

Abstract
The c-Myc oncoprotein (Myc) controls cell fate by regulating gene transcription in association with a DNA-binding partner, Max. While Max lacks a transcription regulatory domain, the N terminus of Myc contains a transcription activation domain (TAD) that recruits cofactor complexes containing the histone acetyltransferases (HATs) GCN5 and Tip60. Here, we report a novel functional interaction between Myc TAD and the p300 coactivator-acetyltransferase. We show that p300 associates with Myc in mammalian cells and in vitro through direct interactions with Myc TAD residues 1 to 110 and acetylates Myc in a TAD-dependent manner in vivo at several lysine residues located between the TAD and DNA-binding domain. Moreover, the Myc:Max complex is differentially acetylated by p300 and GCN5 and is not acetylated by Tip60 in vitro, suggesting distinct functions for these acetyltransferases. Whereas p300 and CBP can stabilize Myc independently of acetylation, p300-mediated acetylation results in increased Myc turnover. In addition, p300 functions as a coactivator that is recruited by Myc to the promoter of the human telomerase reverse transcriptase gene, and p300/CBP stimulates Myc TAD-dependent transcription in a HAT domain-dependent manner. Our results suggest dual roles for p300/CBP in Myc regulation: as a Myc coactivator that stabilizes Myc and as an inducer of Myc instability via direct Myc acetylation.
The c-Myc oncoprotein (Myc) is the ubiquitous member of a small family of highly related DNA-binding transcription factors (including L-Myc and N-Myc) that regulate a wide variety of genes involved in the control of cell growth, proliferation, differentiation, and apoptotic cell death. Myc is essential for embryonic development and both Myc expression and activity are tightly regulated by mitogens and other physiological stimuli in normal somatic cells. Notably, unregulated Myc expression is tumorigenic in mice and has been associated with most types of cancer in humans. Myc binds to E-box DNA elements having the core consensus sequence CACGTG as a heterodimer with an obligatory partner protein called Max. Myc and Max dimerize and bind DNA via their respective basic-helix-loop-helix-leucine zipper (bHLHZip) domains. While Max does not have a transcription regulatory domain, Myc has a phylogenetically conserved N-terminal transcription activation domain (TAD) that is also essential for oncogenic cellular transformation (reviewed in reference 10).
Several proteins have been shown to interact with Myc N-terminal TAD and are potential regulators or mediators of Myc transactivating and transforming activities (10, 30). Among these, the TRRAP protein has been shown to contribute to the transformation activity of Myc through interactions with the conserved Myc box 1 (MB1) and MB2 regions within the TAD (23) and is a subunit of various transcription regulatory cofactors complexes that have histone acetyltransferase (HAT) activity. These TRRAP-HAT complexes include the GCN5 HAT-containing complexes STAGA (21, 22) and TFTC (3), the related PCAF HAT-containing complex (25), and the Tip60 HAT-containing complex (14). Although the paralogous mammalian GCN5 and PCAF HATs preferentially acetylate histone H3, the preferred substrate for the Tip60 HAT is histone H4 (29). Significantly, Myc-stimulated gene transcription in vivo correlates with an increased acetylation of both histones H3 and H4 in the vicinity of E-box elements within regulatory regions of Myc target genes (2, 8, 24). This suggests that Myc-induced transcription in vivo might involve the recruitment by Myc of the TIP60 complex and GCN5/PCAF-containing complexes. Consistent with this, Myc associates with STAGA components in vivo and directly interacts through its TAD with the purified STAGA complex in vitro, TRRAP and GCN5 synergistically stimulate Myc TAD transactivating functions, and components of the TIP60 complex, including the Tip60 HAT subunit, are recruited to Myc target promoters in vivo during Myc-activated transcription (9, 19). Although these results suggest that TIP60 and GCN5 might mediate Myc transactivating functions via acetylation of histones on target promoters, additional nonhistone protein substrates for these HATs have been described (reviewed in reference 35). In particular, GCN5, Tip60, and CBP were reported recently to induce Myc acetylation in mammalian cells (26, 31). Thus, coactivator-HATs might regulate Myc functions at multiple levels and perhaps differentially.
