

Abstract
The matrilins are a family of four noncollagenous oligomeric extracellular matrix proteins with a modular structure. Matrilins can act as adapters which bridge different macromolecular networks. We therefore investigated the effect of collagen IX deficiency on matrilin-3 integration into cartilage tissues. Mice harboring a deleted Col9a1 gene lack synthesis of a functional protein and produce cartilage fibrils completely devoid of collagen IX. Newborn collagen IX knockout mice exhibited significantly decreased matrilin-3 and cartilage oligomeric matrix protein (COMP) signals, particularly in the cartilage primordium of vertebral bodies and ribs. In the absence of collagen IX, a substantial amount of matrilin-3 is released into the medium of cultured chondrocytes instead of being integrated into the cell layer as in wild-type and COMP-deficient cells. Gene expression of matrilin-3 is not affected in the absence of collagen IX, but protein extraction from cartilage is greatly facilitated. Matrilin-3 interacts with collagen IX-containing cartilage fibrils, while fibrils from collagen IX knockout mice lack matrilin-3, and COMP-deficient fibrils exhibit an intermediate integration. In summary, the integration of matrilin-3 into cartilage fibrils occurs both by a direct interaction with collagen IX and indirectly with COMP serving as an adapter. Matrilin-3 can be considered as an interface component, capable of interconnecting macromolecular networks and mediating interactions between cartilage fibrils and the extrafibrillar matrix.
In cartilage, the extracellular matrix occupies the major volume fraction of the tissue and is responsible for its main functions, i.e., load bearing and, in the case of joint cartilage, allowing smooth articulation of long bones. These functions are engendered by two supramolecular systems, the collagen-containing fibrils and the extrafibrillar matrix which comprises the large, cartilage-specific proteoglycan aggrecan. Both aggrecan and the collagens occurring in cartilage fibrils self-assemble into their respective aggregates. In order to warrant tissue stability, a mutual interaction of the two supramolecular compartments is required. Thus, the molecular nature of the fibrillar periphery is of substantial interest, and in this respect, further cartilage matrix components are likely to be of particular biological importance. These further components include matrilins and cartilage oligomeric matrix protein (COMP), which are neither collagens nor proteoglycans. The matrilins constitute a family of four modular proteins belonging to the von Willebrand A-like domain superfamily. All matrilins contain up to 2 von Willebrand A-like domains, up to 10 epidermal growth factor-like domains, and 1 C-terminal coiled-coil α-helix mediating the oligomerization of single matrilin subunits (9, 49). Native matrilin-1 and matrilin-3 extracted from fetal calf epiphyseal cartilage are able to form homo- and hetero-oligomers with a varying stoichiometry (27, 51). All matrilins are expressed during mouse limb development. Matrilin-1 and -3 show a skeletal expression mainly in cartilaginous tissues (28), whereas matrilin-2 and -4 have a broad tissue distribution (26, 39). In mice, matrilin-1 and -3 are often colocalized, e.g., in the cartilage primordium of the vertebral bodies, costal cartilage, sternum, ilium, the cranial bones, and the joints of developing bones (28).
Matrilin-3, strongly expressed in growing skeletal tissues, as the epiphyseal growth plate, or in bone undergoing growth or remodeling, has never been found outside skeletal tissues. It can be integrated into filamentous networks as shown in vitro for Swarm rat chondrosarcoma cells which assemble filaments of variable thickness containing matrilin-3. These filaments often form branches and can connect cells over a distance of several cell diameters, preferentially cells which appear to have recently undergone mitosis. In addition to matrilin-3, the filaments contain collagens II or VI, matrilin-1, and the small leucine-rich proteoglycans decorin and biglycan (27, 50). Matrilin-3-containing filamentous structures are observed in sternal, costal, and tracheal cartilage. In the latter two cases, the fibrils are situated perpendicular to the perichondrium, and they possibly represent the in vivo counterpart of the fibrils observed in cell culture (27).
In vitro binding assays have demonstrated that matrilin-3 can interact directly with COMP (32); however, a specific binding partner for matrilin-3 in fibrillar networks in vivo has not yet been identified. In particular, cartilage fibrils constitute complex structural aggregates containing at least collagens II and XI and, optionally, collagen IX (14, 33) or XVI (25). In addition, some fibril populations are associated with small leucine-rich proteins or proteoglycans, such as decorin, biglycan, and fibromodulin (14, 20, 34). Collagen IX, a member of the fibril-associated collagens with interrupted triple helices (FACIT) family of collagens (42), is a component of D-periodically banded cartilage fibrils. It is assembled with collagens II and XI in antiparallel direction (12) and points its N-terminal triple helical domain 3 (COL3) and noncollagenous domain 4 (NC4) away from the body toward the periphery of the fibril (52). The interaction sites of collagen IX within the collagenous fibril body include the triple helical domain 1 (COL1), the most conserved sequence occurring in all FACITs. Aggregation is also effected through the noncollagenous domain 1 (NC1), which is C-terminally adjacent to COL1 (12). In adult tissues, collagen IX is associated with a subpopulation of thin fibrils preferentially occurring in the territorial matrix and mostly lacking decorin. In embryonic cartilage, collagen IX is distributed in homogenously occurring fibrils with a uniform width of about 20 nm (14, 35). Collagen IX stabilizes the individual fibrils (11) and is thought to direct the organization at the tissue level of the fibrillar network in cartilage. Deletion of the α1 (IX) chain of the heterotrimeric molecule leads to a functional knockout of the entire collagen IX protein (15). These animals are born with no conspicuous skeletal abnormalities but may develop early osteoarthritis postnatally (13).
Early-onset osteoarthritis and irregular ossification of the epiphyses are the characteristics of a relatively mild and clinically variable osteochondrodysplasia, called multiple epiphyseal dysplasia (MED) (47). Interestingly, not only do mutations in all three collagen IX genes (Col9a1, Col9a2, and Col9a3) cause this disease, but MED displays a genetic heterogeneity. Recently, it has been demonstrated that mutations in the genes encoding COMP and matrilin-3 also result in MED (1, 4, 7, 8, 38). For COMP it is well known that it interacts with collagen IX in a zinc-dependent manner. Binding sites for COMP are located at the ends and within two internal sites of the collagen IX molecule (21, 46).
By employing the α1 collagen IX knockout mouse model and solid-phase binding assays, we demonstrate a specific interaction of matrilin-3 with fibrillar collagen IX. The absence of collagen IX does not alter the gene expression level of matrilin-3. However, stable integration of matrilin-3 into the extracellular matrix of cartilage tissues is compromised.
MATERIALS AND METHODS
Animals.
Wild-type and Col9a1 knockout mice were maintained and bred under specific-pathogen-free conditions in single ventilated cages. Genotyping and controls for the presence of common infectious agents were routinely performed. Wild-type and transgenic mice with a similar genetic background were used for all studies.
Antibodies.
Polyclonal antibodies against murine matrilin-3 (27) and bovine COMP (10) were described elsewhere. Polyclonal antibodies prepared against the NC4 domain of murine collagen IX are described below. Polyclonal affinity-purified antibodies against bovine and human collagen II were purchased from DPC-Biermann, Germany. Gold-conjugated secondary antibodies (mouse anti-rabbit and -chicken immunoglobulin Gs [IgGs]), bridging antibodies (rabbit anti-mouse antibody), and tertiary antibodies (mouse monoclonal anti-alkaline phosphatase) used for alkaline phosphatase plus mouse monoclonal alkaline phosphatase-anti-alkaline phosphatase (APAAP) staining were purchased from DAKO, Denmark. Secondary donkey anti-rabbit and anti-mouse antibodies conjugated with horseradish peroxidase used for immunoblotting analysis were purchased from Sigma, Germany. Gold-labeled goat anti-rabbit (18-nm particles) and donkey anti-chicken (12-nm particles) antibodies used for immunogold labeling of fibril fragments were purchased from Dianova, Germany.
Recombinant expression of the α1 (IX) NC4 domain.
