

Abstract
Lineage specification and cellular maturation require coordinated regulation of gene expression programs. In large part, this is dependent on the activator and repressor functions of protein complexes associated with tissue-specific transcriptional regulators. In this study, we have used a proteomic approach to characterize multiprotein complexes containing the key hematopoietic regulator SCL in erythroid and megakaryocytic cell lines. One of the novel SCL-interacting proteins identified in both cell types is the transcriptional corepressor ETO-2. Interaction between endogenous proteins was confirmed in primary cells. We then showed that SCL complexes are shared but also significantly differ in the two cell types. Importantly, SCL/ETO-2 interacts with another corepressor, Gfi-1b, in red cells but not megakaryocytes. The SCL/ETO-2/Gfi-1b association is lost during erythroid differentiation of primary fetal liver cells. Genetic studies of erythroid cells show that ETO-2 exerts a repressor effect on SCL target genes. We suggest that, through its association with SCL, ETO-2 represses gene expression in the early stages of erythroid differentiation and that alleviation/modulation of the repressive state is then required for expression of genes necessary for terminal erythroid maturation to proceed.
Hematopoiesis is a complex differentiation process that involves commitment of stem cells to progenitors with progressive restriction of their lineage potentiality and proliferative capacity (58). Stochastic events are believed to initiate this highly hierarchical differentiation system in hematopoietic stem cells (HSCs) and progenitors, where multilineage priming is observed (30, 38). Progressive establishment of highly specialized transcriptional programs then takes place in the HSC progeny. Several lines of evidence show that, among the hematopoietic lineages, the erythroid and megakaryocytic lineages are closely related. First, erythroleukemic and megakaryoblastic cell lines were shown to coexpress erythroid and megakaryocytic markers (45, 60, 68). Second, bipotent primary progenitors were characterized in culture assays (18) and isolated by cell surface phenotype (1, 74, 78). Finally, analysis of mouse knockout models has revealed that some tissue-specific transcriptional regulators such as SCL/Tal-1, GATA-1, FOG1, and NF-E2 are required for differentiation of both lineages (see below) (72, 73, 75, 82). However, although these lineages emerge from a common progenitor and share common transcription factors, terminal differentiation results in production of cellular entities (erythrocytes and platelets) with very distinct biological properties. Understanding the different cellular and molecular events that govern differentiation along these two cell types might give insight into binary decision pathways and lineage specification/maturation. We chose to analyze the molecular basis of these differences by characterizing multiprotein complexes nucleated by a key transcription factor, SCL/Tal-1 (hereafter referred as to SCL), that plays crucial roles in erythroid and megakaryocytic development.
The basic helix-loop-helix (bHLH) transcription factor SCL, involved in up to 60% of T-cell acute lymphoblastic leukemia (T-ALL) cases, is required at different stages of normal hematopoietic development (for a review, see reference 42). In addition to its crucial role in the specification, but not maintenance, of hematopoietic stem cells (27, 51, 63, 65, 66), SCL is essential for maturation of the erythroid and megakaryocytic lineages. This was initially suggested by overexpression experiments with an erythroid cell line (6). More recently, analysis of hematopoiesis derived from SCL−/− embryonic stem (ES) cells rescued with an SCL DNA-binding mutant showed differentiation blocks in erythroid and megakaryocytic maturation (62). Finally, conditional knockout studies have revealed that erythroid and megakaryocytic precursors do not develop in the bone marrow of mice upon deletion of SCL (27, 51).
To exert its functions, SCL acts through both DNA-binding-dependent and -independent mechanisms (62) and interacts with protein partners. Heterodimerization of SCL with the ubiquitously expressed E proteins (E2A, HEB, and E2-2) is a prerequisite for all its functions (62). SCL is also part of multiprotein complexes. It was initially described in a pentameric complex comprising SCL, E2A, the LIM-only protein LMO2, the LIM-binding protein Ldb-1, and the zinc finger transcription factor GATA-1 in vitro in erythroid cells (81). These proteins were later shown to co-occupy regulatory sequences of GATA-1, α-globin, and glycophorin A (GPA) genes in vivo (5, 40, 76). In a recent study, we have characterized several functionally critical residues in the SCL HLH domain. These amino acids presumably promote protein-protein interactions that remain to be characterized (70).
Like most transcription factors (22, 83, 85), SCL is both a positive and a negative transcriptional regulator. SCL positively regulates erythroid differentiation when overexpressed in mouse erythroleukemia (MEL) cells (6). Because SCL/E2A heterodimers are less potent activators than E2A homodimers, it was, however, postulated that SCL could exert repressor effects on gene expression (29, 61). SCL also appeared to function as a repressor of erythroid differentiation in the human cell line K562 in antisense experiments (26) and can block differentiation when ectopically expressed in normal and leukemic myeloid precursors (14). Finally, the pentameric complex is believed to positively regulate expression of GPA and protein 4.2 (40, 87) and to both activate and repress expression of the c-kit receptor depending on the cellular context and nature of additional partners (41). Interestingly, in T-ALL cells where it is ectopically expressed, SCL is also believed to have activator (56, 57) and repressor (28, 55) functions.
Activation and repression of gene expression are achieved through multiple mechanisms including recruitment of chromatin-remodelling factors and histone-modifying proteins like histone acetyltransferases (HATs) and histone deacetylases (HDACs) (9, 39). Interestingly, coimmunoprecipitation (coIP) of SCL with HATs CBP/p300 and pCAF (33, 34) as well as with the corepressor mSin3A and associated HDACs (31) has been reported in an erythroid cell line, thereby reinforcing the hypothesis that SCL might have a dual function, as an activator and a repressor. A recent study also indicated that SCL binds pericentromeric DNA and mediates repression by chromatin remodelling in K562 cells (84).
In order to undertake a full characterization of the nature of SCL protein partners, we have isolated SCL-containing multiprotein complexes upon biotin/streptavidin affinity purification and identified the associated factors by mass spectrometry (19). We have analyzed both erythroid and megakaryoblastic cell lines as a first step towards the characterization of protein complexes nucleated by SCL in two related but distinct hematopoietic cell types. Here, we show that SCL interacts with important corepressors ETO-2 and Gfi-1b (growth factor independence 1b) and that the composition of the SCL/ETO-2 protein complexes in erythroid cells differs from that in megakaryocytes. Functional studies suggest that these multiprotein complexes are involved in transcriptional repression of genes important for hematopoietic differentiation.
MATERIALS AND METHODS
Constructs.
The Escherichia coli BirA biotin ligase gene (19) was inserted as a PCR fragment downstream of the human EF1α promoter in a vector bearing a puromycin resistance gene (pEF1α-BirA). Wild-type SCL cDNA was tagged at the 5′ end with an oligonucleotide sequence encoding a 23-amino-acid biotinylation tag (19) using a PCR strategy and cloned downstream of the EF1α promoter in a vector bearing a Zeocin resistance gene (pEF1α-bio-SCL). The truncated form of SCL (bHLH domain only), swapped and point mutants (SES, SNS, SMS, RER, FL, and H2(F-G) (62, 70), and mouse SSDP2 cDNA (kind gift from D. van Meyel) (77) were subcloned into the pEF1α-bio-SCL vector in place of the wild-type SCL sequence using a PCR-based strategy. To achieve constitutive overexpression of ETO-2 and E2A cDNAs (kind gifts from S. Meyers and T. Hoang, respectively), Flag-tagged coding sequences were subcloned into the pHR-SIN-CSGW-EGFP lentiviral expression construct (20) (kind gift from Adrian Thrasher). To generate the E2A mutant form impaired in its ability to bind ETO-2 (called DM), point mutations were engineered in the E2A cDNA that resulted into mutation of two residues in activation domain 1: D19A and D22A (89). All PCR fragments were generated using Pfu polymerase (Stratagene) or Expand Long Template (Roche) and verified by sequencing.
Cell lines and transfections.
MEL (clone 585) and NIH 3T3 cells were maintained in RPMI 1640 supplemented with 10% fetal calf serum (FCS) and 2 mM l-glutamine. MEL cells were induced to terminally differentiate by addition of 2% dimethyl sulfoxide to the media for 3 to 5 days. Benzidine and May-Grunwald-Giemsa (MGG) stainings were performed according to standard protocols. L8057 cells (murine megakaryoblastic cell line) (37) were grown in 50% Iscove's modified Dulbecco medium-50% RPMI 1640 in the presence of 15% FCS and 2 mM l-glutamine. MEL or L8057 cells (5 × 106) were electroporated with 20 μg of linearized pEF1α-BirA (220 V, 960 μF for MEL cells; 160 V, 250 μF for L8057 cells). Stable clones were selected under puromycin (Sigma; 2 μg/ml). For each cell line, a clone expressing high levels of BirA mRNA (not shown) was chosen and subsequently transfected with all (MEL cells) or only some (L8057 cells) of the following constructs: pEF1α-bioSCL (wild-type and mutants) and -bioSSDP2. Stable clones were selected using puromycin (as above) and Zeocin (Invitrogen; 200 μg/ml). 293T cells maintained in Dulbecco's modified Eagle medium-Glutamax (Gibco) plus 10% FCS, 50 U ml−1/50 μg ml−1 penicillin/streptomycin, and 2 mM l-glutamine were used to package lentiviral expression constructs into infectious particles.
Nuclear extract preparations.
Small-scale nuclear extracts were prepared from 106 to 107 cells as described previously (4). Large-scale nuclear extracts were prepared from 3 liters of uninduced MEL and L8057 cells expressing BirA only or BirA and bio-SCL (wild type or mutants) using salt extraction (21). Cells were harvested by centrifugation at 3,500 rpm and washed once in phosphate-buffered saline. The cell pellets were resuspended in 10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, protease inhibitors (Complete, EDTA free; Roche), and 0.5 mM dithiothreitol. Cells were spun down, resuspended in the same buffer, and transferred into a loose Dounce homogenizer. After ultracentrifugation of homogenates for 1 h at 20,000 rpm and 4°C, the nuclear pellets were resuspended in 8 ml of 150 mM Heng buffer (150 mM KCl, 20 mM HEPES, 20% glycerol, 0.25 mM EDTA, 0.05% NP-40). One milliliter of 2.2 M KCl Heng buffer was added dropwise to achieve a final salt concentration of 420 mM KCl, and nuclei were homogenized using a tight Dounce homogenizer. The homogenates were ultracentrifugated at 40,000 rpm for 1 h at 4°C and supernatants aliquoted and stored at −80°C.
Gel filtration.
Crude nuclear extracts prepared from wild-type MEL and L8057 cells (25 mg total protein) were precipitated with 55% saturated ammonium sulfate and the precipitated proteins redissolved in 20 mM Tris, pH 8, 200 mM NaCl, 10% glycerol, and 0.05% Triton. Concentrated nuclear extracts were fractionated on a Superose 6H/R column (Amersham Biosciences) in the same buffer. Fractions (0.5 ml) were collected at a flow rate of 0.4 ml/min. Individual fractions were then subjected to Western blotting.
Western blotting.
Western blot analyses were performed as described previously (62) using precast NuPAGE gels (4 to 12% Bis-Tris; Invitrogen) according to manufacturer's instructions. For primary antibodies used for detection see the supplementary material. Secondary antibodies were conjugated protein A- or protein G-HRP (horseradish peroxidase). Detection was performed with the ECL kit (Amersham/Pharmacia). For quantitative analyses, signal intensities were measured with Quantity One software (Bio-Rad).
Biotin-streptavidin pull-downs.
Pull-downs were performed with 5 mg of nuclear extracts as described previously (19) in 50 mM Tris-150 mM NaCl-0.3% Nonidet P-40 (NP-40) followed by washes in 50 mM Tris-250 mM NaCl-0.3% NP-40, unless otherwise indicated. Twenty-nine microliters (of 30 μl) of eluate was run on a gel for colloidal-Coomassie blue staining and subsequent mass spectrometry analysis. For Western blot analyses, crude nuclear extracts (input [IN], 10 μg), pull-down products (PD, 1/30 of the eluate), and unbound fractions (UN, 10 μg) were resolved on a 4 to 12% Bis-Tris NuPAGE gel in MOPS (morpholinepropanesulfonic acid) buffer (Invitrogen).
