt1p) modifies poly(A)+-RNA-binding proteins. Mol. Cell. Biol., 16, 3668–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvai A.E., Xu,L., Korzus,E., Brard,G., Kalafus,D., Mullen,T.M., Rose,D.W., Rosenfeld,M.G. and Glass,C.K. (1997) Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl Acad. Sci. USA, 94, 1074–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei Y. et al. (1996) A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell, 85, 403–414. [DOI] [PubMed] [Google Scholar]
- Korzus E., Torchia,J., Rose,D.W., Xu,L., Kurokawa,R., McInerney,E.M., Mullen,T.-M., Glass,C.K. and Rosenfeld,M.G. (1998) Transcription factor specific requirements for coactivators and their acetyltransferase functions. Nature, 279, 703–707. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J., 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2002) Histone methylation in transcriptional control. Curr. Opin. Genet. Dev., 12, 198–209. [DOI] [PubMed] [Google Scholar]
- Kwok R.P.S., Lundblad,J.R., Chrivia,J.C., Richards,J.P., Bächinger,H.P., Brennan,R.G., Roberts,S.G.E., Green,M.R. and Goodman,R.H. (1994) Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature, 370, 223–226. [DOI] [PubMed] [Google Scholar]
- Lin W.J., Gary,J.D., Yang,M.C., Clarke,S. and Herschman,H.R. (1996) The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J. Biol. Chem., 271, 15034–15044. [DOI] [PubMed] [Google Scholar]
- Ma H. et al. (2001) Hormone-dependent, CARM1-directed, arginine-specific methylation of histone H3 on a steroid-regulated promoter. Curr. Biol., 11, 1981–1985. [DOI] [PubMed] [Google Scholar]
- Martinez-Balbas M.A., Bannister,A.J., Martin,K., Haus-Seuffert,P., Meisterernst,M. and Kouzarides,T. (1998) The acetyltransferase activity of CBP stimulates transcription. EMBO J., 17, 2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowen K.A., Tang,J., Zhu,W., Schurter,B.T., Shuai,K., Herschman,H.R. and David,M. (2001) Arginine methylation of STAT1 modulates IFNα/β-induced transcription. Cell, 104, 731–741. [DOI] [PubMed] [Google Scholar]
- Ogryzko V.V., Schiltz,R.L., Russanova,V., Howard,B.H. and Nakatani,Y. (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- Pollack B.P., Kotenko,S.V., He,W., Izotova,L.S., Barnoski,B.L. and Pestka,S. (1999) The human homologue of the yeast proteins Skb1 and Hsl7p interacts with Jak kinases and contains protein methyltransferase activity. J. Biol. Chem., 274, 31531–31542. [DOI] [PubMed] [Google Scholar]
- Shang Y., Hu,X., DiRenzo,J., Lazar,M.A. and Brown,M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell, 103, 843–852. [DOI] [PubMed] [Google Scholar]
- Smith C.L., Onate,S.A., Tsai,M.J. and O’Malley,B.W. (1996) CREB binding protein acts synergistically with steroid receptor coactivator-1 to enhance steroid receptor-dependent transcription. Proc. Natl Acad. Sci. USA, 93, 8884–8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl B.D. et al. (2001) Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol., 11, 996–1000. [DOI] [PubMed] [Google Scholar]
- Torchia J., Glass,C. and Rosenfeld,M.G. (1998) Co-activators and co-repressors in the integration of transcriptional responses. Curr. Opin. Cell Biol., 10, 373–383. [DOI] [PubMed] [Google Scholar]
- Vandel L., Nicolas,E., Vaute,O., Ferreira,R., Ait-Si-Ali,S. and Trouche,D. (2001) Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol. Cell. Biol., 21, 6484–6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N. and Goodman,R.H. (2001) CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem., 276, 13505–13508. [DOI] [PubMed] [Google Scholar]
- Wang H. et al. (2001) Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science, 293, 853–857. [DOI] [PubMed] [Google Scholar]
- Xu W., Chen,H., Du,K., Asahara,H., Tini,M., Emerson,B.M., Montminy,M. and Evans,R.M. (2001) A transcriptional switch mediated by cofactor methylation. Science, 294, 2507–2511. [DOI] [PubMed] [Google Scholar]
- Yuan W., Condorelli,G., Caruso,M., Felsani,A. and Giordano,A. (1996) Human p300 protein is a coactivator for the transcription factor MyoD. J. Biol. Chem., 271, 9009–9013. [DOI] [PubMed] [Google Scholar]
- Zanger K., Radovick,S. and Wondisford,F.E. (2001) CREB binding protein recruitment to the transcription complex requires growth factor-dependent phosphorylation of its GF box. Mol. Cell, 7, 551–558. [DOI] [PubMed] [Google Scholar]
- Zhang Y. and Reinberg,D. (2001) Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev., 15, 2343–2360. [DOI] [PubMed] [Google Scholar]