Total RNA was prepared by lithium chloride (LiCl)-urea extraction of whole rib cages of neonatal mice (day 2), proteinase K digestion, and subsequent isopropanol precipitation (45) and reverse transcribed into cDNA using Stratascript reverse transcriptase (Stratagene). A 762-bp fragment encoding the entire NC4 domain of the α1 chain of collagen IX was generated by PCR using the primers NC4F (5′-TGA CTG CTA GCA ACT CTT AAG CGT CGC GCA A-3′) and NC4R (5′-ACG ATG CGG CCG CTG CTC ACC CGG AGG ACC TCT CT-3′). NC4F contained an artificial NheI restriction site, while NC4R included a NotI site. After PCR amplification, purification, and digestion with the corresponding restriction enzymes (New England Biolabs), the construct was ligated into the pCEP-Pu vector (43) containing a BM40 signal peptide and a C-terminal six-His sequence, resulting in a fusion protein of α1 (IX) NC4 and a His epitope. The vector construct was then transiently transfected into 293-EBNA cells (Invitrogen) using the lipofection reagent FuGENE 6 (Roche) according to the protocol of the manufacturer. Selection for successfully transfected cells was performed by culturing in selection medium: Dulbecco's modified Eagle medium (DMEM)/F12 (1:1) with final concentrations of 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10% (vol/vol) fetal calf serum, 175 μg/ml Geneticin (all by Gibco), and 0.5 μg/ml puromycin (Sigma, Germany). Positive clones were kept for three to four passages in selection medium followed by culturing in serum-free DMEM/F12 (1:1) medium. The cells were viable and produced large amounts (ca. 0.5 mg/liter of medium) of NC4 protein for 18 to 21 days. Media conditioned for 2 days were pooled, dialyzed against 20 mM sodium phosphate, pH 6.0, and applied to cationic-exchange chromatography (carboxymethyl cellulose CM-52; Whatman, England). The protein was eluted by a sodium chloride gradient (0.05 to 0.30 M NaCl, with a main protein peak at 0.14 M). The NC4-containing fractions were pooled and further purified by a TALON cobalt affinity resin which allows high-affinity binding of the six-His tag introduced at the C terminus (Clontech).
Production of a polyclonal antiserum against the NC4 domain.
The purified NC4 protein was used to raise a polyclonal antiserum in rabbit. The crude serum was affinity purified using a column prepared with purified NC4 protein covalently coupled to Affi-Gel 10 Gel (Bio-Rad, Hercules, Calif.). The antibodies were eluted with 100 mM glycine, pH 2.5, neutralized with 1 M Tris, dialyzed against phosphate-buffered saline (PBS), and concentrated using VIVASPIN 15R concentrators (molecular weight cutoff, 15,000; Vivascience, Germany). The titer and specificity of the purified antibodies were assessed by dot blot analysis, immunoblotting, and enzyme-linked immunosorbent assay (ELISA).
Isolation and culture of primary murine chondrocytes.
Rib cages of 2- to 3-day-old mice were dissected and carefully cleaned of adherent nonskeletal tissue, and matrix-free chondrocytes were prepared by digestion for 16 h with collagenase P (1 mg/ml; Roche) dissolved in DMEM/F12 containing 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco, Germany), and 1 mM l-cysteine (Fluka, Germany). The cell suspension was passed through 100-μm and 40-μm filters (Becton Dickinson Biosciences, Germany), and cells were plated in either 6- or 24-well plates and cultured in monolayer for 5 days at 37°C in a humidified atmosphere containing 95% air and 5% CO2. The culture medium (DMEM/F12, 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin) was changed every second day. To enhance collagen biosynthesis, the medium was supplemented with 100 μM ascorbate.
Cartilage preparation for matrilin-3 extraction studies.
Rib cages of newborn (1- to 2-day-old) wild-type and collagen IX knockout mice were isolated and freed from surrounding noncartilaginous tissue. Fibrillar suspensions were prepared by homogenizing the rib cages with a Polytron (Kinematica, Switzerland) in 10 volumes of DMEM buffered with 10 mM HEPES, pH 7.4, containing 20 μM ZnCl2, 0.3 μM MnCl2, 20 μM CuCl2, 1 mM benzamidine HCl, 5 mM 6-aminocaproic acid, 1 mM phenylmethylsulfonyl fluoride, and 1 mM N-ethylmaleimide (NEM) (HEPES extraction buffer). The homogenates were agitated for 15 min at 4°C and centrifuged for 15 min at 14,000 × g and 4°C. The pellets were resuspended in 10 volumes of the HEPES extraction buffer described above, agitated overnight, and centrifuged as described above. The extraction procedure was consecutively repeated with HEPES extraction buffer supplemented with 10 mM EDTA and 2 M urea and finally with HEPES extraction buffer containing 10 mM EDTA and 4 M guanidinium hydrochloride (GuHCl). The final pellets were resuspended in PBS supplemented with 1 mM NEM and 60 U/ml collagenase CLSPA (Cooper Biomedical) and digested overnight at 37°C. All supernatants in these extraction procedures were separately subjected to analysis of matrilin-3 content by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting. The guanidinium hydrochloride-containing supernatants were dialyzed against Tris-buffered saline (TBS) prior to analysis by SDS-PAGE.
Cartilage preparation for immunoelectron microscopy.
Rib cages of newborn (2-day-old) wild-type COMP and collagen IX knockout mice were isolated and freed from surrounding noncartilaginous tissue. Fibril suspensions were prepared by homogenizing the rib cages with a Polytron (Kinematica, Switzerland) in 10 volumes of 50 mM Tris HCl, pH 7.4, 100 mM NaCl, 1 mM benzamidine HCl, 5 mM 6-aminocaproic acid, 1 mm phenylmethylsulfonyl fluoride, and 1 mm NEM. The homogenates were centrifuged at 14,000 × g for 15 min at 4°C. This procedure was repeated twice. The final supernatant was analyzed by immunoelectron microscopy as described previously (17). The primary antibodies, a rabbit serum against matrilin-3 and the affinity-purified antibody against the NC4 domain, were used at a dilution of 1:50. Secondary gold-conjugated antibodies were used at a dilution of 1:30.
Northern blot analysis.
Total RNA of rib cage cartilage of newborn mice (2 days old) was prepared using the LiCl-urea method, and RNA samples were electrophoresed on 1% (wt/vol) agarose gels in the presence of 2 M formaldehyde and blotted to Hybond N membranes (Amersham Biosciences, Germany). Probes were labeled with [32P]dCTP (ICN) using a Prime-a-Gene labeling kit (Promega, Germany). The filters were hybridized for 16 h using QuikHyb hybridization solution (Stratagene) at 60°C, washed, and exposed to PhosphoImager screens (Amersham Biosciences, Germany). The following cDNA fragments were used as hybridization probes: mouse matrilin-3, 430-bp cDNA fragment; rat COMP, 1,172-bp cDNA fragment; and rat GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 1-kbp cDNA fragment (friendly gift from H. Kresse, Münster, Germany). Quantitation of gene expression was performed with ImageQuant software (Amersham Biosciences, Germany).
SDS-PAGE and immunoblotting.
Cartilage extracts were precipitated with 4 volumes of ethanol, resuspended, boiled in SDS sample buffer containing 5% β-mercaptoethanol for reduction of samples, and run on 4.5 to 15% polyacrylamide gradient gels (30). Alternatively, for the extraction studies, the extracts were boiled in nonreducing SDS sample buffer.
After 5 days of cell culture, supernatants and cell layers of wild-type and collagen IX- and COMP-deficient chondrocytes were harvested. The supernatant was collected, and the remaining cell layer was washed twice with PBS. After addition of sample buffer, the cell layer was collected using a cell scraper. The supernatant was trichloroacetic acid precipitated and resuspended in the same amount of sample buffer as that used for the harvest of the cell layer to ensure comparable loading.
All gels were electrotransferred onto nitrocellulose membranes, blocked with 5% skim milk, and hybridized overnight at 4°C with an affinity-purified rabbit NC4 antibody (1:5,000), a polyclonal COMP antibody (1:1,500), an affinity-purified rabbit anti-matrilin-3 antibody (1:1,000) (27), or a monoclonal mouse antibody to GAPDH (1:200; Chemicon, Germany). After washing, the membrane was incubated with a 1:10,000 diluted secondary horseradish peroxidase-conjugated donkey anti-rabbit IgG for 1 h at room temperature (RT) (Amersham Biosciences, Germany). Antibody detection was performed by using 2.5 mM luminol, 0.4 mM p-coumaric acid, 0.01% H2O2 as luminescent agent (Fluka, Germany) visualized by exposure on X-ray films.
Immunohistochemistry employing the APAAP complex method.
Newborn mice (2 days old) were sacrificed with CO2, divided into two halves, and fixed with 4% paraformaldehyde in PBS (pH 7.4) for at least 2 days at 4°C. For paraffin embedding, samples were rehydrated for 24 h at 4°C and subsequently dehydrated in increasing ethanol concentrations at 4°C. For immunohistochemistry, sections were deparaffinized in Rotihistol (Roth, Germany) at RT. After rehydration, sections were briefly postfixed in 3% paraformaldehyde in PBS for 5 min at RT and sequentially digested for 8 min at 37°C with 0.05% Protease XXIV (Sigma, Germany) in PBS and for 90 min at 37°C with 0.1% hyaluronidase (Sigma, Germany) in 0.1% acetate buffer (pH 6.0). After extensive washing with PBS, sections were blocked overnight at 4°C with blocking buffer consisting of 1% bovine serum albumin and 10% normal goat serum in PBS including 10% Complete Mini solution (protease inhibitor tablets; Roche Diagnostics, Germany). Thereafter, sections were incubated with the appropriate primary antibodies for 90 min at 37°C in blocking buffer. Dilution for NC4 antibody was 1:200, that for collagen II antibody was 1:150, that for COMP antibody was 1:350, and that for matrilin-3 antibody was 1:1,000. Subsequently, samples were incubated with the secondary mouse anti-rabbit antibody diluted 1:100 for 30 min at RT followed by incubation with the “bridging” rabbit anti-mouse antibody diluted 1:200 for 30 min at RT. Finally the APAAP complex solution (alkaline phosphatase plus mouse monoclonal anti-alkaline phosphatase antibody) was added, and slides were developed with NBT-BCIP (5-bromo-4-chloro-3-indolylphosphate-nitroblue tetrazolium from Sigma, Germany) solution including 10% levamisole (inhibition of endogenous alkaline phosphatase) for 30 min at RT. Between addition of antibodies, sections were each time washed twice with TBS plus 0.05% Tween 20 (Sigma, Germany). The sections were counterstained with 1% methylene green in 20% ethanol and mounted with Kaiser's glycerin gelatin (Merck, Germany).