Coimmunoprecipitation and depletion experiments.
Nuclear extracts were diluted in 50 mM Tris-0.3% NP-40 to obtain a final concentration of 150 mM NaCl and precleared at 4°C with normal immunoglobulin G (IgG; Santa Cruz; rat sc-2026, rabbit sc-2027, and goat sc-2028) and protein G beads (Fastflow; Sigma). Beads were pelleted and kept as the IgG control. The supernatant was immunoprecipitated overnight at 4°C with primary antibodies and protein G beads. For immunoprecipitation (IP) with anti-GATA-1 N6 antibody, a rabbit anti-rat bridging antibody (Jackson Immunodiagnostics) bound to beads was used. IgG and IP fractions were washed in 50 mM Tris-250 mM NaCl-0.3% NP-40 and boiled in 1× Laemmli buffer together with IN (10-μg) and UN (10-μg) fractions. For depletion experiments, the supernatant of the first immunoprecipitation was immunoprecipitated again. The four fractions (IN, IP, UN, and IgG) were loaded onto NuPAGE gels. For immunoprecipitation with anti-Flag antibodies, the anti-Flag affinity gel was used according to the manufacturer's instructions (Sigma).
Note that migration of ETO-2 in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels is sensitive to salt concentrations. We sometimes observe differences in ETO-2 mobility in IN/UN fractions versus IP or PD fractions.
Immunolabeling and colocalization.
MEL and L8057 cells were fixed on coverslips in 4% paraformaldehyde and antigens indirectly immunolabeled with monoclonal mouse antibodies directed against SCL (1/100 dilution) and SC35 (1/100 dilution), rabbit polyclonal anti-Ldb-1 antibody (1/100 dilution), or goat anti-ETO-2 antibody (1/20 dilution). Secondary antibodies used were Alexa Fluor 488-conjugated donkey anti-mouse (Molecular Probes), Alexa Fluor 546-conjugated donkey anti-goat (Molecular Probes), and Cy-3-conjugated donkey anti-rabbit (Jackson Laboratory) antibodies. Nucleic acids were counterstained with DAPI (4′,6′-diamidino-2-phenylindole; not shown), monochrome images collected, and fluorescence intensities measured as described previously (36). Images were exported into Adobe Photoshop and contrast stretched and pseudocolored images generated.
Mass spectrometry.
Proteins eluted from the beads were separated on a 4 to 12% SDS-PAGE gel and stained with colloidal Coomassie blue (Invitrogen). The entire lane was cut into 25 slices and subjected to trypsin digestion using either a MWG Roboseq 4204 or a QIAGEN 3000 robot. Analysis of tryptic digests was performed on a MicroMass Q-TOF Global with a capillary high-pressure liquid chromatography system with a nanospray probe. Five microliters of sample was run on a long column (15 cm by 75 μm C18, 3 μm, 100 A) to separate out peptides. Database searches were performed with MASCOT using the following settings: fixed modifications, carbamidomethyl (C); variable modifications, oxidation (M), peptide charge 2+ and 3+. The data format was pkl (www.matrixscience.com).
Transactivation assays.
Twenty-four hours after being plated in 24-well plates at a density of 7 × 104 cells/well, NIH 3T3 cells were transfected using the Lipofectamine 2000 reagent (Invitrogen). Expression vectors for members of the pentameric complex and ETO-2 were kind gifts from T. Hoang and S. Meyers, respectively. Luciferase reporter gene expression was under the control of the human GATA-1 IE promoter, HS-3.5, and HS+14 (P. Vyas, unpublished data) (76). Amounts of transfected DNA were adapted from reference 41. Twenty-four hours posttransfection, cells were lysed and luciferase and β-galactosidase (for transfection efficiencies) activities measured using standard procedures (Roche). Each transfection was performed in duplicate, and data presented are from three to five independent experiments.
Purification of mouse primary hematopoietic cells. (i) Ter119+ splenocytes.
C57BL/6 mice were injected intraperitoneally with phenylhydrazine (Sigma; 0.04 mg/g body weight) three times at 12-hour intervals. Ter119+ cells were isolated at day 6 by labeling splenocytes with biotin-conjugated rat anti-mouse Ter119 antibody (BD Pharmingen) and incubating them with antibiotin microbeads (Miltenyi Biotec). The positive fraction was recovered on AUTOMACS (MiltenyiBiotec). Purity was assessed by May-Grunwald-Giemsa staining.
(ii) Primary megakaryocytes.
C57BL/6 mice were treated with 5-fluorouracil (150 mg/kg, intraperitoneal). Eight days postinjection, bone marrow was replated at a density of 5 × 106 cells/ml in serum-free medium (StemPro-34; Invitrogen) with thrombopoietin (1% conditioned medium). After 3 days of culture, megakaryocytes were isolated by immunodepletion of Ter119-, Mac1-, Gr1-, and B220-positive cells using biotin-conjugated rat anti-mouse antibodies (BD Pharmingen) and streptavidin microbeads (Miltenyi Biotec). The positive and negative fractions were then separated on magnetically activated cell sorting columns for large cells, according to the manufacturer's instructions (Miltenyi Biotec). Purity of the negative fraction was assessed by May-Grunwald-Giemsa staining and flow cytometry (data not shown). The cell population was at least 95% CD61+ megakaryocytes (data not shown).
(iii) Expansion and erythroid differentiation of day 12.5 wild-type fetal liver cells.
Fetal liver cells from day 12.5 wild-type mouse embryos (CBA) were expanded and subsequently differentiated towards the erythroid lineage as described previously (13, 80). Cells were harvested before induction of differentiation (day 0) and after 24 h (day 1) and 48 h (day 2) for May-Grunwald-Giemsa staining, flow cytometry analysis, and preparation of nuclear extracts.
Lentiviral packaging and infection.
For each lentiviral construct, Gag-Pol expression (pCMVΔR8.9) and vesicular stomatitis virus G expression plasmids (pMDG, both plasmids kind gifts from Didier Trono, EPFL, Lausanne, Switzerland) were added to a pHR-SIN-CSGW-derived gene expression construct in the ratio 1:1:1.5. Seventy to 95% confluent 293T cells in 75-cm2 tissue culture flasks were transfected using FuGENE 6 (Roche). Seventy-two hours posttransfection, virus-containing supernatants were concentrated via centrifugation at 1,500 rpm for 90 min at 15°C through Amicon Centricon Plus-70 centrifugal filter units (Millipore). Viral concentrates were recovered on spinning at 1,500 rpm for 5 min at 15°C. Concentrated viral particles were stored at −80°C. MEL cells were transduced by incubation with viral supernatants for 24 h, after which the cells were washed three times and returned to culture for a further 48 h before sorting green fluorescent protein (GFP)-positive cells on a MoFlo FACsorter (DakoCytomation). Sorted cells were utilized directly for their destination assays or returned to culture. The following cellular and molecular analyses were performed at 72 h postinfection (sorting day; see above) and repeated at day 10 after sorting. For proliferation assays, cells were seeded at 5 × 104 cells/ml and viable cells counted over a period of 6 to 8 days. A fraction of transduced MEL cells were induced to differentiate and nuclear extracts prepared from uninduced (day 0) and induced (day 1 to day 3) cells for analyses of the level of expression of the exogenous proteins compared to the endogenous proteins. RNA was also extracted for SCL target gene expression analyses. Morphology of the transduced cells was checked by MGG staining. Finally, unsorted cells were regularly analyzed by flow cytometry on a CyAn analyzer (DakoCytomation) to follow the GFP+ fraction over time. Two independent lentiviral infections were performed.
Real-time PCR.
RNA was extracted from induced and uninduced infected MEL cells using the RNA extraction Mini kit (QIAGEN) and cDNA synthesized using the Sensiscript kit (QIAGEN) according to the manufacturer's instructions. c-kit, GPA, α-globin, and band 4.2 cDNA sequences were analyzed in duplicate with ready-made primers and probe mix from Applied Biosystems (ABI) using an ABI Prism 7000 sequence detection system. Results were related to a control GAPDH (glyceraldehyde-3-phosphate dehydrogenase) cDNA sequence.
For chromatin immunoprecipitation (ChIP) experiments, primers and 5′-6-carboxyfluorescein-3′-6-carboxy tetramethylrhodamine-labeled probes were selected from unique sequences in the murine α-globin locus and appropriate external controls using Primer Express (5). Input and immunoprecipitated materials were analyzed in duplicate as described previously (5).
ChIP experiments.
Day 12.5 fetal liver cells were expanded for 3 days and magnetically purified using Ter119 antibodies (BD Pharmingen) as described previously (80). Protein-DNA cross-linking was performed on 1 × 107 Ter119+ or Ter119− cells with 1% formaldehyde for 15 min at room temperature. Glycine (0.125 M) was added to quench the reaction. Cells were washed twice in phosphate-buffered saline containing protease inhibitors (Complete; Roche). Nuclei were lysed in lysis buffer (ChIP assay kit; Upstate Biotechnology) and sheared by sonication to reduce the chromatin fragments to 200 to 500 base pairs. Immunoprecipitation of cross-linked chromatin was performed using anti-SCL, anti-ETO-2, and IgG control antibodies and ready-made buffers (ChIP assay kit; Upstate Biotechnology).
RESULTS
In vivo biotinylation of SCL.
To demonstrate that SCL is detected in multimeric protein complexes in cells chosen for purification of its partners, we first performed gel filtration analyses of nuclear extracts with a Superose 6 column from uninduced MEL cells. Collected fractions were subjected to Western blot analysis (Fig. 1A). SCL elutes in fractions 6 to 10, which represent an apparent molecular mass of 670 kDa, and is therefore present in one or more high-molecular-mass multiprotein complexes. A small proportion of SCL is also detected in a lower-molecular-mass fraction (fraction 18). Interestingly, two previously characterized partners of SCL, GATA-1 and Ldb-1, coeluted with SCL in the high-molecular-weight fractions. Very similar results were obtained after fractionation of nuclear extracts purified from the L8057 megakaryoblastic cell line (data not shown). Therefore, in erythroid cells and megakaryocytes, SCL, GATA-1, and Ldb-1 associate with other nuclear protein partners to form multiprotein complexes. A smaller proportion of SCL may also exist as heterodimers with E proteins or in small protein complexes.
FIG. 1.

Setting up the biotin/streptavidin system in MEL and L8057 cells. (A) Gel filtration analysis. MEL cell nuclear extracts were fractionated on a Superose 6 H/R gel filtration column. The indicated fractions were analyzed by Western blotting for SCL, GATA-1, and Ldb1. Protein molecular masses in kilodaltons are indicated by arrows. In the bottom panel, the UV profile shows the void and two elution peaks containing the majority of the protein complexes. (B) Scheme of the biotin/streptavidin protein purification. (C) Nuclear extracts were prepared from wild-type MEL cells and from independent MEL and L8057 clones coexpressing bio-SCL or the bio-H2(F-G) mutant and the BirA biotin ligase. Western blots were performed using anti-SCL (αSCL) antibodies (top panel; note that endogenous SCL is not detected in MEL and L8057 cells expressing wild-type [wt] bio-SCL). Incubation with streptavidin-HRP conjugate (bottom panel) confirms biotinylation of SCL (wild type and mutant). (D) Quantitation of SCL protein expression in transfected MEL clones by Western blot analysis. Ratios represent SCL expression levels in nuclear extracts from wild-type MEL cells and clones expressing bio-SCL [wild type, top panel; H2(F-G) mutant, bottom panel] relative to expression levels of loading control GRB2. Triangles represent serial dilutions of the input.
To identify SCL protein partners in both erythroid cells and megakaryocytes, we used a recently described strategy relying on in vivo biotinylation of the protein of interest, precipitation of protein complexes by streptavidin affinity, and analysis of purified products by Western blotting and mass spectrometry (19) (Fig. 1B).
Before characterizing SCL-containing multiprotein complexes, we first tested the functionality of biotinylated SCL. We took advantage of the rescue assay of SCL-null ES cells, whereby introduction of a wild-type SCL cDNA fully restores hematopoietic development from these cells (63). We expressed biotinylated SCL in SCL−/− ES cells and submitted the cells to in vitro differentiation into hematopoietic lineages (see the supplemental material). Development of both primitive and definitive hematopoiesis was observed in a manner similar to that observed from control wild-type ES cells (see Fig. S1 in the supplemental material). We concluded that biotinylation of SCL did not perturb hematopoietic specification and lineage maturation from SCL-null ES cells.
MEL and L8057 cells were stably transfected with the bacterial BirA biotin ligase. Clones expressing high levels of the ligase transcripts were selected (data not shown) and subsequently transfected with a wild-type SCL cDNA fused to a biotin tag (hereafter named bio-SCL). A variant form of SCL bearing a single point mutation in helix 2 [phenylalanine to glycine, mutant SCL H2(F-G)] that is impaired in its ability to rescue hematopoiesis from SCL-null ES cells (70) was also studied. Expression of bio-SCL [wild type and mutant H2 (F-G)] in independent MEL and L8057 cell clones was then checked by Western blot analysis (Fig. 1C, top panel). Endogenous SCL was not detected in MEL and L8057 clones transfected with wild-type bio-SCL, as only the slow-migrating biotin-tagged protein was observed. In contrast, MEL and L8057 cells transfected with bio-SCL H2(F-G) retained expression of endogenous SCL, as bands corresponding to both tagged and untagged SCL proteins are detected (Fig. 1C, top panel). Efficient biotinylation of bio-SCL was then checked using streptavidin-HRP conjugate (Fig. 1C, bottom panel). Down-regulation of expression of the endogenous SCL gene in MEL and L8057 clones transfected with wild-type bio-SCL (and possible perturbation in clones expressing the bio-SCL variant) precluded any direct comparison of levels of expression of the transgenes versus the endogenous gene. Therefore, to select MEL and L8057 clones with levels of bio-SCL comparable to that of endogenous SCL, levels of expression of bio-SCL and of a loading control (GRB2) in transfected cells, and of wild-type SCL and GRB2 in untransfected cells, were compared and ratios calculated. Transfected clones that showed ratios comparable to that of untransfected cells were chosen for further analyses (Fig. 1D and data not shown).
Previous studies reported that SCL overexpression in MEL cells induced spontaneous terminal maturation (6). To check whether this applied to MEL clones expressing bio-SCL or bio-H2(F-G), benzidine staining of the cells was performed before and after exposure to dimethyl sulfoxide, an inducer of terminal differentiation. Cells did not show enhanced differentiation, either in the presence or absence of inducer (see Fig. S2 in the supplemental material).
In conclusion, the function of SCL in hematopoietic cells is not affected by biotinylation. Moreover, we generated MEL and L8057 clones expressing biotinylated SCL (wild type and mutant) at levels similar to that of the endogenous protein in wild-type cells, thus preserving normal protein stoichiometry.
Purification of SCL-containing multiprotein complexes reveals newly identified protein interactions.
Cells contain endogenously biotinylated proteins or peptide sequences that might be recognized by biotin ligase BirA. Therefore, to identify proteins specifically pulled down upon SCL biotinylation, crude nuclear extracts were prepared from MEL and L8057 clones coexpressing bio-SCL and BirA ligase as well as from those expressing BirA alone. Biotinylated multiprotein complexes were precipitated by streptavidin beads. After elution, complexes were analyzed by Western blotting. The presence of precipitated bio-SCL and bio-H2(F-G) specifically from MEL and L8057 clones transfected with bio-SCL was confirmed (Fig. 2A, PD fractions). Most biotinylated SCL bound to the streptavidin beads with very little or no product being present in the UN fraction. We next checked whether known partners had copurified with wild-type bio-SCL. We were able to detect E2A, HEB, Ldb-1, LMO2, and GATA-1 in PD fractions, thereby validating the strategy. GATA-1 was pulled down only when the salt concentration in the washing buffer was decreased from 250 to 150 mM NaCl. These proteins were never detected in the PD fraction from cells transfected with BirA only, confirming that they had been specifically pulled down upon biotinylation of SCL. Interestingly, pull-down profiles observed from MEL and L8057 cells were similar, with substantial enrichment for E2A (E12/47), HEB, LMO2, and Ldb-1. Enrichment of GATA-1 in both cell types was not as prominent, suggesting that only a small fraction of total GATA-1 might be present in SCL-containing protein complexes. Using the biotin system, we previously reported that the H2(F-G) SCL mutant, although able to heterodimerize with E2A and HEB, was impaired in its ability to bind LMO2 in MEL cells (70). We now show that, in MEL cells, the binding of Ldb-1 is also affected by this mutation. Enrichment of Ldb-1 in the PD fraction is not as strong with the H2(F-G) mutant as with wild-type bio-SCL (Fig. 2A, left panel). Interestingly, the mutation seems to have a more dramatic effect on the composition of an SCL-containing multiprotein complex in L8057 cells compared to MEL cells. The binding of SCL heterodimerization partners E2A and HEB was also reduced. Presumably as a consequence of this, the binding of other partners was also either absent or reduced (Fig. 2A, right panel, PD fractions).
FIG. 2.

Two newly identified partners of SCL. (A) Nuclear extracts from MEL and L8057 cells transfected with BirA biotin ligase only (negative control) or BirA and bio-SCL or bio-H2(F-G) were analyzed by Western blot after streptavidin pull-down for SCL and known partners as indicated on the left. (B) Protein complexes pulled down from MEL and L8057 clones transfected with BirA only or expressing biotinylated wild-type SCL (BirA+bio-SCL) were resolved by SDS-PAGE. Proteins were revealed by colloidal-Coomassie blue staining. Biotinylated SCL is indicated. (C) The numbers of peptides identified in the mass spectrometry analysis for SCL, its known partners (E12/E47, E2.2, and Ldb-1), and two newly identified partners, SSDP2 and ETO-2, are shown.
To identify additional putative partners, SCL-containing complexes from MEL and L8057 cells expressing BirA and wild-type bio-SCL or BirA only were separated on a colloidal-Coomassie blue-stained gel prior to mass spectrometry. Analysis of the gel revealed different patterns between control cells and cells expressing bio-SCL (Fig. 2B). From MEL cells expressing bio-SCL, there was a significant enrichment in bands not present in the BirA-only control. From L8057 cells, although the control lane showed more background biotinylated proteins, there were also significant differences compared to cells expressing bio-SCL. The background proteins identified by mass spectrometry were similar to those reported previously (19). They consisted mainly of naturally biotinylated proteins such as carboxylases and their coenzymes, splicing factors, ribosomal proteins, and cytoskeleton proteins (actin and tubulin) (data not shown). By contrast, analysis of proteins purified from cells expressing bio-SCL identified transcription factors, cofactors, and chromatin-remodelling proteins that were absent from the control BirA-only experiments (see Table S1 in the supplemental material).
Figure 2C shows the identity of known partners of SCL detected in mass spectrometry analysis as well as two newly identified partners we chose for further analysis, SSDP2 and ETO-2. SCL was the most abundant protein detected, followed by its heterodimerization partners (E12/E47 and E2.2). Another member of the pentameric complex, Ldb-1, was found in both MEL and L8057 cells. SSDP2 (recently identified as a cofactor of Ldb-1 during development [11, 77]) and ETO-2 (a corepressor protein involved in acute myeloid leukemia [17]) were identified as partners of SCL in both MEL and L8057 cells.
To validate the mass spectrometry results, we confirmed the interaction between SCL and SSDP2 or ETO-2 by Western blot analysis from pulled-down fractions (data not shown). We also expressed biotinylated SSDP2 in MEL cells and isolated copurifying proteins with streptavidin beads. We showed that SSDP2 interacts with the SCL core complex (SCL, E12/E47, Ldb-1, and LMO2) by Western blotting (see Fig. S3 in the supplemental material). We then exclusively studied the interaction between SCL and ETO-2.
SCL interacts with the corepressor ETO-2 in erythroid cells and megakaryocytes.
We first validated the interaction between endogenous SCL and ETO-2 using immunoprecipitation experiments. Increasing amounts of ETO-2 antibodies precipitated increasing amounts of SCL from MEL nuclear extracts, suggesting specific interaction (Fig. 3A, top panel). SCL also coimmunoprecipitated with ETO-2 from L8057 nuclear extracts (Fig. 3A, bottom panel). To exclude the possibility that ETO-2 could have been purified because of nonspecific binding to nucleic acids, nuclear extracts were treated with DNase before coIP with ETO-2 antibodies. SCL was still detected in the immunoprecipitated fraction (data not shown), confirming the interaction between ETO-2 and SCL.
FIG. 3.

Validation of the SCL/ETO-2 interaction in cell lines and primary cells. (A) Anti-ETO-2 (αETO-2) antibodies (Ab) immunoprecipitate SCL from MEL (top panel) and L8057 (bottom panel) nuclear extracts. The triangle represents increasing amounts of αETO-2 antibodies. IgG, negative control. (B) Nuclear colocalization of SCL and ETO-2 in MEL cells was detected by immunofluorescence (upper panel, first row, merge image). Incubation with αSCL/αLdb-1 antibodies served as a positive control (second row). Incubation with αLdb-1/αSC35 antibodies served as a negative control (third row). (C) Colocalization is revealed by antibody blocking in MEL (left panel) and L8057 (right panel) nuclei. Anti-ETO-2 and anti-Ldb-1 antibodies prevent access of anti-SCL antibodies to the antigen and vice versa. Anti-SC35 antibodies have no blocking effect on anti-Ldb-1 (and vice versa). Antibodies used for blocking and detection are shown. *, P value <0.05. (D and E) Nuclear extracts prepared from Ter119+ mouse splenocytes (D) and primary megakaryocytes (E) were subjected to coimmunoprecipitation with anti-SCL or anti-ETO-2 (splenocytes only) antibodies. Immunoprecipitated proteins were detected by Western blot. Morphology of Ter119+ cells and megakaryocytes used for nuclear extract preparation was assessed by May-Grunwald-Giemsa staining. Ter119+ cells represent proerythroblasts to mature erythrocyte and enucleated stages. Primary megakaryocytes comprise immature megakaryocyte precursors (dark cytoplasm) and mature megakaryocytes (large cells with a granular cytoplasm and a polyploid nucleus).
To further document this association, we performed colocalization experiments. MEL and L8057 cells were fixed and immunolabeled with anti-SCL and anti-ETO-2 antibodies. For both proteins, the staining appeared nuclear when compared with DAPI staining (data not shown) and showed a punctate pattern (Fig. 3B and data not shown) as previously reported for other cell types (17, 64). In a first attempt to demonstrate colocalization of the proteins, we undertook a conventional analysis. Dual staining of the cells showed that a substantial amount of SCL and ETO-2 colocalizes in the nuclei of both MEL (Fig. 3B, merge) and L8057 (data not shown) cells. As controls, we also show colocalization of SCL and known partner Ldb-1, but not of an abundant unrelated nuclear protein (splicing factor SC35 [32]) with a component of the SCL complex such as Ldb-1 (Fig. 3B).
To further analyze the colocalization and exclude the possibility that the overlap observed in the merge images resulted simply from the presence of very large areas stained by both antibodies, we used a high-resolution approach that takes advantage of the ability of an antibody to block access of another antibody to its antigen. In contrast to the conventional analysis indicating that two targets lie within 200 nm, this approach reveals targets that lie within a few nanometers (36, 46) and, therefore, are very likely to interact with each other. In the absence of blocking antibodies, the intensity of the fluorescence detected in MEL and L8057 nuclei upon incubation with anti-SCL, -ETO2, -Ldb-1, and -SC35 antibodies was arbitrarily set to 100 (Fig. 3C). The intensity of the signal emitted from anti-SCL antibodies was significantly reduced by coincubation with anti-ETO-2 or anti-Ldb-1 antibodies. So were those emitted from anti-ETO-2 and anti-Ldb-1 antibodies when coincubated with anti-SCL antibodies, confirming, at a very high resolution, that these proteins do interact with each other. No interaction could be detected between Ldb-1 and the control protein SC35.