Alcian blue staining.
For alcian blue staining, sections were deparaffinized in Rotihistol, rehydrated, adjusted to 0.1 M HCl (pH 1.0) for 3 min, and subsequently incubated for 30 min in 1% alcian blue 8GX (Sigma, Germany) solubilized in 0.1 M HCl. After rinsing sections in 0.1 M HCl, slides were dehydrated with xylol and mounted with Kaiser's glycerin gelatin (Merck, Germany).
Immunofluorescence analysis of costal chondrocytes of newborn mice.
Cells were plated on coverslips in 24-well plates and grown for 5 days. After fixation with 2% paraformaldehyde, permeabilization using 0.2% Triton X-100 in PBS, and three washes with PBS, the cultures were treated with 10% normal goat serum in PBS as blocking reagent. Cells were incubated for 90 min with primary antibodies directed against the NC4 domain of α1 (IX), COMP (46), type II collagen (Calbiochem, Germany), and matrilin-3 at dilutions of 1:2,000, 1:800, 1:2,000, and 1:1,000, respectively, followed by detection with a secondary antibody (goat anti-rabbit IgG Alexa Fluor 488 and anti-mouse Cy3-conjugated antibodies, both obtained from Molecular Probes, The Netherlands) for a further 60 min. For nuclear counterstaining, bisbenzimide (Sigma, Germany) was added to the cells for 3 min at a final concentration of 0.05 μg/ml. Coverslips were finally mounted in DAKO fluorescent mounting medium and examined under an Axiophot fluorescence microscope (Zeiss, Germany).
Solid-phase binding assay (ELISA).
Recombinant murine matrilin-3 and recombinant human collagen IX were produced and isolated as described elsewhere (32, 40). Ninety-six-well flat-bottom plates (F96; Nunc, Wiesbaden, Germany) were coated with 1 μg human recombinant collagen IX or its digestion products in 100 μl TBS (pH 7.4) overnight at 4°C. After blocking with 2% skim milk powder in 150 μl TBS for 4 h, plates were washed three times with TBS containing 0.05% Tween 20. Then 0.1 to 40 μg/ml mouse recombinant matrilin-3 in 100 μl TBS was added and incubated at 4°C for 2 h. After washing four times with TBS containing 0.05% Tween 20, affinity-purified anti-matrilin-3 antiserum was added at 1:1,000 in 100 μl TBS containing 2% skim milk powder and incubated overnight at 4°C. Then, the plates were washed four times with TBS containing 0.05% Tween 20 and incubated with a horseradish peroxidase-coupled anti-rabbit immunoglobulin antibody at 1:1,000 in TBS containing 2% skim milk powder for 1 h at 4°C. After washing four times with TBS containing 0.05% Tween 20, binding was detected using 100 μl 0.04% (wt/vol) o-phenylenediamine, 24.3 mM citric acid, 51.4 mM Na2HPO4, and 0.012% (vol/vol) H2O2. The reaction was stopped by adding 50 μl of 2.5 M H2SO4. Absorbance at 490 nm was measured using a microplate reader (Dynatech Laboratories, Sussex, United Kingdom; software, MikroWIN V2.38, Mikrotek, Overath). All buffers contained 1 mM CaCl2 and 1 mM MgCl2. When indicated, buffers were supplemented with 10 mM EDTA.
Enzymatic treatment of human recombinant collagen IX.
For a limited pepsin digestion (6), 1 μg of collagen IX was incubated with 100 μg/ml pepsin (Serva, Heidelberg, Germany) in 0.5 M acetic acid overnight at 4°C; for collagenase digestion, 1 μg of collagen IX was incubated with 1 mU/μl collagenase CLSPA (Worthington) in PBS (pH 7.4) overnight at 37°C in the presence of 1 mM NEM; and for chondroitinase digestion, 1 μg of collagen IX was incubated with 2 mU chondroitinase ABC for 3 h at 37°C according to the manufacturer's protocol (Seikagaku, Tokyo, Japan).
RESULTS
Characterization of a novel polyclonal antibody specific for the α1 (IX) chain.
To generate an antibody against the α1 chain of mouse collagen IX, we prepared the N-terminal NC4 domain of the long form by recombinant expression in 293-EBNA cells. After purification, the recombinant NC4 domain was used for generation of polyclonal antibodies in rabbit. The specificity of the affinity-purified antibody was tested by immunoblotting using protein extracts from costal cartilage of newborn wild-type and collagen IX-deficient mice (Fig. 1, lanes 1 and 2). In wild-type cartilage extracts, affinity-purified NC4 antibodies recognized the α1 (IX) chain in addition to three additional polypeptides with high molecular mass (Fig. 1, lane 1), which presumably were covalent cross-linking products containing α1 (IX) chains. These polypeptides are typical for collagen IX, since bands with similar molecular weights are also recognized by other antibodies independently raised against collagen IX. None of these bands were detected in cartilage extracts of collagen IX knockout mice (Fig. 1, lane 2). Also, when culture medium of transfected 293-EBNA cells was used, only the recombinant NC4 protein at about 30 kDa was recognized by the antibody (Fig. 1, lane 4).
FIG. 1.
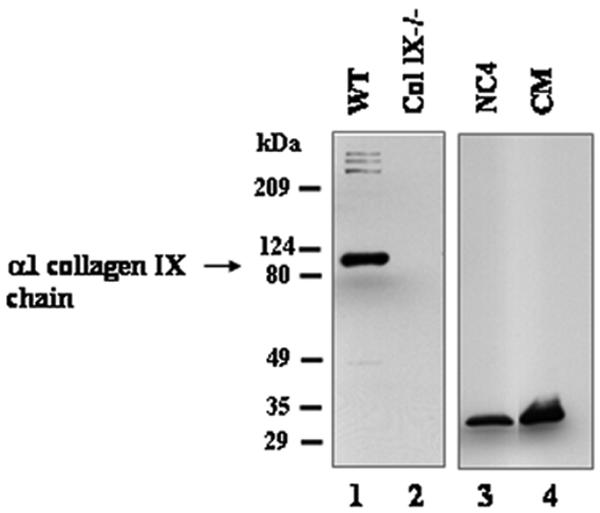
Immunoblot analysis of the anti-NC4 antiserum. Lanes 1and 2 were loaded with extracts from rib cages of newborn wild-type (WT) and collagen IX knockout (Col IX−/−) mice. Lane 3 contains 50 ng of purified recombinant NC4 protein, and lane 4 was loaded with culture medium (CM) from transfected 293-EBNA cells. All lanes were incubated with affinity-purified antibodies to NC4.
Immunohistochemical analysis of newborn collagen IX knockout mice.
For immunohistochemical analysis, serial sagittal sections of paraffin-embedded newborn mice of each genotype were prepared. In order to assess cartilage formation in newborn collagen IX knockout mice, we stained the sections for collagen IX, collagen II, aggrecan, COMP, and matrilin-3.
Staining for collagen IX on control sections of vertebral bodies and ribs produced a positive signal and showed a specific distribution pattern of the protein in the cartilage primordium of the vertebral bodies and ribs (Fig. 2G and J). The strongest collagen IX signal was detected in the extracellular matrix (ECM) of proliferating and hypertrophic chondrocytes (Fig. 2G), while immunostaining for collagen IX in the matrix around resting chondrocytes appeared to be comparable to that for COMP and matrilin-3 (Fig. 3E and F and Fig. 4E and F). However, it is uncertain whether these staining discontinuities reflect differences in the abundance of collagen IX protein or whether they are due to differential epitope masking. At any rate, cartilage of collagen IX-deficient mice lacked a specific signal for collagen IX (Fig. 2A and D).
FIG. 2.

Histological and immunohistological analysis of newborn mice. Paraffin-embedded sagittal sections of newborn collagen IX knockout (A to F) and wild-type (G to L) mice were stained with antibodies to collagen IX (A, D, G, and J) or collagen II (B, E, H, and K) and with alcian blue (C, F, I, and L). Panels A to C and G to I show vertebral bodies, and panels D to F and J to L display ribs. All cartilaginous and cartilage primordial tissues were stained similarly for collagen II and with alcian blue regardless of genotype (B, C, E, and F; H, I, K, and L). However, collagen IX-deficient cartilage did not show any staining with antibodies to collagen IX (A and D). Bar, 550 μm.