To confirm the interaction between SCL and ETO-2 in primary cells, we isolated splenic Ter119+ erythroid cells from phenylhydrazine-treated mice and megakaryocytes from adult mouse bone marrow (Fig. 3D and E show cell morphology) and prepared nuclear extracts. From both cell types, coIP with SCL antibodies revealed the SCL core complex (SCL, E2A, Ldb-1, and LMO2) but failed to detect interaction with GATA-1 (Fig. 3D and E). Importantly, interaction with ETO-2 was confirmed. Reverse coimmunoprecipitation with ETO-2 antibodies from Ter119+ extracts confirmed the interaction with the SCL core complex (E2A, SCL, and LMO2; Fig. 3D, right panel).
Finally, in vivo mapping shows that the bHLH region of SCL is sufficient for interaction with ETO-2 and that heterodimerization with E proteins is required (see the supplemental text and Fig. S4 in the supplemental material). Consistent with this, recent data suggest that ETO-2 interacts with bHLH dimers through direct binding to E2A (89).
Taken together, these results validate the interaction between SCL and ETO-2 in nontransfected MEL and L8057 cells as well as primary erythroid cells and megakaryocytes.
Compositions of SCL- and ETO-2-containing complexes differ in erythroid cells and megakaryocytes.
To more precisely characterize the nature of SCL-containing protein complexes, we performed immunodepletion experiments. Nuclear extracts from MEL and L8057 cells were first subjected to immunoprecipitation with antibodies against ETO-2 and GATA-1. After characterization of the pulled-down complexes by Western blotting, proteins in the depleted supernatants were immunoprecipitated with antibodies against SCL to identify the remaining complexes (the scheme is summarized in Fig. 4A).
FIG. 4.

Characterization of multiple protein complexes in MEL and L8057 cells. (A) Scheme of the immunodepletion experiments. (B and C) Immunodepletion experiments. Wild-type MEL and L8057 cell nuclear extracts were first immunodepleted of ETO-2 (B) or GATA-1 (C). Immunoprecipitated complexes were analyzed by Western blot to detect proteins indicated on the left (B and C, left panels). Note that all depletions (apart from GATA-1 depletion in MEL cells) were complete, as no ETO-2 nor GATA-1 products were detected in the UN fractions. A second immunoprecipitation was performed on the depleted supernatant using anti-SCL (αSCL) antibodies to analyze the nature of the remaining complexes (B and C, right panels). (D) Coimmunoprecipitation from MEL and L8057 cell nuclear extracts with αGfi-1b (top) or αSCL antibodies (bottom). Detection of the proteins indicated on the left was performed by Western blot analysis. IgG, negative control. (E) Model representing the possible nature of protein complexes revealed in the immunodepletion and coIP experiments given the limitations of the technique as discussed in the text. As E2A is an obligate partner of SCL (62), we have represented SCL as a heterodimer although we did not specifically check its presence in the complexes in these experiments.
Immunoprecipitation with ETO-2 antibodies from L8057 and MEL nuclear extracts confirmed the interaction of ETO-2 with the SCL core complex (SCL, Ldb-1, and LMO2) observed in primary erythroid cells (Fig. 4B, left panel, IP lanes). In MEL cells, ETO-2 was also found to interact with GATA-1. MTG16, the human homolog of ETO-2, interacts with the zinc finger oncoprotein Gfi-1 in in vitro assays (16). As the highly related protein Gfi-1b is crucial for erythropoiesis and megakaryopoiesis (69), we reasoned that it may interact with ETO-2 in mouse hematopoietic cells. We show here that Gfi-1b is found in the ETO-2-immunoprecipitated fraction (Fig. 4B). Importantly, reverse immunoprecipitation with Gfi-1b antibodies indicated that Gfi-1b is also likely to be part of SCL-containing complexes in MEL cells (Fig. 4D, upper panel, MEL IP lane). ETO-2 is known to interact with the corepressor N-CoR, while ETO binds to N-CoR and mSin3A (16). Because mSin3A has been shown to interact with SCL (31), we tested whether we could copurify it with ETO-2 and SCL from MEL nuclear extracts. We found that mSin3A interacts with ETO-2 (Fig. 4B, left panel) and SCL (Fig. 4D, lower panel). N-CoR was not detected in the precipitated products (data not shown).
The ETO-2-depleted supernatant was then immunoprecipitated with SCL antibodies. This showed presence of the core SCL complex (SCL, LMO-2, and Ldb-1). No interaction with Gfi-1b was detected, suggesting that Gfi-1b binds SCL through ETO-2 (Fig. 4B, right panel, MEL).
Upon immunodepletion with GATA-1 antibodies, SCL (data not shown), Ldb-1, and LMO-2 were undetectable in the IP fractions (Fig. 4C, left panel). In contrast, there was enrichment of ETO-2, Gfi-1b, and mSin3A. coIP of the depleted supernatant with SCL antibodies showed the presence of the core complex and interaction with ETO-2 (Fig. 4C, right panel). As GATA1 depletion was not complete (Fig. 4C legend) and although the SCL core complex was not detected in the first immunoprecipitate, we cannot exclude the possibility that GATA1 might be necessary for SCL/ETO-2 interaction. Separately, we show that an ETO-2 complex comprising the corepressor proteins mSin3A and Gfi-1b may also involve GATA-1.
In L8057 cells, ETO-2 does not interact with GATA-1, Gfi-1b, or mSin3A (Fig. 4B left panel; data confirmed by reverse coIP for GATA-1 interaction [Fig. 4C, left panel] and for Gfi-1b interaction [Fig. 4D, upper panel]). The second immunoprecipitation with anti-SCL antibodies showed, as in MEL cells, the presence of the SCL core complex in the absence of ETO-2. There was no detectable interaction between GATA-1 and the SCL core complex or with the repressor proteins (ETO-2, Gfi-1b, and mSin3A; Fig. 4C, left panel). A second immunoprecipitation with anti-SCL antibodies from GATA-1-depleted supernatant showed the presence of the SCL core complex and ETO-2 in the absence of GATA-1 (Fig. 4C, right panel) as seen in MEL cells. In order to exclude the possibility that absence of the SCL/Gfi-1b interaction in L8057 cells was a cell line-specific phenomenon, we performed coIP from nuclear extracts prepared from primary murine megakaryocytes with anti-SCL antibodies. This confirmed the absence of interaction between SCL and Gfi-1b (data not shown).
One possible representation of the data from depletion experiments is shown in Fig. 4E. It is very likely that additional complexes exist, that complexes vary in composition, and that proteins represented in a given complex may not always interact altogether. Nevertheless, the major finding is that, although ETO-2 binds the SCL core complex in both MEL and L8057 cells, the corepressor proteins Gfi-1b and mSin3A are recruited to that complex in MEL cells but not L8057 cells.
Taken together, these results lead to the conclusion that the natures of the SCL and ETO-2 complexes are likely to be different in erythroid cells versus megakaryocytes. We then concentrated on understanding the functional role of the SCL/ETO-2 interaction in an erythroid cell environment.
ETO-2 represses the activator function of the pentameric complex in heterologous cells.
As a first step in assessing the possible function of the SCL/ETO-2 interaction, we performed transactivation studies using GATA-1 regulatory sequences in a reporter assay. We showed recently by ChIP assay that the DNase I-hypersensitive sites (DHS) situated 3.5 kb upstream (hHS-3.5) and 14 kb downstream (hHS+14) of the human GATA-1 promoter (IE) bind members of the SCL pentameric complex in MEL cells (76). These sequences were used to direct expression of a luciferase reporter gene. NIH 3T3 cells were transfected with the reporter vector alone or with combinations of vectors expressing members of the pentameric complex (SCL, E47, LMO2, Ldb-1, and GATA-1). A sevenfold increase in luciferase levels upon coexpression of the five proteins was observed (Fig. 5A). Each of these factors was necessary for maximal activation, as expression levels decreased when they were individually omitted. We then cotransfected increasing concentrations of an ETO-2 expression plasmid together with the components of the pentameric complex. Levels of expression of members of the SCL complex were not affected by expression of ETO-2 (Fig. 5B). We observed a 2.4- to 7-fold reduction of the levels of activation of the reporter gene (Fig. 5A) and concluded that ETO-2 represses the activator function of the SCL complex in this setting. These results suggest that ETO-2 might mediate repression of SCL target genes that have to be silenced for hematopoietic differentiation to proceed.
FIG. 5.

ETO-2 confers repressor function to the SCL complex in transactivation assays. (A) NIH 3T3 cells were transiently transfected with luciferase reporter constructs (pGL3, promoterless; hHS-3.5-IE-hHS+14, under control of the enhancer/promoter regions of the human GATA-1 locus [see schematic representation at the top]) and vectors expressing the indicated transcription factors. The triangle represents increasing amounts of ETO-2 expression vector (75, 150, and 300 ng). Transfection of pGL3 alone provides the baseline reporter activity. (B) Western blot analysis of expression of members of the pentameric complex in the absence (+P) or presence (+P+E) of the ETO-2 expression plasmid. p300 serves as a loading control.
The SCL/ETO-2/Gfi-1b interaction is lost during terminal erythroid differentiation.
To further characterize the role of the SCL/ETO-2 proteins in erythroid differentiation, we studied the kinetics of complex formation during erythropoiesis. We analyzed the composition of SCL-containing complexes during in vitro erythroid differentiation of primary wild-type fetal liver cells. We isolated benzidine-negative, c-kit+ Ter119− erythroid precursors (day 0) that subsequently underwent differentiation to obtain, after 24 h, CD71+ Ter119+ cells (day 1) and, after 48 h, benzidine-positive, CD71+ Ter119+ or CD71− Ter119+ erythrocytes (day 2) (see Fig. S5 in the supplemental material). Western blot analyses of nuclear extracts isolated from these populations showed maintenance of the levels of expression of all proteins we tested at day 1 of differentiation compared to the undifferentiated population (data not shown). In contrast, a general decrease in levels of expression was observed at day 2 of differentiation (Fig. 6, input lanes). In erythroid precursors (day 0), SCL was found to interact with members of the core complex as well as GATA-1, ETO-2, Gfi-1b, and mSin3A (Fig. 6, IP αSCL D0 lane). At day 1, the same interactions were observed (data not shown). In terminally differentiated erythrocytes (day 2), although levels of expression of the proteins tested had generally decreased (Fig. 6, input D2 lanes), we detected specific differences in the nature of protein-protein interactions. Whereas SCL still bound to members of the core complex (E2A and LMO2) and GATA-1, the interaction with ETO-2 and Gfi-1b was lost (Fig. 6, IP αSCL D2 lane). Interestingly, mSin3A still coimmunoprecipitated with SCL, suggesting that this protein-protein interaction could occur independently of ETO-2 and Gfi-1b binding.
FIG. 6.
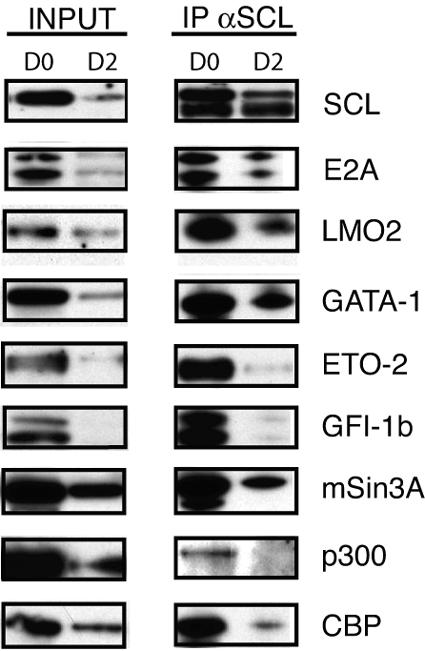
The SCL/ETO-2/Gfi-1b interaction is lost upon erythroid differentiation of day 12.5 fetal liver cells. An analysis of SCL-containing protein complexes upon in vitro erythroid differentiation of day 12.5 wild-type primary fetal liver cells was performed. Shown are expression profiles of proteins indicated on the right of the figure in crude nuclear extracts prepared from fetal liver cells on day 0 (D0) and day 2 (D2) of differentiation (input, equal amounts of extracts were loaded in each lane) and immunoprecipitation (IP) profiles after incubation with anti-SCL (αSCL) antibodies (amounts of precipitated proteins were compared at each time point rather than between time points).
The coactivator and intrinsic histone acetylase CBP/p300 was previously shown to interact with SCL (34). Recent work by Zhang et al. (89) suggests that, in HeLa cells, E2A can act both as a repressor or activator by exchanging members of the ETO family for the coactivator CBP/p300. We therefore tested the hypothesis that CBP/p300 binding to SCL might increase in terminal differentiation. However, in primary erythroid cells, SCL/CBP/p300-containing complexes decreased with differentiation (Fig. 6).
In summary, we conclude that SCL binds to ETO-2 and Gfi-1b primarily in erythroid precursors and that this interaction is lost in terminal differentiation whereas mSin3A continues to bind to SCL. Furthermore, we show that the mechanism underlying ETO-2-directed repression in immature erythroid cells is not likely to be inhibition of CBP/p300 binding.
ETO-2 binds to regulatory sequences of an SCL target gene in vivo.
We next sought to determine whether ETO-2 was present at cis-regulatory elements of SCL target genes. Recent data on the transcriptional regulation of the mouse α-globin gene showed that, although activation of the gene occurs only in the late stages of erythroid differentiation, several hypersensitive sites upstream of the α-globin gene bind the SCL core complex in early nonexpressing progenitors such as CFU-Es and uninduced MEL cells (5). We therefore hypothesized that, in nonexpressing erythroid cells, SCL might recruit a repressor to prevent recruitment of activators and maintain the α-globin gene silent. To test whether ETO-2 might be that repressor, we performed ChIP experiments with early erythroid progenitors (Ter119−) and late normoblasts (Ter119+) isolated from day 12.5 primary fetal liver cells (Fig. 7C shows cell morphology). As previously described (5), we document in vivo binding of SCL on several DHS along the α-globin locus in both Ter119+ and Ter119− cells (Fig. 7A). Remarkably, occupancy by ETO-2 was reproducibly observed on one DHS interacting with SCL, HS-12, in early Ter119− cells, but not in more mature Ter119+ cells (Fig. 7B). Therefore, we suggest that the SCL/ETO-2 repressor complex binds at least one of the identified α-globin regulatory elements in early erythroid progenitors and that ETO-2 dissociates from the DNA-bound complex as erythroid differentiation progresses.
FIG. 7.

ETO-2 and SCL co-occupy hypersensitive site HS-12 on the α-globin locus in Ter119− primary fetal liver cells. ChIP was performed using chromatin isolated from Ter119− and Ter119+ embryonic day 12.5 fetal liver cells and antibodies directed against SCL (A), ETO-2 (B), and IgG as controls (A and B). Immunoprecipitated material was analyzed by real-time PCR. The y axis represents the factor of enrichment in selected sequences in the α-globin locus in the ChIP fractions over the input fractions. On the x axis are shown the regions of the α-globin locus that were analyzed. 14k, sequence within the 14k gene located upstream of the α-globin locus; HS, hypersensitive sites; pr, promoter; int α, intergenic region. Error bars correspond to ±1 standard deviation from two independent experiments. At the bottom of each graph, coordinates of the α-globin locus (5) are shown. (C) Benzidine/MGG staining of Ter119− and Ter119+ cells.
Transcriptional regulation of SCL target genes is altered upon constitutive expression of an E2A mutant or ETO-2 during erythroid differentiation.
To further dissect the function of ETO-2/E2A/SCL interaction during erythroid differentiation of MEL cells, we constitutively expressed Flag-tagged versions of ETO-2, E2A, and an E2A mutant (called DM) defective for ETO-2 binding (see Materials and Methods) using bicistronic, GFP-containing, lentiviral vectors. Analysis of protein lysates from transfected 293T producer cells confirmed that all vectors expressed full-length protein (see Fig. S6A in the supplemental material). Seventy-two hours after lentiviral infection, transduced GFP-expressing MEL cell populations were purified and either used for immediate analyses or kept in culture. The cultures expanded rapidly and maintained transgene expression (data not shown).
We first demonstrated that DM was unable to bind ETO-2 in transduced MEL cells. Immunoprecipitation of nuclear extracts with anti-Flag antibodies and Western blot analyses showed binding of Flag-tagged E2A to ETO-2 and SCL. In contrast, mutant DM did not bind ETO-2 but was able to interact with SCL (Fig. 8A, compare E2A and DM IP lanes). Though previous data suggested that binding of ETO-2 and binding of CBP/p300 to E2A are mutually exclusive in HeLa cells (89), we did not observe increased binding of p300 to DM compared to wild-type E2A in MEL cells (Fig. 8A, top panel, compare E2A and DM IP lanes). This is in agreement with our coIP data for fetal liver cells (Fig. 6). Finally, we confirmed that Flag-ETO-2 behaved like wild-type ETO-2 and bound SCL (Fig. 8A).
FIG. 8.

Constitutive expression of E2A mutant DM and ETO-2 affects expression of SCL target genes during erythroid maturation. (A) Nuclear extracts were prepared from transduced MEL cells stably expressing GFP only, E2A, DM, or ETO-2. Immunoprecipitation was carried out with anti-Flag antibodies and the precipitated products analyzed by Western blotting with antibodies indicated on the right. We used the anti-Flag tag antibody to confirm that all exogenous Flag-tagged proteins were expressed. They are not detected in the input lane as the blot had been stripped. MEL cells served as a control. (B) The MEL cell populations described for panel A were induced to terminally differentiate over 3 days. Levels of expression of E2A and ETO-2 in uninduced cells (day 0 [D0]) and at days 1 to 3 of erythroid maturation were analyzed by Western blotting. p300 served as loading control. Note the difference in protein loading in the D0 ETO-2 lane compared to the D0 GFP lane, as judged by p300 levels (bottom panel). (C) Changes in expression of selected SCL target genes during erythroid maturation were quantitated by real-time PCR in the MEL cell populations characterized in panel B. For each target gene, the y axis represents the enrichment in cDNA sequences over the GAPDH gene control sequences. The x axis represents day 0 to day 3 of erythroid differentiation. Error bars correspond to ±1 standard deviation from two or three independent experiments. (D) MGG staining showing the morphology of the transduced MEL cells (GFP, E2A, DM, and ETO-2) at day 0 and day 1 to day 5 of erythroid maturation. At day 0 to day 4, no morphological differences between the cell populations were detected. A representative picture is shown for each day. At day 5, morphological differences were observed between samples. White arrowheads, late normoblasts with a small peripheral nucleus; black arrowheads, proerythroblasts with a high nucleus/cytoplasm ratio.
Before analyzing possible biological perturbation in transduced MEL cell populations, we first confirmed that the levels of total E2A and ETO-2 proteins were increased in cells expressing exogenous E2A (wild type or mutant DM) and ETO-2 proteins, before and after induction of terminal erythroid maturation. In Fig. 8B, Western blot analysis shows increased E2A expression in MEL cells expressing exogenous wild-type E2A or mutant DM when compared to these expressing GFP alone. This is most marked 3 days after induction of erythroid differentiation (compare D0 to D3, Fig. 8B, top panel). Levels of ETO-2 were slightly increased at day 0 in the population expressing exogenous ETO-2 when compared to the GFP-expressing population, and were consistently higher throughout differentiation, particularly so at day 3 (Fig. 8B, bottom panel).
Cellular analyses were performed on uninduced GFP+ MEL cells. Overexpression of E2A (wild type or DM mutant) or ETO-2 did not affect cell proliferation or apoptosis or induce spontaneous differentiation (see the supplemental text and Fig. S6B and C in the supplemental material).
We then performed more-detailed analyses of the differentiation kinetics of transduced MEL cell populations. In particular, we performed molecular analyses on RNA extracted from the transduced populations from day 0 to day 3 of erythroid differentiation and examined expression of select SCL target genes by real-time PCR. In MEL cells expressing GFP alone (Fig. 8C), expression of GPA increases steadily from day 1 to 3. In contrast, c-kit expression is down-regulated in the early stages of erythroid maturation (day 1) as would be expected. Band 4.2 mRNA levels are up-regulated at day 1 and decrease by day 2. Finally, a dramatic increase of α-globin transcripts is observed over the induction period.
Importantly, upon overexpression of the ETO-2-nonbinding E2A mutant (DM), mRNA levels of c-kit, protein band 4.2, and, to a lesser extent, GPA were significantly up-regulated at day 3 compared to cells expressing GFP alone (Fig. 8C, top panel). This result suggests that a repressor effect on SCL target genes has been lost, presumably through loss of binding to ETO-2. As a control, overexpression of wild-type E2A did not alter transcriptional regulation of the genes tested (Fig. 8C, top panel). To complement these data, we then analyzed the effects of constitutive overexpression of ETO-2 and found that levels of α-globin were significantly reduced at days 2 and 3 of erythroid differentiation (Fig. 8C, bottom panel). Maintenance of high levels of ETO-2 through terminal maturation therefore impairs transcriptional activation of α-globin.
The biological consequences suggested by these molecular changes were best observed morphologically at day 5 of terminal erythroid maturation (Fig. 8D). This is not surprising, as gene expression precedes morphological changes. We noticed a higher proportion of late normoblasts in the population overexpressing DM when compared to populations expressing GFP alone or wild-type E2A, indicative of an accelerated differentiation. In contrast, there is persistence of immature proerythroblasts in the populations overexpressing ETO-2, indicative of retarded erythroid maturation.
DISCUSSION
Transcriptional regulators need to both activate and repress gene expression programs of the lineages they specify in a timely fashion and suppress programs of alternate lineages. SCL is a good example of such a regulator, as it has been described both as an activator and repressor of transcription in normal and malignant hematopoiesis. The molecular mechanisms underpinning SCL function are largely unknown but are likely to involve interaction with protein partners. In this study, we have purified SCL-containing multiprotein complexes and describe the interaction of SCL with the cofactor SSDP2 and with important corepressors ETO-2 and Gfi-1b in erythroid cells and megakaryocytes.
SSDP proteins were originally identified as sequence-specific single-stranded DNA-binding proteins (7) and subsequently characterized as essential cofactors of Ldb-1 (11, 77). In Drosophila, SSDP is a functional component of Chip(Ldb-1)/Ap(LIM-HD) complexes, important for development of the wing (11, 77). Drosophila SSDP can also synergize with Ldb-1 and LIM-homeodomain (LIM-HD) proteins to induce secondary axes in Xenopus embryos (11). In vertebrates, related family members have been described. In mice, SSDP1 and SSDP2 are widely expressed (77). SSDP1 is believed to activate a Lim1-Ldb-1 complex essential for head morphogenesis during embryonic development (53). Here, we show for the first time that SSDP proteins are involved in formation of hematopoietic multiprotein complexes in vertebrates. This is of particular interest as all members of the SSDP family have been localized to translocation breakpoints and deletions in myeloid malignancies (10). The specific role of SSDP2 in SCL-containing complexes remains to be determined.
SCL interacts with the transcriptional corepressor ETO-2 in erythroid cells and megakaryocytes.
ETO-2 is a member of the family of corepressor proteins comprising ETO and MTGR1 (homologs of Drosophila Nervy). Both ETO and ETO-2 were originally identified by characterization of chromosomal breakpoints in acute myeloid leukemia (for a review, see reference 16). Although ETO family members are widely expressed during embryogenesis and adult life (17, 23), the pattern of their expression in hematopoiesis suggests a potential role in hematopoietic differentiation (43). In humans, ETO is detected in CD34+ progenitor cells and up-regulated in erythroblasts (GPA+ CD71+ cells) while ETO-2 (also called MTG16) is expressed at high levels in CD34+ cells and down-regulated during erythroid and granulocytic differentiation (43). In the mouse, ETO-2 is expressed in myeloid (FDCP-mix) and erythroid (MEL) cell lines (17). Evidence for a corepressor function of ETO proteins is based on characterization of their protein partners (for a review, see reference 35). In vitro assays and overexpression in heterologous cell lines suggested that ETO interacts with HDAC-1, -2, and -3 either directly or in corepressor complexes with mSin3A, N-CoR, and SMRT. In contrast, ETO-2 is believed to interact with HDAC-1, -2, -3, -6, and -8, N-CoR, and SMRT but not mSin3A.