FIG. 3.

Immunohistological localization of COMP in newborn mice. Paraffin-embedded sagittal sections of newborn collagen IX knockout (A to D) and wild-type (E to H) mice were stained with a COMP-specific polyclonal antibody. All wild-type cartilaginous and cartilage primordial tissues were stained. (E) Growth plates of vertebral bodies. (F) Higher-magnification image of boxed area in panel E. (G) Ribs. (H) Higher-magnification image of boxed area in panel G. Contrary to the collagen II and alcian blue staining, the COMP signal is reduced in collagen IX (A to D)-deficient cartilage compared to wild-type mice. Abbreviations: VB, vertebral bodies; TP, transverse processes; NP, nucleus pulposus; RC, resting chondrocytes; PC, proliferating chondrocytes; HC, hypertrophic chondrocytes. Bars, 550 μm (A, C, E, and G) and 90 μm (B, D, F, and H).
FIG. 4.

Immunohistological localization of matrilin-3 in newborn mice. Matrilin-3 staining is virtually absent in mutant cartilage (A to D). In contrast, wild-type cartilage exhibits a strong matrilin-3 signal (E to H). For details, see the legend to Fig. 3.
Staining for collagen II, the major fibrillar collagen in cartilage tissues, revealed positive signals for all cartilaginous areas in both control and knockout sections. Collagen II distribution in the growth plate zone of vertebral bodies (Fig. 2B and H) and ribs (Fig. 2E and K) resembled the alcian blue staining pattern (Fig. 2C, I, F, and L). No alteration in signal intensity and staining pattern occurred in the absence of collagen IX. Higher magnification of selected areas of vertebral bodies and ribs did not reveal differences between knockout and control mice (data not shown).
Alcian blue, which binds to acidic glycosaminoglycans and thus mainly to aggrecan, the major proteoglycan in cartilage, revealed no differences in the absence of collagen IX. Alcian blue staining was found in the cartilage primordium of vertebrae and their transverse processes, hip joint, tarsal and carpal bones of the limbs, nasal cavity, and different cartilaginous parts of the skull. No alterations in alcian blue staining were apparent in collagen IX knockout mice (Fig. 2C and F). Cartilage primordia of vertebral bodies (Fig. 2C and I) and ribs (Fig. 2F and L) were stained, regardless of the presence of collagen IX. Even at higher magnification of selected areas of vertebral bodies and ribs, differences between knockout and control mice were not observed (data not shown).
In contrast, the distribution of COMP in the cartilage primordium of vertebral bodies and ribs was strongly influenced by the absence of collagen IX (Fig. 3). Cartilage of collagen IX-deficient mice exhibited a clear decrease of the COMP signal (Fig. 3A to D). Specifically, the zone of resting and partly that of proliferating chondrocytes in the growth plate of vertebral bodies (Fig. 4A and B) as well as the ECM of rib cartilage (Fig. 3C and D) retained only little COMP protein. Residual COMP is seen in the matrix surrounding part of the proliferating zone and the entire hypertrophic zone. By contrast, the corresponding cartilage tissues of wild-type mice contain COMP protein, demonstrated by a strong COMP antibody reaction (Fig. 3E to H). Notably, the matrix zone adjacent to the nucleus pulposus area revealed a clear COMP signal in the absence of collagen IX (Fig. 3B), an area which was less intensely stained in the control tissue (Fig. 3F).
An even more striking staining pattern emerged for matrilin-3 (Fig. 4). In the absence of collagen IX, basically all of matrilin-3 protein disappeared from the ECM of the entire growth zone of vertebral bodies (Fig. 4A and B) and ribs (Fig. 4C and D). Some matrilin-3 staining remained within the matrix of chondrocytes at the boundary between hypertrophic cartilage and the newly formed bone of the vertebral bodies (Fig. 4A and B). The corresponding cartilage tissues of control sections exhibited a strong signal for matrilin-3 (Fig. 4E to H). Similar to COMP, the matrix adjacent to the nucleus pulposus seemed to lack a matrilin-3 signal (Fig. 4E and F).
Immunohistochemical analysis of costal chondrocytes from newborn mice.
Costal chondrocytes were isolated from newborn wild-type, collagen IX-deficient, and COMP-deficient mice and were cultured in monolayer for 5 days. The cultures were then analyzed for collagen II, collagen IX, COMP, and matrilin-3 by immunofluorescence microscopy (Fig. 5). The intensity and distribution of the signals for collagens II and IX and matrilin-3 were comparable in COMP-deficient and wild-type cells. However, chondrocytes from collagen IX knockout mice exhibited an intracellularly confined matrilin-3 signal while lacking extracellular matrilin-3. In contrast, the collagen II antibody reaction appeared similar to that observed in wild-type and COMP knockout chondrocytes. COMP was densely integrated into the matrix adjacent to wild-type chondrocytes, whereas COMP staining was more diffuse at the periphery of collagen IX-deficient cells, indicating direct interaction with collagen IX. Taken together, these observations indicate that COMP can also undergo direct interactions with fibril components other than collagen IX, such as collagens II or XI. Matrilin-3, however, probably lacks the capacity for such interactions. Finally, matrix deposition of collagen IX does not depend on the presence of COMP.
FIG. 5.

Immunohistological analysis of costal chondrocytes from wild-type, collagen IX−/−, and COMP−/− newborn mice. Costal chondrocytes were isolated from wild-type and collagen IX and COMP knockout mice and cultured for 5 days before staining the cell layer with antibodies to collagen II, collagen IX, COMP, and matrilin-3. Detection was performed by immunofluorescence, and bisbenzimide was used as nuclear counterstain. Bars are 20 μm.
Quantitation of matrilin-3 and COMP gene expression in costal cartilage of newborn mice.
Total RNA was extracted from rib cages of newborn mice and analyzed by Northern blotting. GAPDH gene expression was used as a loading control. Matrilin-3 and COMP gene expression in wild-type mice was adjusted to GAPDH mRNA levels and arbitrarily set to 1 U using the densitometric analysis program ImageQuanNT (Amersham Biosciences, Germany). Matrilin-3 and COMP mRNA levels in collagen IX-deficient and wild-type cartilage did not differ significantly (Fig. 6). Thus, deficiency of Col9a1 mRNA or collagen IX protein did not change expression levels of the Matn-3 or COMP genes.
FIG. 6.

Gene expression levels of matrilin-3 and COMP in costal cartilage of newborn mice. Thirty micrograms of total RNA prepared from costal cartilage of newborn mice was blotted onto nylon filters and hybridized with probes specific for matrilin-3 (A) and COMP (B). Lanes 1, RNA extracted from costal cartilage from wild-type mice; lanes 2, RNA extracted from collagen IX knockout mice. Shown is a densitometric analysis of matrilin-3 and COMP gene expression levels normalized to GAPDH gene expression. Bars demonstrate the relative gene expression level of matrilin-3 and COMP in collagen IX knockout (IX−/−) mice compared to wild-type (wt) mice (n = 4 experiments each).
Determination of matrilin-3 protein content and solubility in costal cartilage and cultured chondrocytes of newborn mice.
In order to compare protein contents in mutant and wild-type tissues, we extracted matrilin-3 from rib cages of newborn mice and subjected the extracts to 4.5 to 15% SDS-PAGE under nonreducing conditions, followed by immunoblot analysis using affinity-purified antibodies against matrilin-3 (27) (Fig. 7A). To allow evaluation of both protein solubility and protein yield, three different extraction buffer systems were employed sequentially. The first, mild extraction buffer, was applied twice. The soluble fraction of matrilin-3 was isolated with 10 mM HEPES, and the insoluble fraction was consecutively extracted with 2 M urea and with 4 M GuHCl, both in the presence of 10 mM EDTA. SDS-PAGE of these extracts followed by immunoblot detection displayed two bands of different molecular masses. The lower faint band migrated at 175 kDa, the main band around 200 kDa, which is in agreement with earlier data on matrilin-3 structure and size (27). The fractions obtained under mild extraction conditions from rib cages of newborn collagen IX-deficient animals contained distinctly more matrilin-3 protein than the controls (Fig. 7A, lanes 1 to 4), while total protein of the HEPES extracts of wild-type and collagen IX knockout animals appear equal according to GAPDH protein signal used as an internal control (Fig. 7B). Subsequent extraction of the insoluble protein fractions with urea-EDTA and GuHCl-EDTA gave clearly higher yields of matrilin-3 from wild-type rib cages (Fig. 7A, lanes 5 to 8). To assess if additional matrilin-3 occurs covalently bound to collagens, we performed collagenase digestion of the final pellet obtained after GuHCl extraction. This enzyme degrades triple helical regions of collagens only and thereby potentially releases undigested noncollagenous proteins bound to the fibrillar collagens by covalent cross-linking. However, our results provided no indication for residual matrilin-3 which had not already been extracted before with denaturing buffers (Fig. 7A, lanes 9 and 10).
FIG. 7.