It was shown recently that ETO proteins are recruited by the conserved N-terminal activation domain (AD1) of E proteins (89). Our data are consistent with this observation in that, to see an SCL/ETO-2 interaction (i) the bHLH domain of SCL is sufficient, (ii) heterodimerization of SCL with the E2A proteins is necessary, and (iii) interaction with LMO2/Ldb-1 is not required. Although binding to the SES construct was weak, we detected ETO-2 interaction with heterodimers formed by E2A and various chimeric proteins with unrelated HLH domains (see Fig. S4 in the supplemental material). Therefore, ETO-2 binds both class I (homodimers) and class II (heterodimers) bHLH proteins, and we speculate that it will function as a corepressor for both classes. bHLH transcription factors can be added to the growing list of regulators that interact with ETO family members (12, 47, 50, 86).
SCL-containing repressor complexes differ in erythroid cells and megakaryocytes.
Although the same complex (comprising SCL, E2A, or HEB; LMO2; Ldb-1; and ETO-2) is detected in erythroid cells and megakaryocytes, several lines of evidence suggest differences in the overall composition of the SCL-containing complexes between the two cell types. First, mutation of a critical residue in SCL destabilizes the SCL core complexes in different ways (Fig. 2A), suggesting that protein-protein interactions vary in the two cell types. Second, few SCL-interacting proteins were identified by mass spectrometry analysis from L8057 cells compared to MEL cells, implying that the composition of protein complexes might differ (see Table S1 in the supplemental material). Third, the immunodepletion experiments suggest that only a subset of protein-protein interactions detected in MEL cells are present in L8057 cells (Fig. 4).
In erythroid cells, we describe the presence of one or more multiprotein repressor complexes containing SCL and ETO-2.
First, we document interactions between the corepressor mSin3A and SCL (previously reported [31]) and between mSin3A and ETO-2 in MEL cells. Recently published data suggest that ETO, but not ETO-2, interacts with mSin3A (2). These conflicting results may reflect differences in experimental conditions used and suggest that the interaction detected between mSin3A and ETO-2 might not be direct but mediated by tissue-specific transcription factors such as SCL or GATA-1 (Fig. 4). Interestingly, we show from material isolated from fetal liver cells that the interaction between SCL and mSin3A, detected in proerythroblasts, persists in the late stages of erythroid differentiation in the absence of detectable interaction between SCL and ETO-2 (Fig. 6). Thus, SCL recruits mSin3A independently of ETO-2.
Second, our data indicate that the oncoprotein Gfi-1b, SCL, and ETO-2 coimmunoprecipitate in MEL cells (Fig. 4) and primary erythroid precursors (not shown). Gfi-1b and its highly related homolog Gfi-1 are DNA-binding transcription factors with C-terminal zinc fingers and an N-terminal SNAG repressor domain (for a review see reference 52). Gfi-1 proteins interact directly with ETO proteins (47). Interestingly, gain-of-function studies have indicated a role for Gfi-1b in terminal erythropoiesis (59). Loss-of-function studies have established an essential requirement for Gfi-1b in erythroid and megakaryocytic maturation (69). Moreover, the SNAG domain seems required for Gfi-1b activity in erythroid maturation, implying that Gfi-1b acts as a repressor (24). Several lines of evidence suggested that bHLH and Gfi-1 proteins could be part of the same transcriptional pathway or could physically interact in multiprotein complexes. (i) Ablation of SCL and Gfi-1b in adult hematopoiesis produces similar phenotypes (27, 51, 69). (ii) In T cells, Gfi-1 influences E-protein dosage by down-regulating expression of HLH proteins Id1 and Id2, inhibitors of E2A and HEB (88). (iii) Gfi-1 cooperates with the bHLH-leucine zipper protein Myc in T-cell lymphomagenesis (71). (iv) In Drosophila, there is a synergistic interaction between Senseless (Gfi-1 homolog) and bHLH proneural transcription factors (54). Here, we formally demonstrate interaction between SCL and Gfi-1b in erythroid cells and show that this association is mediated by ETO-2.
ETO-2 confers repressor functions on SCL.
We first established the repressive effect of ETO-2 on the activator function of the pentameric complex in heterologous cells (Fig. 5). Upon in vitro differentiation of fetal liver cells, we then showed that the SCL/ETO-2/Gfi-1b interaction is lost in the late stages of erythroid differentiation, whereas the SCL core complex including GATA-1 remains intact and suggested that ETO-2 may function in the early stages of erythroid differentiation to repress expression of SCL target genes (Fig. 6). Down-regulation of ETO-2 expression, possibly by GATA-1 (83), and/or dissociation of ETO-2 from the SCL core complex might then lead to the release or modulation of repression, allowing expression of genes important for terminal erythroid maturation.
Functional studies with MEL cells confirmed these hypotheses. The lentiviral expression system we used led to low-level, constitutive protein overexpression that unveiled important aspects of ETO-2 function in the regulation of erythroid cell-specific genes. Prevention of ETO-2 interaction with the SCL core complex through the use of the E2A mutant DM led to alleviation of transcriptional repression on the c-kit and band 4.2 genes and more-pronounced GPA expression when compared to control cells. ETO-2 might therefore have different functions depending on the nature of the target genes: to maintain low-level gene expression or to slow down transcriptional activation. In complementary experiments, constitutive expression of ETO-2 down-regulated α-globin expression.
Confirming a role for ETO-2 in the transcriptional regulation of erythroid cell-specific genes, we demonstrated an in vivo interaction of ETO-2 with one of the DHS (HS-12) of the α-globin locus that interacts with the SCL core complex. This binding was detected in early erythroid progenitors but not in more mature erythroid cells. Independently, we have shown that, in fetal liver cells expressing a DNA-binding mutant form of SCL, binding of SCL to HS-12 is dramatically decreased and results in up-regulation of α-globin mRNA levels in early progenitors (M. Kassouf, P. Vyas, and C. Porcher, unpublished data). This suggested that SCL nucleates a repressor complex on the α-globin locus to silence gene expression in early erythroid progenitors. Taken together, our findings very strongly suggest that ETO-2 is likely to provide this repressor function to the SCL core complex.
Possible mechanisms of ETO-2-mediated repression.
ETO-2-driven transcriptional repression may be mediated by recruitment of HDACs. Our preliminary data suggest that ETO-2 and SCL interact with HDAC-3 in MEL cells (data not shown). Importantly, although not verified in hematopoietic cells, Gfi-1 proteins also associate with HDACs (47). Alternatively, ETO-2 could prevent interaction of SCL with a coactivator. Cofactor exchange is a mechanism commonly used to regulate transcription levels. Different classes of transcription factors can recruit both corepressors and coactivators (44, 48), as recently exemplified by NF-E2p18/MafK, whose dimerization partner switches from corepressor to coactivator during MEL cell differentiation (8). Interestingly, recent data suggest that the binding of ETO to E2A might lead to dissociation of the E2A/p300 interaction (89). However, we could not confirm that SCL/CBP/p300-containing complexes increased with terminal erythroid differentiation as ETO-2 levels decrease, nor did we detect increased binding of p300 to the E2A mutant form DM when compared to wild-type E2A. Prevention of CBP/p300 binding is therefore not likely to be the mechanism underlying ETO-2-mediated repression in erythroid cells. Finally, in megakaryocytes, interaction of ETO-2 with the SCL core complex may lead to recruitment of unidentified corepressors or may just reflect passive repressive functions by either sequestration mechanisms or prevention of DNA binding.
SCL and GATA-1.
SCL and GATA-1 are thought to transcriptionally coregulate a number of hematopoietic genes. GATA-1/SCL interaction is likely to activate expression of erythroid target genes such as the GPA (40), α-globin (5), protein 4.2 (87), and EKLF (3) genes. Moreover, an increasing number of studies also show co-occupancy of cis elements by SCL and GATA-1 (3, 76, 81), and synergy between the two proteins has been suggested (25, 49).
Surprisingly, the SCL/GATA-1 association did not appear as a robust interaction in this study. We detected GATA-1 in fractions pulled down by bio-SCL from MEL and L8057 nuclear extracts (Fig. 2A), but coIP experiments failed to detect the interaction in MEL and L8057 cells (Fig. 4C) and in primary splenocytes and megakaryocytes (Fig. 3D and E). However, GATA-1 was immunoprecipitated with SCL from fetal liver cells (Fig. 6). The discrepancy between these results is likely to be due to one or more of the different experimental conditions: primary cells versus cell lines, fetal versus adult stage, mass spectrometry versus immunoprecipitation, and, finally, salt concentration in washing buffers. However, what is most likely is that only a small fraction of GATA-1 is associated with SCL, as suggested upon pull-down from MEL and L8057 cell nuclear extracts (Fig. 2A).
Whereas functional data in this paper and from other studies (see above) argue that SCL/GATA-1 activates transcription, we also describe an erythroid complex consisting of ETO-2, Gfi-1b, mSin3A, and GATA-1, but without SCL, suggesting that GATA-1 has repressor functions independently of its interaction with SCL. These data are strengthened by a complementary study we recently completed. Using the same proteomic approach, we have isolated GATA-1 protein partners from induced MEL cells. We showed that GATA-1/SCL complexes are of very low abundance and that GATA-1 is part of several repressor complexes (MeCP1, ACF/WCRF, and Gfi-1b) in the absence of SCL (67).
Corroborating these data, a differential role for SCL protein complexes in presence or absence of GATA-1 has been reported. In gain-of-function studies of zebra fish embryos, SCL/LMO-2-induced hemangioblasts develop into endothelial cells with no observed myeloid differentiation. In contrast, in the presence of GATA-1, erythroid development is induced (25). In independent reports about the regulation of c-kit expression in hematopoietic differentiation, the SCL core complex associated with pRb represses transcription whereas it activates transcription when associated with GATA-1 (41, 79). The GATA-1/SCL interaction is reminiscent of a GATA/bHLH protein association in cardiac myocytes, where GATA-4 and dHAND interact and synergistically activate specific programs of gene expression (15).
Concluding remarks.
The likely function of SCL is to mark genetic loci for activation, repression, or chromatin remodelling (5, 83, 40, 41, 78). In this study, we have begun to dissect the mechanisms by which SCL could lead to transcriptional repression of target genes. These mechanisms seem to differ in erythroid cells versus megakaryocytes. We are now analyzing the interactions between SCL and other putative partners isolated in this study and are focusing on the function of SCL-containing multimeric complexes in the expression of key target genes during erythroid differentiation. To fully understand the role of SCL in megakaryopoiesis, SCL target genes in this lineage will need to be identified. Lastly, in the light of recent reports suggesting a repressor function for SCL in leukemogenesis (27, 54), it will be of interest to determine whether the SCL/ETO-2 complex is detected in SCL-expressing T-ALL cells, as this would shed new light on the mechanisms involved in T-ALL and possibly on therapeutic approaches for this disease.
Supplementary Material
Acknowledgments
We are very grateful to Anthony Jones and his team (Functional Genomics and Proteomics Laboratories, University of Birmingham) for mass spectrometry analyses; to Hedia Chagraoui, Mira Kassouf, and Douglas Vernimmen for invaluable advice; to Lauren Aronson for technical expertise; to Craig Waugh for FACS sorting; and to Ian Hickson and Roger Patient for critical reading of the manuscript.
A.H.S. is a recipient of a Leukemia Research Fund Clinical Scientist Award. T.E.'s laboratory is supported by a specialist program from the Leukemia Research Fund. P.V. is a Wellcome Trust Senior Clinical Fellow. C.P. is funded by the Medical Research Council.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Akashi, K., D. Traver, T. Miyamoto, and I. L. Weissman. 2000. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404:193-197. [DOI] [PubMed] [Google Scholar]
- 2.Amann, J. M., J. Nip, D. K. Strom, B. Lutterbach, H. Harada, N. Lenny, J. R. Downing, S. Meyers, and S. Hiebert. 2001. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol. Cell. Biol. 21:6470-6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson, K. P., S. C. Crable, and J. B. Lingrel. 2000. The GATA-E box-GATA motif in the EKLF promoter is required for in vivo expression. Blood 95:1652-1655. [PubMed] [Google Scholar]
- 4.Andrews, N. C., and D. V. Faller. 1991. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19:2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anguita, E., J. Hughes, C. Heyworth, G. A. Blobel, W. G. Wood, and D. R. Higgs. 2004. Globin gene activation during haemopoiesis is driven by protein complexes nucleated by GATA-1 and GATA-2. EMBO J. 23:2841-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aplan, P. D., K. Nakahara, S. H. Orkin, and I. R. Kirsch. 1992. The SCL gene product: a positive regulator of erythroid differentiation. EMBO J. 11:4073-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayarsaihan, D., R. J. Soto, and L. N. Lukens. 1998. Cloning and characterisation of a novel sequence-specific single-stranded-DNA-binding protein. Biochem. J. 331:447-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brand, M., J. A. Ranish, N. T. Kummer, J. Hamilton, K. Igarashi, C. Francastel, T. H. Chi, G. R. Crabtree, R. Aebersold, and M. Groudine. 2004. Dynamic changes in transcription factor complexes during erythroid differentiation revealed by quantitative proteomics. Nat. Struct. Mol. Biol. 11:73-80. [DOI] [PubMed] [Google Scholar]
- 9.Carrozza, M. J., R. T. Utley, J. L. Workman, and J. Cote. 2003. The diverse functions of histone acetyltransferase complexes. Trends Genet. 19:321-329. [DOI] [PubMed] [Google Scholar]
- 10.Castro, P., H. Liang, J. C. Liang, and L. Nagarajan. 2002. A novel, evolutionarily conserved gene family with putative sequence-specific single-stranded DNA-binding activity. Genomics 80:78-85. [DOI] [PubMed] [Google Scholar]
- 11.Chen, L., D. Segal, N. A. Hukriede, A. V. Podtelejnikov, D. Bayarsaihan, J. A. Kennison, V. V. Ogryzko, I. B. Dawid, and H. Westphal. 2002. Ssdp proteins interact with the LIM-domain-binding protein Ldb1 to regulate development. Proc. Natl. Acad. Sci. USA 99:14320-14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chevallier, N., C. M. Corcoran, C. Lennon, E. Hyjek, A. Chadburn, V. J. Bardwell, J. D. Licht, and A. M. Melnick. 2004. ETO protein of t(8;21) AML is a corepressor for Bcl-6 B-cell lymphoma oncoprotein. Blood 103:1454-1463. [DOI] [PubMed] [Google Scholar]
- 13.Clark, A. J., K. M. Doyle, and P. O. Humbert. 2004. Cell-intrinsic requirement for pRB in erythropoiesis. Blood 104:1324-1326. [DOI] [PubMed] [Google Scholar]
- 14.Condorelli, G. L., A. Tocci, R. Botta, F. Facchiano, U. Testa, L. Vitelli, M. Valtieri, C. M. Croce, and C. Peschle. 1997. Ectopic TAL-1/SCL expression in phenotypically normal or leukemic myeloid precursors: proliferative and antiapoptotic effects coupled with a differentiation blockade. Mol. Cell. Biol. 17:2954-2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai, Y.-S., P. Cserjesi, B. E. Markham, and J. D. Molkentin. 2002. The transcription factors GATA4 and dHAND physically interact to synergistically activate cardiac gene expression through a p300-dependent mechanism. J. Biol. Chem. 277:24390-24398. [DOI] [PubMed] [Google Scholar]
- 16.Davis, J. N., L. McGhee, and S. Meyers. 2003. The ETO (MTG8) gene family. Gene 303:1-10. [DOI] [PubMed] [Google Scholar]
- 17.Davis, J. N., B. J. Williams, J. T. Herron, F. J. Galiano, and S. Meyers. 1999. ETO-2, a new member of the ETO-family of nuclear proteins. Oncogene 18:1375-1383. [DOI] [PubMed] [Google Scholar]
- 18.Debili, N., L. Coulombel, L. Croisille, A. Katz, J. Guichard, J. Breton-Gorius, and W. Vainchenker. 1996. Characterisation of a bipotent erythro-megakaryocytic progenitor in human bone marrow. Blood 88:1284-1296. [PubMed] [Google Scholar]
- 19.de Boer, E., P. Rodriguez, E. Bonte, J. Krijgsveld, E. Katsantoni, A. Heck, F. Grosveld, and J. Strouboulis. 2003. Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. USA 100:7480-7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demaison, C., K. Parsley, G. Brouns, M. Scherr, K. Battmer, C. Kinnon, M. Grez, and A. J. Trasher. 2002. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum. Gene Ther. 13:803-813. [DOI] [PubMed] [Google Scholar]
- 21.Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fosset, N., K. Hyman, K. Gajewski, S. H. Orkin, and R. Schulz. 2003. Combinatorial interactions of serpent, lozenge and U-shaped regulate crystal cell lineage commitment during Drosophila hematopoiesis. Proc. Natl. Acad. Sci. USA 100:11451-11456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gamou, T., E. Kitamura, F. Hosoda, K. Shimizu, K. Shinohara, Y. Hayashi, T. Nagase, Y. Yokoyama, and M. Ohki. 1998. The partner gene of AML1 in t(16;21) myeloid malignancies is a novel member of the MTG8(ETO) family. Blood 91:4028-4037. [PubMed] [Google Scholar]
- 24.Garcon, L., C. Lacout, F. Svinartchouk, J.-P. Le Couedic, J.-L. Villeval, W. Vainchenker, and D. Dumenil. 2005. Gfi-1B plays a critical role in terminal differentiation of normal and transformed erythroid progenitor cells. Blood 105:1448-1455. [DOI] [PubMed] [Google Scholar]
- 25.Gering, M., Y. Yamada, T. H. Rabbits, and R. K. Patient. 2003. Lmo2 and Scl/Tal-1 convert non-axial mesoderm into haemangioblasts which differentiate into endothelial cells in the absence of Gata1. Development 130:6187-6199. [DOI] [PubMed] [Google Scholar]
- 26.Green, A. R., E. DeLuca, and C. G. Begley. 1991. Antisense SCL suppresses self-renewal and enhances spontaneous erythroid differentiation of the human leukaemic cell line K562. EMBO J. 10:4153-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall, M. A., D. J. Curtis, D. Metcalf, A. G. Elefanty, K. Sourris, L. Robb, J. R. Gothert, S. M. Jane, and C. G. Begley. 2003. The critical regulator of embryonic hematopoiesis, SCL, is vital in the adult for megakaryopoiesis, erythropoiesis, and lineage choice in CFU-S12. Proc. Natl. Acad. Sci. USA 100:992-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herblot, S., A.-M. Steff, P. Hugo, P. D. Aplan, and T. Hoang. 2000. SCL and LMO1 alter thymocyte differentiation: inhibition of E2A-HEB function and pre-Tα chain expression. Nat. Immunol. 1:138-144. [DOI] [PubMed] [Google Scholar]
- 29.Hsu, H.-L., I. Wadman, J. T. Tsan, and R. Baer. 1994. Positive and negative transcriptional control by the TAL1 helix-loop-helix protein. Proc. Natl. Acad. Sci. USA 91:5947-5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu, M., D. Krause, M. Greaves, S. Sharkis, M. Dexter, C. Heyworth, and T. Enver. 1997. Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 11:774-785. [DOI] [PubMed] [Google Scholar]
- 31.Huang, S., and S. J. Brandt. 2000. mSin3A regulates murine erythroleukemia cell differentiation through association with the TAL1 (or SCL) transcription factor. Mol. Cell. Biol. 20:2248-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang, S., A. Mayeda, A. R. Krainer, and D. L. Spector. 1997. RCC1 and nuclear organization. Mol. Biol. Cell 8:1143-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang, S., Y. Qiu, Y. Shi, Z. Xu, and S. J. Brandt. 2000. P/CAF-mediated acetylation regulates the function of the basic helix-loop-helix transcription factor TAL1/SCL. EMBO J. 19:6792-6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang, S., Y. Qiu, R. W. Stein, and S. J. Brandt. 1999. p300 functions as a transcriptional coactivator for the TAL1/SCL oncoprotein. Oncogene 18:4958-4967. [DOI] [PubMed] [Google Scholar]
- 35.Hug, B. A., and M. A. Lazar. 2004. ETO interacting proteins. Oncogene 23:4270-4274. [DOI] [PubMed] [Google Scholar]
- 36.Iborra, F. J., A. E. Escargueil, K. Y. Kwek, A. Akoulitchev, and P. R. Cook. 2003. Molecular cross-talk between the transcription, translation, and nonsense-mediated decay machineries. J. Cell Sci. 117:899-906. [DOI] [PubMed] [Google Scholar]
- 37.Ishida, Y., J. Levin, G. Baker, P. E. Stenberg, Y. Yamada, H. Sasaki, and T. Inoue. 1993. Biological and biochemical characteristics of murine megakaryoblastic cell line L8057. Exp. Hematol. 21:289-298. [PubMed] [Google Scholar]
- 38.Ivanova, N. B., J. T. Dimos, C. Schaniel, J. A. Hackney, K. A. Moore, and I. R. Lemischka. 2002. A stem cell molecular signature. Science 298:601-604. [DOI] [PubMed] [Google Scholar]
- 39.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science 293:1074-1080. [DOI] [PubMed] [Google Scholar]
- 40.Lahlil, R., E. Lecuyer, S. Herblot, and T. Hoang. 2004. SCL assembles a multifactorial complex that determines glycophorin A expression. Blood 24:1439-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lecuyer, E., S. Herblot, M. Saint-Denis, R. Martin, C. G. Begley, C. Porcher, S. H. Orkin, and T. Hoang. 2002. The SCL complex regulates c-kit expression in hematopoietic cells through functional interaction with Sp1. Blood 100:2430-2440. [DOI] [PubMed] [Google Scholar]
- 42.Lecuyer, E., and T. Hoang. 2004. SCL: from the origin of hematopoeisis to stem cells and leukemia. Exp. Hematol. 32:11-24. [DOI] [PubMed] [Google Scholar]
- 43.Lindberg, S. R., A. Olsson, A.-M. Persson, and I. Olsson. 2005. The leukemia-associated ETO homologues are differently expressed during hematopoietic differentiation. Exp. Hematol. 33:189-198. [DOI] [PubMed] [Google Scholar]
- 44.Mannervik, M., Y. Nibu, H. Zhang, and M. Levine. 1999. Transcriptional coregulators in development. Science 284:606-609. [DOI] [PubMed] [Google Scholar]
- 45.Martin, D. I. K., L. I. Zon, G. Mutter, and S. H. Orkin. 1990. Expression of an erythroid transcription factor in megakaryocytic and mast cell lineages. Nature 344:444-446. [DOI] [PubMed] [Google Scholar]
- 46.Mason, D. W., and A. F. Williams. 1986. Kinetics of antibody reactions and the analysis of cell surface antigens, p. 38.1-38.17. In D. M. Weir (ed.), Handbook of experimental immunobiology. Blackwell Scientific Publications, Oxford, United Kingdom.