Immunoblot analysis of extracts from costal cartilage and costal chondrocyte cultures of newborn mice. (A) Matrilin-3 was extracted from costal cartilage of newborn mice using three different buffer systems in sequence. Protein extracts were subjected to 4 to 15% gradient SDS-PAGE followed by immunoblot analysis with affinity-purified matrilin-3 antibodies. Lanes 1 to 4 contain HEPES extracts, lanes 5 and 6 contain HEPES/EDTA/urea extracts, and lanes 7 and 8 contain HEPES/EDTA/GuHCl extracts. The last two lanes (9 and 10) were loaded with samples extracted from the final pellet after GuHCl treatment and subsequently digested with collagenase. (B) For protein standardization, the first extracts were also incubated with a monoclonal antibody recognizing GAPDH. (C) Analysis of extracted cell layers (CL) and supernatants (SN) of costal chondrocytes isolated from newborn wild-type (WT), collagen IX knockout (IX), and COMP knockout (COMP) mice using a matrilin-3 antibody. (D) Analysis of extracted cell layers and supernatants of costal chondrocytes isolated from newborn wild-type, collagen IX knockout, and COMP knockout mice using a COMP antibody. Molecular weight standards are indicated in thousands.
Since immunofluorescence studies indicated that collagen IX-deficient chondrocytes incorporate much less matrilin-3 into their newly deposited extracellular matrix in vitro (Fig. 5), we analyzed matrilin-3 distribution in extracts of cell layers and in culture medium by immunoblotting (Fig. 7C). Chondrocytes derived from wild-type and COMP-deficient mice retained virtually all matrilin-3 within the cell layer, consisting of cells and extracellular matrix (Fig. 7C, lanes 1, 2, 5, and 6). Notably, a substantial amount of matrilin-3 was found in the supernatant of chondrocyte cultures isolated from collagen IX knockout mice (Fig. 7C, lanes 3 and 4). However, the majority of COMP was secreted into the supernatant after 5 days of culture, and only little protein was retained in the cell layer from both wild-type and collagen IX-deficient chondrocytes (Fig. 7D, lanes 1 to 4). Apparently, the matrix-associated pool of COMP is rather small, even though it is detectable with immunofluorescence microscopy (Fig. 5), and not critically dependent on collagen IX for anchorage. These observations corroborated our conclusions that, in the absence of collagen IX, matrilin-3 is produced in normal quantities but is less stably integrated into cartilage matrix.
Collagen IX-deficient cartilage fibrils lack matrilin-3.
Next, we assessed the composition of isolated fibril fragments from costal cartilage of newborn mice. These mechanically generated fragments retain their authentic tissue properties. Their strongly incorporated components can be identified by immunoelectron microscopy, while loosely associated material is eliminated during the isolation procedure. Uranyl acetate-stained fragments of thin D-periodically banded fibrils are clearly distinguishable from more amorphous-appearing extrafibrillar aggregates. Matrilin-3 and collagen IX were identified by indirect immunogold labeling (Fig. 8).
FIG. 8.

Ultrastructural localization of matrilin-3 and collagen IX on fibril fragments from costal cartilage by indirect immunogold electron microscopy. Thin cartilage fibrils from wild-type mice are heavily labeled with collagen IX (18-nm gold particles) (A). Similarly sized fibrils from collagen IX knockout mice lack collagen IX labeling (D), while fibrils from COMP knockout mice resemble wild-type fibrils (G). Cartilage fibrils from wild-type mice are labeled with matrilin-3 (12-nm gold particles). Matrilin-3 is not restricted to fibrils but was also localized in extrafibrillar aggregates (B). Similar-appearing fibrils from collagen IX knockout mice lack matrilin-3 labeling; however, matrilin-3 is still present in extrafibrillar aggregates (E). Fibrils from COMP knockout mice are associated with matrilin-3, which is also found in extrafibrillar structures (H). Matrilin-3 and collagen IX colocalize at the same region of cartilage fibrils (C). Cartilage fibrils from collagen IX knockout mice are devoid of collagen IX and matrilin-3 (F), while cartilage fibrils from COMP knockout mice (I) show no difference in collagen IX integration compared to wild-type fibrils and exhibit a similar labeling pattern for matrilin-3. Open arrows indicate matrilin-3.
Fibril fragments from wild-type cartilage displayed a heavy labeling with the NC4-specific antibody. Collagen IX is densely packed along the fibrils of wild-type and COMP knockout mice and was exclusively associated with these (Fig. 8A and G). Fibrils from collagen IX-deficient mice were devoid of specific collagen IX labeling (Fig. 8D). Similarly sized fibrillar fragments from wild-type mice exhibited single as well as distinctly clustered matrilin-3 molecules along the fibril axis. Matrilin-3 labeling was restricted mainly to a population of thin, D-banded fibrils derived from the territorial matrix. However, matrilin-3 immunogold labeling was not exclusively confined to these fibrils but also formed extrafibrillar clusters associated to very thin filamentous structures (Fig. 8B). Fibrils from collagen IX-deficient mice displayed a strongly reduced matrilin-3 content (Fig. 8E), and those from COMP-deficient cartilage had an intermediate matrilin-3 labeling (Fig. 8H). To assess fibrillar localization of matrilin-3 in relation to collagen IX, we performed double immunogold labeling. As expected, we observed a heavy labeling of cartilage fibrils from control and COMP-deficient mice with collagen IX (18-nm gold particles). Matrilin-3 (12-nm gold particles) colocalized with collagen IX along the fibril. In COMP-deficient fibrils, matrilin-3 labeling was somewhat less dense. On both fibril types, matrilin-3 had a tendency for clustering, as seen in Fig. 8B (Fig. 8C and I). In the absence of collagen IX, cartilage fibrils lack collagen IX staining and have a strongly decreased matrilin-3 labeling (Fig. 8F). Notably, the occurrence of extrafibrillar clusters of matrilin-3 was not influenced and can thus be considered independent of collagen IX.
Statistical analysis of gold-labeled fibrils confirmed a reduced number of matrilin-3-positive particles on COMP-deficient fibrils and, much more drastically, on collagen IX-deficient fibrils (Fig. 9A). Wild-type and COMP-deficient fibrils were similarly labeled for collagen IX (Fig. 9B). Notably, collagen IX-deficient fibrils were somewhat wider (mean fibril diameter, 68.7 nm) than wild-type (61.9 nm) or COMP-deficient (63.4 nm) fibrils (Fig. 9C and D).
FIG. 9.

Statistical analysis of fibrillar matrilin-3 and collagen IX distribution. Fibrillar costal cartilage extracts of neonatal wild-type and collagen IX- and COMP-deficient mice were immunolabeled with anti-matrilin-3 (A) and anti-NC4 (B) antibodies. Electron micrographs were generated randomly using a Philips EM410 (60 kV) at a magnification of ×21,000. Fibril length and diameter were analyzed using image processing software (Image J v1.31) (C and D); gold particles were counted manually (A and B). Only gold particles clearly connected to the fibril by extrafibrillar material and not further than 100 nm from the fibril surface were counted. For each fibril, the number of gold particles per μm was determined. Arithmetic means and 99% confidence intervals were calculated for each sample group. The means of different sample groups were compared using the unpaired, heteroscedastic, two-tailed Student t test. Data shown are arithmetic means ± confidence intervals (P = 99%). *, P < 0.05; **, P < 0.01; ***, P < 0.005.
Matrilin-3 directly interacts with the collagenous domains of collagen IX.
By using a solid-phase binding assay (ELISA), we analyzed direct interaction between recombinant matrilin-3 and recombinant collagen IX. Immobilized collagen IX was titrated with indicated concentrations of soluble matrilin-3 (Fig. 10A). Saturation of binding was achieved at a concentration of 1.0 μg/ml matrilin-3, resulting in a calculated Kd value of 2 nM. Interaction with collagen IX proved to be strongly dependent on divalent cations. Addition of EDTA effectively abolished binding activity of matrilin-3 (Fig. 10B). In order to define matrilin-binding domains in collagen IX, we enzymatically eliminated either the noncollagenous domains (pepsin), the collagenous regions (collagenase), or a potentially attached glycosaminoglycan chain (chondroitinase ABC) (Fig. 10B). Binding activity of matrilin-3 to collagen IX was not affected by eliminating either the noncollagenous domains or putative GAG chains. However, the interaction was strongly diminished after removal of the collagenous domains, indicating that the recognition site(s) for matrilin-3 must be located within triple helical regions of collagen IX.
FIG. 10.

Analysis of the collagen IX/matrilin-3 interaction by solid-phase binding assay (ELISA). Recombinant matrilin-3 was added at the indicated concentrations to immobilized intact recombinant collagen IX coated at a concentration of 1 μg. The resulting saturation curve was used for calculation of an apparent Kd value (A). Binding of matrilin-3 to immobilized intact collagen IX was assayed in the presence of EDTA (Col IX + EDTA), and the interaction with immobilized enzymatically treated collagen IX was tested. For the latter application, collagen IX was treated with either pepsin (Col IX + pepsin), collagenase (Col IX + collagenase), or chondroitinase ABC (Col IX + ABC) prior to immobilization onto microtiter plates (B).