- 47.McGhee, L., J. Bryan, L. Elliott, H. L. Grimes, A. Kazanjian, J. N. Davis, and S. Meyers. 2003. Gfi-1 attaches to the nuclear matrix, associates with ETO (MTG8) and histone deacetylase proteins, and represses transcription using a TSA-sensitive mechanism. J. Cell Biochem. 89:1005-1018. [DOI] [PubMed] [Google Scholar]
- 48.McKenna, N. J., and B. W. O'Malley. 2002. Combinatorial control of gene expression by nuclear receptors and regulators. Cell 108:465-474. [DOI] [PubMed] [Google Scholar]
- 49.Mead, P. E., A. E. Deconinck, T. L. Huber, S. H. Orkin, and L. I. Zon. 2001. Primitive erythropoiesis in the Xenopus embryo: the synergistic role of LMO2, SCL and GATA-binding proteins. Development 128:2301-2308. [DOI] [PubMed] [Google Scholar]
- 50.Melnick, A. M., J. J. Westendorf, A. Polinger, G. W. Carlile, S. Arai, H. J. Ball, B. Lutterbach, S. W. Hiebert, and J. D. Licht. 2000. The ETO protein disrupted in t(8;21)-associated acute myeloid leukemia is a corepressor for the promyelocytic leukemia zinc finger protein. Mol. Cell. Biol. 20:2075-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mikkola, H. K., J. Klintman, H. Yang, H. Hock, T. M. Schlaeger, Y. Fujiwara, and S. H. Orkin. 2003. Haematopoietic stem cells retain long-term repopulating activity and multipotency in the absence of stem-cell leukaemia SCL/tal-1 gene. Nature 421:547-551. [DOI] [PubMed] [Google Scholar]
- 52.Moroy, T. 2005. The zinc finger transcription factor growth factor independence 1 (Gfi-1). Int. J. Biochem. Cell Biol. 37:541-546. [DOI] [PubMed] [Google Scholar]
- 53.Nishioka, N., S. Nagano, R. Nakayama, H. Kiyonari, T. Ijiri, K. Taniguchi, W. Shawlot, Y. Hayashizaki, H. Westphal, R. Behringer, Y. Matsuda, S. Sakoda, H. Kondoh, and H. Sasaki. 2005. Ssdp1 regulates head morphogenesis of mouse embryos by activating the Lim1-Ldb1 complex. Development 132:2535-2546. [DOI] [PubMed] [Google Scholar]
- 54.Nolo, R., L. A. Abbott, and H. J. Bellen. 2000. Senseless, a Zn finger transcription factor, is necessary and sufficient for sensory organ development in Drosophila. Cell 102:349-362. [DOI] [PubMed] [Google Scholar]
- 55.O'Neil, J., J. Shank, N. Cusson, C. Murre, and M. Kelliher. 2004. TAL1/SCL induces 90 leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell 5:587-596. [DOI] [PubMed] [Google Scholar]
- 56.Ono, Y., N. Fukuhara, and O. Yoshie. 1998. TAL1 and LIM-only proteins synergistically induce retinaldehyde dehydrogenase 2 expression in T-cell acute lymphoblastic leukemia by acting as cofactors for GATA3. Mol. Cell. Biol. 18:6939-6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ono, Y., N. Fukuhara, and O. Yoshie. 1997. Transcriptional activity of TAL1 in T cell acute lymphoblastic leukemia (T-ALL) requires RBTN1 or -2 and induces TALLA1, a highly specific tumor marker of T-ALL. J. Biol. Chem. 272:4576-4581. [DOI] [PubMed] [Google Scholar]
- 58.Orkin, S. 2000. Diversification of haematopoietic stem cells to specific lineages. Nat. Rev. Genet. 1:57-64. [DOI] [PubMed] [Google Scholar]
- 59.Osawa, M., T. Yamaguchi, Y. Nakamura, S. Kaneko, M. Onodera, K.-I. Sawada, A. Jegalian, H. Wu, H. Nakauchi, and A. Iwama. 2002. Erythroid expansion mediated by the Gfi-1B zinc finger protein: role in normal hematopoiesis. Blood 100:2769-2777. [DOI] [PubMed] [Google Scholar]
- 60.Papayannopoulou, T., E. Raines, S. Collins, B. Nakamoto, M. Tweeddale, and R. Ross. 1987. Constitutive and inducible secretion of platelet-derived growth factor analogs by human leukemic cell lines coexpressing erythroid and megakaryocytic markers. J. Clin. Investig. 79:859-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park, S. T., and X. H. Sun. 1998. The Tal1 oncoprotein inhibits E47-mediated transcription. Mechanism of inhibition. J. Biol. Chem. 273:7030-7037. [DOI] [PubMed] [Google Scholar]
- 62.Porcher, C., E. C. Liao, Y. Fujiwara, L. I. Zon, and S. H. Orkin. 1999. Specification of hematopoietic and vascular development by the bHLH transcription factor SCL without direct DNA binding. Development 126:4603-4615. [DOI] [PubMed] [Google Scholar]
- 63.Porcher, C., W. Swat, K. Rockwell, Y. Fujiwara, F. W. Alt, and S. H. Orkin. 1996. The T-cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell 86:47-57. [DOI] [PubMed] [Google Scholar]
- 64.Pulford, K., N. Lecointe, K. Leroy-Viard, M. Jones, D. Mathieu-Mahul, and D. Y. Mason. 1995. Expression of TAL-1 proteins in human tissues. Blood 85:675-684. [PubMed] [Google Scholar]
- 65.Robb, L., N. J. Elwood, A. G. Elefanty, F. Kontgen, R. Li, L. D. Barnett, and C. G. Begley. 1996. The scl gene product is required for the generation of all hematopoietic lineages in the adult mouse. EMBO J. 15:4123-4129. [PMC free article] [PubMed] [Google Scholar]
- 66.Robertson, S. M., M. Kennedy, J. M. Shannon, and G. Keller. 2000. A transitional stage in the commitment of mesoderm to hematopoiesis requiring the transcription factor SCL/tal-1. Development 127:2447-2459. [DOI] [PubMed] [Google Scholar]
- 67.Rodriguez, P., E. Bonte, J. Krijgsveld, K. Kolodziej, B. Guyot, A. Heck, P. Vyas, E. de Boer, F. Grosveld, and J. Strouboulis. 2005. GATA-1 forms distinct activating and repressive complexes in erythroid cells. EMBO J. 24:2354-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Romeo, P.-H., M.-H. Prandini, V. Joulin, V. Mignotte, M. Prenant, W. Vainchenker, G. Marguerie, and G. Uzan. 1990. Megakaryocytic and erythrocytic lineages share specific transcription factors. Nature 344:447-449. [DOI] [PubMed] [Google Scholar]
- 69.Saleque, S., S. Cameron, and S. H. Orkin. 2002. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev. 16:301-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schlaeger, T. M., A. Schuh, S. Flitter, A. Fisher, H. Mikkola, S. H. Orkin, P. Vyas, and C. Porcher. 2004. Decoding hematopoietic specificity in the helix-loop-helix domain of the transcription factor SCL/Tal-1. Mol. Cell. Biol. 24:7491-7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmidt, T., H. Karsunky, E. Gau, B. Zevnik, H. P. Elsasser, and T. Moroy. 1998. Zinc finger protein GFI-1 has low oncogenic potential but cooperates strongly with pim and myc genes in T-cell lymphomagenesis. Oncogene 17:2661-2667. [DOI] [PubMed] [Google Scholar]
- 72.Shivdasani, R., and S. H. Orkin. 1995. Erythropoiesis and globin gene expression in mice lacking the transcription factor NF-E2. Proc. Natl. Acad. Sci. USA 92:8690-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shivdasani, R. A. 2001. Molecular and transcriptional regulation of megakaryocyte differentiation. Stem Cells 19:397-407. [DOI] [PubMed] [Google Scholar]
- 74.Traver, D., T. Miyamoto, J. Christensen, J. Iwasaki-Arai, K. Akashi, and I. L. Weissman. 2001. Fetal liver myelopoiesis occurs through distinct, prospectively isolatable progenitor subsets. Blood 98:627-635. [DOI] [PubMed] [Google Scholar]
- 75.Tsang, A. C., J. E. Visvader, C. A. Turner, Y. Fujiwara, C. Yu, M. J. Weiss, M. Crossley, and S. H. Orkin. 1997. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell 90:109-119. [DOI] [PubMed] [Google Scholar]
- 76.Valverde-Garduno, V., B. Guyot, E. Anguita, I. Hamlett, C. Porcher, and P. Vyas. 2004. Differences in the chromatin structure and cis-element organisation of the human and mouse GATA1 loci: implications for cis-element identification. Blood 104:3106-3116. [DOI] [PubMed] [Google Scholar]
- 77.van Meyel, D. J., J. B. Thomas, and A. D. Agulnick. 2003. Ssdp proteins bind to LIM-interacting co-factors and regulate the activity of LIM-homeodomain protein complexes in vivo. Development 130:1915-1925. [DOI] [PubMed] [Google Scholar]
- 78.Vannucchi, A. M., F. Paoletti, S. Linari, C. Cellai, R. Caporale, P. R. Ferrini, M. Sanchez, G. Migliaccio, and A. R. Migliaccio. 2000. Identification and characterisation of a bipotent (erythroid and megakaryocytic) cell precursor from the spleen of phenylhydrazine-treated mice. Blood 95:2559-2568. [PubMed] [Google Scholar]
- 79.Vitelli, L., G. Condorelli, V. Lulli, T. Hoang, L. Luchetti, C. M. Croce, and C. Peschle. 2000. A pentamer transcriptional complex including tal-1 and retinoblastoma protein downmodulates c-kit expression in normal erythroblasts. Mol. Cell. Biol. 20:5330-5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.von Lindern, M., E. M. Deiner, H. Dolznig, M. Parren-Van Amelsvoort, M. J. Hayman, E. W. Mullner, and H. Beug. 2001. Leukemic transformation of normal murine erythroid progenitors: v-and c-ErbB act through signaling pathways activated by the EpoR and c-Kit in stress erythropoiesis. Oncogene 20:3651-3664. [DOI] [PubMed] [Google Scholar]
- 81.Wadman, I. S., H. Osada, G. G. Grutz, A. D. Agulnick, H. Westphal, A. Forster, and T. H. Rabbitts. 1997. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which include TAL1, E47, GATA-1, and Ldb1/NL1 proteins. EMBO J. 16:3145-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weiss, M. J., G. Keller, and S. H. Orkin. 1994. Novel insights into erythroid development revealed through in vitro differentiation of GATA-1-embryonic stem cells. Genes Dev. 8:1184-1197. [DOI] [PubMed] [Google Scholar]
- 83.Welch, J. J., J. A. Watts, C. R. Vakoc, Y. Yao, H. Wang, R. C. Hardisson, G. A. Blobel, L. A. Chodosh, and M. J. Weiss. 2004. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 104:3136-3147. [DOI] [PubMed] [Google Scholar]
- 84.Wen, J., S. Huang, S. D. Pack, X. Yu, S. J. Brandt, and C. T. Noguchi. 2005. Tal1/SCL binding to pericentromeric DNA represses transcription. J. Biol. Chem. 280:12956-12966. [DOI] [PubMed] [Google Scholar]
- 85.Westman, B. J., J. P. Mackay, and D. Gell. 2002. Ikaros: a key regulator of haematopoiesis. Int. J. Biochem. Cell Biol. 34:1304-1307. [DOI] [PubMed] [Google Scholar]
- 86.Wood, J. D., F. C. Nucifora, Jr., K. Duan, C. Zhang, J. Wang, Y. Kim, G. Schilling, N. Sacchi, J. M. Liu, and C. A. Ross. 2000. Atrophin-1, the dentato-rubral and pallido-luysian atrophy gene product, interacts with ETO/MTG8 in the nuclear matrix and represses transcription. J. Cell Biol. 150:939-948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu, Z., S. Huang, L.-H. Chang, A. D. Agulnick, and S. J. Brandt. 2003. Identification of a TAL1 target gene reveals a positive role for the LIM domain-binding protein Ldb1 in erythroid gene expression and differentiation. Mol. Cell. Biol. 23:7585-7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yucel, R., H. Karsunky, L. Klein-Hitpass, and T. Moroy. 2003. The transcriptional repressor Gfi1 affects development of early, uncommitted c-Kit+ T cell progenitors and CD4/CD8 lineage decision in the thymus. J. Exp. Med. 197:831-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang, J., M. Kalkum, S. Yamamura, B. T. Chait, and R. G. Roeder. 2004. E protein silencing by the leukemogenic AML1-ETO fusion protein. Science 305:1286-1289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.