DISCUSSION
The stability of the cartilage extracellular matrix and, thereby, normal tissue function critically depend on strong interactions between the two suprastructural compartments of cartilage matrix. These are the collagen fibrils, as the major tensile elements, and the extrafibrillar matrix, mainly resisting compressive forces and comprising the hydrated hyaluronan-aggrecan network. In this context, the periphery of fibrils, its components, and their interactions are of considerable interest. In the present report, we provide evidence for an interaction of matrilin-3 and, independently, of COMP with collagen IX-containing cartilage fibrils in vivo. We took advantage of the Col9a1 knockout mouse deficient in the α1 (IX) chain (13), which leads to a functional knockout of the entire collagen IX protein (15). This model allowed us to investigate alterations in stability and integration of other structural components within the ECM of cartilage when collagen IX protein is missing. The main role assigned to collagen IX is in tissue organization. In addition to directing fibril organization, collagen IX stabilizes characteristic cartilage fibrils in vitro and, presumably, also in vivo (11).
In humans, mutations in cartilage matrix proteins often cause osteochondrodysplasias with various degrees of severity. Mutations in each of the three collagen IX α chains can cause the human disease multiple epiphyseal dysplasia (MED) (8, 36, 38). However, the complete loss of collagen IX, as in the mouse knockout, does not lead to the development of MED. Notably, MED does not result from mutations in only collagen IX but does result from mutations in collagen IX and matrilin-3 (7) and COMP (4). Matrilin-3 is the only member of the matrilin family known to be associated with MED (24). Interestingly, targeted disruption of the Matn-3 gene produces viable mice with no detectable skeletal abnormalities. Development of long bones, ribs, vertebral bodies, and intervertebral disks is normal. In addition, chondrocyte differentiation and endochondral bone formation are not affected by the knockout (29). These results imply that, similar to collagen IX deficiency, it is not the overall loss but rather structural alterations in matrilin-3 that result in MED, indicating that the disorder is most probably due to a dominant-negative effect (3, 7). Recent results indicate that the mutations in the Matn-3 gene cause MED by leading to a retention of matrilin-3 protein in the rough endoplasmic reticulum (37).
COMP has been shown to bind directly to both collagen IX and matrilin-3 (21, 32, 46). Thus, a loss of COMP from collagen IX-deficient cartilage is expected, given that an important structural anchor integrating COMP into cartilage matrix is absent. By contrast, the distributions of collagen II and aggrecan were normal in the absence of collagen IX. Particularly in ribs and cartilage primordium of vertebral bodies, COMP signal levels were much lower in collagen IX-deficient mice than in wild-type mice. However, residual COMP staining around proliferating and hypertrophic chondrocytes either results from interactions with matrix components other than collagen IX, e.g., collagen II (41), or is due to remnant COMP protein that will eventually be lost during further development. This assumption is consistent with the observation that collagen IX-deficient chondrocytes exhibit small, if any, alterations in COMP staining in vitro after culturing for 5 days.
Evidence for a direct interaction between matrilin-3 and collagen IX has been lacking. We have shown here that immunohistochemical staining for matrilin-3 is strongly decreased in collagen IX-deficient cartilage in vivo and in cultures of costal chondrocytes. Since matrilin-3 mRNA levels in mutant cartilage were normal, we surmised that matrilin-3 synthesis also was normal. However, the protein was readily extracted under very mild conditions from collagen IX-deficient cartilage, which implies an important interaction between matrilin-3 and collagen IX. Likewise, matrilin-3 is stably integrated into the cell layer of cultured wild-type chondrocytes, while soluble protein was hardly detected. In the absence of collagen IX, a substantial amount of matrilin-3 is soluble and found in the culture medium. In analogy, in vivo matrilin-3 which is stably integrated into cartilage fibrils probably diffuses out of the tissue of collagen IX-deficient mice and is then subsequently degraded. After exhaustive extraction with denaturing buffers, no additional matrilin-3 could be liberated by collagenase digestion, regardless of the presence of collagen IX. This precludes covalent interactions of matrilin-3 within cartilage collagen fibrils. Proof for the direct interaction of matrilin-3 with collagen IX in vivo comes from immunoelectron microscopy of matrilin-3 in fibril fragments from COMP-deficient cartilage (44). The signal for matrilin-3 is somewhat reduced but by no means absent in isolated fibril fragments when COMP is missing. In agreement, matrilin-3 immunofluorescence staining was comparable in wild-type and COMP-deficient chondrocytes in culture. In summary, the interaction of matrilin-3 with the surface of cartilage fibrils occurs both by a direct interaction with collagen IX and indirectly with COMP serving as an adapter. The former appears to be quantitatively more important.
Incontrovertible evidence for a direct interaction between collagen IX and matrilin-3 came from in vitro binding assays. Complex formation appeared to be of high affinity (apparent Kd of 2 nM) and was dependent on divalent cations, similar to the interaction of matrilin-3 with COMP in vitro (32). The binding site(s) of matrilin-3 resided in the collagenous rather than the noncollagenous regions in collagen IX that are known to harbor the recognition sites for COMP (21). A likely candidate is the COL3 domain which projects from the fibrillar surface into the perifibrillar space (48). This particular structural arrangement would allow interaction with extrafibrillar small macromolecules, thus promoting formation of a fibrillar periphery suitable for the stabilization of some of the interfibrillar connections as well as of contacts with the extrafibrillar aggrecan matrix (Fig. 11) (19).
FIG. 11.

Model for supramolecular assembly of cartilage fibrils and filaments into fibrillar networks. Matrilin-3 and COMP act as adapter molecules to interconnect D-periodically banded fibrils with each other and/or with collagen VI-beaded filaments to generate a heterotypic fibrillar network. The interaction may be mediated either by matrilin-3 binding directly to collagen IX or via COMP, which associates with the noncollagenous domains of collagen IX. Matrilin-3 contacts to collagen VI filaments are mediated by the small proteoglycans decorin and/or biglycan, which preferentially form complexes in the vicinity of the N termini of collagen VI filaments themselves acting as adapter molecules.
The presence of some insoluble matrilin-3 in costal cartilage of collagen IX-deficient mice suggests integration of an additional matrilin-3 population into structural networks independently of the presence of collagen IX, possibly into microfibrils composed of collagen VI or into gels formed by aggrecan aggregates (50). Recent analysis of collagen VI-containing microfibrils derived from the Swarm rat chondrosarcoma tissue by indirect immunogold labeling revealed complexes consisting of biglycan and decorin bound to the N-terminal globular domains of the collagen VI molecule. More distant from the collagen VI fibrils, matrilin-1, matrilin-3, and matrilin-4 were typically bound to these leucine-rich repeat proteoglycans, thus forming sandwich-like complexes associated to the surface of collagen VI microfibrils (50). As expected, association of matrilin-3 with these extrafibrillar thin filaments is not affected by the absence of collagen IX.
In summary, we detected matrilin-3 in two different fibrillar systems. First, we visualized matrilin-3 associated to thin, D-periodically banded cartilage fibrils. This association is critically dependent on the presence of collagen IX and to some extent COMP, as fibrils of similar appearance isolated from Col9a1-deficient mice are almost devoid of matrilin-3 and COMP-deficient fibrils exhibit a reduced matrilin-3 association. Second, matrilin-3 is associated independently of collagen IX to an additional fibrillar population, consisting of very thin, fragile filaments. The nature of these structures has not yet been determined, but as matrilin-3 has been shown to bind to collagen VI, they might well represent collagen VI microfibrils (Fig. 11).
The formation of the two main suprastructural compartments of cartilage, i.e., the extrafibrillar aggrecan matrix and the collagen fibrils, is subject to surprisingly simple control principles. Aggrecan and hyaluronan assemble into large and gel-forming aggregates that, in essence, fulfill all structural and functional requirements of the aggrecan matrix (18). Likewise, collagens II and XI aggregate into prototypic fibrils (2) with most of the suprastructural characteristics seen in the thin fibrils of immature cartilage (23). However, these simple compositions cannot be altered without causing major changes of their structures and functions. For example, substitution of collagen I for collagen II in mixtures with collagen XI results in structurally distinct fibrils with far less stringent growth control (17). Animals lacking normal collagen II or collagen XI have severely abnormal skeletal phenotypes (31), and patients with mutations in collagen II or collagen XI have skeletal or joint cartilage defects with a variable degree of severity (22). Unlike the collagenous fibril bodies and the aggrecan matrix themselves, their interface has a highly complex biochemical composition. The fibril surface is densely populated with several types of macromolecules which are all potential adapters between the fibrils and the extrafibrillar matrix. Sometimes, such molecules occur in a complementary fashion in different regions of the tissues. Collagen IX stabilizes the thin, prototypic cartilage fibrils preferentially found in the territorial matrix as baskets around chondrocytes (5). The protein forms a macromolecular alloy together with collagens II and XI but also projects its amino-terminal domains COL3 and NC4 away from the surface into the fibril-proximal environment, ready to undergo interactions with other adapter proteins or the aggrecan matrix (48). Other macromolecules, including matrilins, COMP, decorin, fibromodulin, and biglycan, bind directly or indirectly to the collagenous bodies of the fibrils and, therefore, are interface components potentially mediating interactions between cartilage fibrils and the extrafibrillar matrix. Deficiencies in such components generally have much less severe consequences than the absence of the core components of the suprastructural compartments. However, mutations that lead to structural abnormalities rather than the absence of these components can cause human or animal disease phenotypes (3). In part, the structural alterations lead to impairment of secretion and to excessive storage in chondrocytes, and thus the disease phenotypes result from cellular rather than matrix insufficiencies. However, in some cases, biogenesis of the fibril periphery occurs normally until structural alterations in the mutated proteins compromise their final functions. Taken together, these facts strongly suggest that, unlike the collagens of the fibril body or the aggrecan aggregates, the macromolecules of the fibril periphery are multiply redundant in their functions. Abnormalities in any one of these components, particularly their absence, are almost completely compensated by several other peripheral macromolecules. However, this compensation is likely to be incomplete under conditions requiring optimal performance of the tissues. Likely scenarios are bone fracture repair (16) or periods of intensified skeletal growth.
Acknowledgments
We thank Andrea Stadtbäumer and Heidi Bracht for their excellent technical assistance, Reinhard Fässler for supporting the study by providing the collagen IX knockout mice, and Ake Oldberg for his helpful suggestions and for providing the COMP knockout mice.
This study was supported by the Deutsche Forschungsgemeinschaft (grants GR 1301/4-2 to S.G. and WA 1338/3 to R.W. and M.P.), by the SFB 492 “Extracellular Matrix” project A2 to P.B., and by the Köln Fortune program of the Medical Faculty of the University of Cologne. K.B. is a student of the International Graduate School in Genetics and Functional Genomics at the University of Cologne.
REFERENCES
- 1.Annunen, S., P. Paassilta, J. Lohiniva, M. Perala, T. Pihlajamaa, J. Karppinen, O. Tervonen, H. Kroger, S. Lahde, H. Vanharanta, L. Ryhanen, H. H. Goring, J. Ott, D. J. Prockop, and L. Ala-Kokko. 1999. An allele of COL9A2 associated with intervertebral disc disease. Science 285:409-412. [DOI] [PubMed] [Google Scholar]
- 2.Blaschke, U. K., E. F. Eikenberry, D. J. Hulmes, H. J. Galla, and P. Bruckner. 2000. Collagen XI nucleates self-assembly and limits lateral growth of cartilage fibrils. J. Biol. Chem. 275:10370-10378. [DOI] [PubMed] [Google Scholar]
- 3.Briggs, M. D., and K. L. Chapman. 2002. Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations. Hum. Mutat. 19:465-478. [DOI] [PubMed] [Google Scholar]
- 4.Briggs, M. D., S. M. Hoffman, L. M. King, A. S. Olsen, H. Mohrenweiser, J. G. Leroy, G. R. Mortier, D. L. Rimoin, R. S. Lachman, E. S. Gaines, J. A. Cekleniak, R. G. Knowlton, and D. H. Cohn. 1995. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat. Genet. 10:330-336. [DOI] [PubMed] [Google Scholar]
- 5.Bruckner, P., M. Mendler, B. Steinmann, S. Huber, and K. H. Winterhalter. 1988. The structure of human collagen type IX and its organization in fetal and infant cartilage fibrils. J. Biol. Chem. 263:16911-16917. [PubMed] [Google Scholar]
- 6.Bruckner, P., and D. J. Prockop. 1981. Proteolytic enzymes as probes for the triple-helical conformation of procollagen. Anal. Biochem. 110:360-368. [DOI] [PubMed] [Google Scholar]
- 7.Chapman, K. L., G. R. Mortier, K. Chapman, J. Loughlin, M. E. Grant, and M. D. Briggs. 2001. Mutations in the region encoding the von Willebrand factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia. Nat. Genet. 28:393-396. [DOI] [PubMed] [Google Scholar]
- 8.Czarny-Ratajczak, M., J. Lohiniva, P. Rogala, K. Kozlowski, M. Perala, L. Carter, T. D. Spector, L. Kolodziej, U. Seppanen, R. Glazar, J. Krolewski, A. Latos-Bielenska, and L. Ala-Kokko. 2001. A mutation in COL9A1 causes multiple epiphyseal dysplasia: further evidence for locus heterogeneity. Am. J. Hum. Genet. 69:969-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deak, F., R. Wagener, I. Kiss, and M. Paulsson. 1999. The matrilins: a novel family of oligomeric extracellular matrix proteins. Matrix Biol. 18:55-64. [DOI] [PubMed] [Google Scholar]
- 10.DiCesare, P. E., M. Morgelin, K. Mann, and M. Paulsson. 1994. Cartilage oligomeric matrix protein and thrombospondin 1. Purification from articular cartilage, electron microscopic structure, and chondrocyte binding. Eur. J. Biochem. 223:927-937. [DOI] [PubMed] [Google Scholar]
- 11.Eikenberry, E. F., and P. Bruckner. 1999. Supramolecular structure of cartilage matrix, p. 289-300. In M. J. Seibel, S. P. Robins, and J. P. Bilezikian (ed.), Bone and cartilage metabolism. Academic Press, San Diego, Calif.
- 12.Eyre, D. R., T. Pietka, M. A. Weis, and J. J. Wu. 2004. Covalent cross-linking of the NC1 domain of collagen type IX to collagen type II in cartilage. J. Biol. Chem. 279:2568-2574. [DOI] [PubMed] [Google Scholar]
- 13.Fassler, R., P. N. Schnegelsberg, J. Dausman, T. Shinya, Y. Muragaki, M. T. McCarthy, B. R. Olsen, and R. Jaenisch. 1994. Mice lacking alpha 1 (IX) collagen develop noninflammatory degenerative joint disease. Proc. Natl. Acad. Sci. USA 91:5070-5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagg, R., P. Bruckner, and E. Hedbom. 1998. Cartilage fibrils of mammals are biochemically heterogeneous: differential distribution of decorin and collagen IX. J. Cell Biol. 142:285-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagg, R., E. Hedbom, U. Mollers, A. Aszodi, R. Fassler, and P. Bruckner. 1997. Absence of the alpha1(IX) chain leads to a functional knock-out of the entire collagen IX protein in mice. J. Biol. Chem. 272:20650-20654. [DOI] [PubMed] [Google Scholar]
- 16.Hankemeier, S., S. Grassel, G. Plenz, H. U. Spiegel, P. Bruckner, and A. Probst. 2001. Alteration of fracture stability influences chondrogenesis, osteogenesis and immigration of macrophages. J. Orthop. Res. 19:531-538. [DOI] [PubMed] [Google Scholar]
- 17.Hansen, U., and P. Bruckner. 2003. Macromolecular specificity of collagen fibrillogenesis: fibrils of collagens I and XI contain a heterotypic alloyed core and a collagen I sheath. J. Biol. Chem. 278:37352-37359. [DOI] [PubMed] [Google Scholar]
- 18.Hascall, V. C., and D. Heinegard. 1974. Aggregation of cartilage proteoglycans. I. The role of hyaluronic acid. J. Biol. Chem. 249:4232-4241. [PubMed] [Google Scholar]
- 19.Hauser, N., M. Paulsson, D. Heinegard, and M. Morgelin. 1996. Interaction of cartilage matrix protein with aggrecan. Increased covalent cross-linking with tissue maturation. J. Biol. Chem. 271:32247-32252. [DOI] [PubMed] [Google Scholar]
- 20.Hedbom, E., and D. Heinegard. 1993. Binding of fibromodulin and decorin to separate sites on fibrillar collagens. J. Biol. Chem. 268:27307-27312. [PubMed] [Google Scholar]
- 21.Holden, P., R. S. Meadows, K. L. Chapman, M. E. Grant, K. E. Kadler, and M. D. Briggs. 2001. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J. Biol. Chem. 276:6046-6055. [DOI] [PubMed] [Google Scholar]
- 22.Horton, W. A., and J. T. Hecht. 2002. Chondrodysplasias: general concepts and diagnostic and management considerations, p. 901-908. In P. M. Royce and B. Steinmann (ed.), Connective tissue and its heritable disorders. Wiley-Liss, New York, N.Y.
- 23.Hunziker, E. B. 1992. Articular cartilage structure in humans and experimental animals, p. 183-199. In K. E. Kuettner, K. E. Schleyerbach, J. G. Peyron, and V. C. Hascall (ed.), Articular cartilage and osteoarthritis. Raven Press, New York, N.Y.
- 24.Jackson, G. C., F. S. Barker, E. Jakkula, M. Czarny-Ratajczak, O. Makitie, W. G. Cole, M. J. Wright, S. F. Smithson, M. Suri, P. Rogala, G. R. Mortier, C. Baldock, A. Wallace, R. Elles, L. Ala-Kokko, and M. D. Briggs. 2004. Missense mutations in the beta strands of the single A-domain of matrilin-3 result in multiple epiphyseal dysplasia. J. Med. Genet. 41:52-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kassner, A., U. Hansen, N. Miosge, D. P. Reinhardt, T. Aigner, L. Bruckner-Tuderman, P. Bruckner, and S. Grassel. 2003. Discrete integration of collagen XVI into tissue-specific collagen fibrils or beaded microfibrils. Matrix Biol. 22:131-143. [DOI] [PubMed] [Google Scholar]
- 26.Klatt, A. R., D. P. Nitsche, B. Kobbe, M. Macht, M. Paulsson, and R. Wagener. 2001. Molecular structure, processing, and tissue distribution of matrilin-4. J. Biol. Chem. 276:17267-17275. [DOI] [PubMed] [Google Scholar]
- 27.Klatt, A. R., D. P. Nitsche, B. Kobbe, M. Morgelin, M. Paulsson, and R. Wagener. 2000. Molecular structure and tissue distribution of matrilin-3, a filament-forming extracellular matrix protein expressed during skeletal development. J. Biol. Chem. 275:3999-4006. [DOI] [PubMed] [Google Scholar]
- 28.Klatt, A. R., M. Paulsson, and R. Wagener. 2002. Expression of matrilins during maturation of mouse skeletal tissues. Matrix Biol. 21:289-296. [DOI] [PubMed] [Google Scholar]
- 29.Ko, Y., B. Kobbe, C. Nicolae, N. Miosge, M. Paulsson, R. Wagener, and A. Aszodi. 2004. Matrilin-3 is dispensable for mouse skeletal growth and development. Mol. Cell. Biol. 24:1691-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 31.Li, Y., D. A. Lacerda, M. L. Warman, D. R. Beier, H. Yoshida, Y. Ninomiya, J. T. Oxford., N. P. Morris, K. Andrikopoulos, F. Ramirez, B. B. Wardell, G. D. Lifferth, C. Teuscher, S. R. Woodward, B. A. Taylor, R. E. Seegmiller, and B. R. Olsen. 1995. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell 80:423-430. [DOI] [PubMed] [Google Scholar]
- 32.Mann, H. H., S. Ozbek, J. Engel, M. Paulsson, and R. Wagener. 2004. Interactions between the cartilage oligomeric matrix protein and matrilins. Implications for matrix assembly and the pathogenesis of chondrodysplasias. J. Biol. Chem. 279:25294-25298. [DOI] [PubMed] [Google Scholar]
- 33.Mendler, M., S. G. Eich-Bender, L. Vaughan, K. H. Winterhalter, and P. Bruckner. 1989. Cartilage contains mixed fibrils of collagen types II, IX, and XI. J. Cell Biol. 108:191-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miosge, N., K. Flachsbart, W. Goetz, W. Schultz, H. Kresse, and R. Herken. 1994. Light and electron microscopical immunohistochemical localization of the small proteoglycan core proteins decorin and biglycan in human knee joint cartilage. Histochem. J. 26:939-945. [PubMed] [Google Scholar]
- 35.Muller-Glauser, W., B. Humbel, M. Glatt, P. Strauli, K. H. Winterhalter, and P. Bruckner. 1986. On the role of type IX collagen in the extracellular matrix of cartilage: type IX collagen is localized to intersections of collagen fibrils. J. Cell Biol. 102:1931-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muragaki, Y., E. C. Mariman, S. E. van Beersum, M. Perala, J. B. van Mourik, M. L. Warman, B. R. Olsen, and B. C. Hamel. 1996. A mutation in the gene encoding the alpha 2 chain of the fibril-associated collagen IX, COL9A2, causes multiple epiphyseal dysplasia (EDM2). Nat. Genet. 12:103-105. [DOI] [PubMed] [Google Scholar]
- 37.Otten, C., R. Wagener, M. Paulsson, and F. Zaucke. 2005. Matrilin-3 mutations that cause chondrodysplasias interfere with protein trafficking while a mutation linked to hand osteoarthritis does not. J. Med. Genet. 42:774-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paassilta, P., J. Lohiniva, S. Annunen, J. Bonaventure, M. Le Merrer, L. Pai, and L. Ala-Kokko. 1999. COL9A3: a third locus for multiple epiphyseal dysplasia. Am. J. Hum. Genet. 64:1036-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piecha, D., S. Muratoglu, M. Morgelin, N. Hauser, D. Studer, I. Kiss, M. Paulsson, and F. Deak. 1999. Matrilin-2, a large, oligomeric matrix protein, is expressed by a great variety of cells and forms fibrillar networks. J. Biol. Chem. 274:13353-13361. [DOI] [PubMed] [Google Scholar]
- 40.Pihlajamaa, T., M. Perala, M. M. Vuoristo, M. Nokelainen, M. Bodo, T. Schulthess, E. Vuorio, R. Timpl, J. Engel, and L. Ala-Kokko. 1999. Characterization of recombinant human type IX collagen. Association of alpha chains into homotrimeric and heterotrimeric molecules. J. Biol. Chem. 274:22464-22468. [DOI] [PubMed] [Google Scholar]
- 41.Rosenberg, K., H. Olsson, M. Morgelin, and D. Heinegard. 1998. Cartilage oligomeric matrix protein shows high affinity zinc-dependent interaction with triple helical collagen. J. Biol. Chem. 273:20397-20403. [DOI] [PubMed] [Google Scholar]
- 42.Shaw, L. M., and B. R. Olsen. 1991. FACIT collagens: diverse molecular bridges in extracellular matrices. Trends Biochem. Sci. 16:191-194. [DOI] [PubMed] [Google Scholar]
- 43.Smyth, N., U. Odenthal, B. Merkl, and M. Paulsson. 2000. Eukaryotic expression and purification of recombinant extracellular matrix proteins carrying the Strep II tag. Methods Mol. Biol. 139:49-57. [DOI] [PubMed] [Google Scholar]
- 44.Svensson, L., A. Aszodi, D. Heinegard, E. B. Hunziker, F. P. Reinholt, R. Fassler, and A. Oldberg. 2002. Cartilage oligomeric matrix protein-deficient mice have normal skeletal development. Mol. Cell. Biol. 22:4366-4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szuts, V., U. Mollers, K. Bittner, G. Schurmann, S. Muratoglu, F. Deak, I. Kiss, and P. Bruckner. 1998. Terminal differentiation of chondrocytes is arrested at distinct stages identified by their expression repertoire of marker genes. Matrix Biol. 17:435-448. [DOI] [PubMed] [Google Scholar]
- 46.Thur, J., K. Rosenberg, D. P. Nitsche, T. Pihlajamaa, L. Ala-Kokko, D. Heinegard, M. Paulsson, and P. Maurer. 2001. Mutations in cartilage oligomeric matrix protein causing pseudoachondroplasia and multiple epiphyseal dysplasia affect binding of calcium and collagen I, II, and IX. J. Biol. Chem. 276:6083-6092. [DOI] [PubMed] [Google Scholar]
- 47.Treble, N. J., F. O. Jensen, A. Bankier, J. G. Rogers, and W. G. Cole. 1990. Development of the hip in multiple epiphyseal dysplasia. Natural history and susceptibility to premature osteoarthritis. J. Bone Joint Surg. Br. 72:1061-1064. [DOI] [PubMed] [Google Scholar]
- 48.Vaughan, L., M. Mendler, S. Huber, P. Bruckner, K. H. Winterhalter, M. I. Irwin, and R. Mayne. 1988. D-periodic distribution of collagen type IX along cartilage fibrils. J. Cell Biol. 106:991-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagener, R., H. W. Ehlen, Y. P. Ko, B. Kobbe, H. H. Mann, G. Sengle, and M. Paulsson. 2005. The matrilins—adaptor proteins in the extracellular matrix. FEBS Lett. 579:3323-3329. [DOI] [PubMed] [Google Scholar]
- 50.Wiberg, C., A. R. Klatt, R. Wagener, M. Paulsson, J. F. Bateman, D. Heinegard, and M. Morgelin. 2003. Complexes of matrilin-1 and biglycan or decorin connect collagen VI microfibrils to both collagen II and aggrecan. J. Biol. Chem. 278:37698-37704. [DOI] [PubMed] [Google Scholar]
- 51.Wu, J. J., and D. R. Eyre. 1998. Matrilin-3 forms disulfide-linked oligomers with matrilin-1 in bovine epiphyseal cartilage. J. Biol. Chem. 273:17433-17438. [DOI] [PubMed] [Google Scholar]
- 52.Wu, J. J., P. E. Woods, and D. R. Eyre. 1992. Identification of cross-linking sites in bovine cartilage type IX collagen reveals an antiparallel type II-type IX molecular relationship and type IX to type IX bonding. J. Biol. Chem. 267:23007-23014. [PubMed] [Google Scholar]