Here we have investigated the possible role of the p300 coactivator-HAT as a mediator of Myc transactivating functions and as a regulator of Myc via direct acetylation. We show that p300 interacts with Myc in vivo and in vitro, and that the N-terminal 1-110 region of Myc TAD is necessary and sufficient for efficient and direct interaction with p300. This is different from the reported interaction of CBP with the C terminus of Myc (31). We further demonstrate that the 1-110 TAD region is also essential for Myc acetylation by endogenous HATs and by ectopic p300 in mammalian cells and that full-length p300 efficiently acetylates both Myc and Max within a Myc:Max complex in vitro. In contrast, GCN5 and the GCN5-containing STAGA complex only acetylate Myc, whereas, unexpectedly, neither Myc nor Max is a direct substrate for acetylation by Tip60 in vitro. We further identify several lysine residues located between the TAD and the HLHZip domains of Myc that are acetylated by p300 in cultured cells and in vitro. Our results suggest that p300-mediated acetylation increases Myc protein turnover but also that p300 (and CBP) can stabilize Myc in mammalian cells independently of acetylation. We further demonstrate that p300 is recruited by Myc to the promoter of the endogenous human telomerase reverse transcriptase (hTERT) gene in human cancer cells and that p300/CBP mediates the transactivating functions of Myc TAD at the hTERT promoter and at an artificial promoter independently of Myc/Max acetylation but in a HAT domain-dependent manner. Altogether, our results reveal a complex regulation of Myc by p300 through interactions with Myc TAD that lead to acetylation-dependent and -independent regulation of Myc protein turnover and stimulation of Myc TAD-mediated transcription activation.
MATERIALS AND METHODS
DNA plasmids.
The bacterial expression vectors for His6-tagged human Max short isoform, pET-His-Max (33), His6-tagged human GCN5 short form, pRSETA-His-GCN5-S (6), and His6-tagged human full-length c-Myc, pRSET-His6-Myc (7) were described previously. The hTERT luciferase reporter vectors p2xEB (hTERT wt) and pBTdel-208 (Del.E2) were described elsewhere (13); p2xEB Mut.1 (Mut.E1) was derived from p2xEB by site-directed mutagenesis of the upstream E-box from CACGTG to CACCTG by using the QuikChange site-directed mutagenesis kit (Stratagene) and verified by DNA sequencing. pG5-E4-Luc was provided by Xuan Liu. Plasmids pSV-Gal4(1-147), pSV-Gal4-Myc(1-262), pSV-Gal4-Myc(1-103), and pSV-Gal4-Myc(41-143) were described elsewhere (16). The plasmids pRc/RSV-CBP (18) and pRc/RSV-CBP(F1541) referred to as CBPΔHAT, in which the Phe1541 essential for CBP HAT activity was substituted with alanine (20), were described previously. The expression vectors for p300 were pCDNA3.1p300 (5), pCMVβ-p300-CHA (gift from R. Goodman), and pCI-p300-FLAG (obtained from Y. Nakatani). The expression vectors for human c-Myc, pUHD-Myc (27), and for Flag-tagged mouse c-Myc, pCbS-Flag-Myc full-length and Δ1-110 (23) were described previously. The expression vectors for the mouse c-Myc K→R mutants (NR, R5, R6, CR, and NRC) were derived from pCbS-Flag-Myc by site-directed mutagenesis and verified by DNA sequencing. The expression vector for Flag-tagged human Max short isoform (pCbS-Flag-Max) was obtained by cloning between the BamHI and Hind III sites of pCbS plasmid a Flag-Max cDNA fragment obtained by PCR of pET-His-Max with the primers 5′-CCTGCCGGATCCACCATGGACTACAAGGACGATGATGACAAGCCCGGGATGAGCGATAAC GATGACATCGAGGTGGAG and 5′-CTGCCCAAGCTTAGCTGGCCTCCATCCGGAG.
The baculovirus transfer/expression vector pFastBac-p300-Flag encodes p300 Flag-tagged at its C terminus and was obtained by cloning the p300 cDNA as a NotI-HindIII fragment from pCMVβ-p300 (Upstate Biotechnology) into pFastBac1 (Invitrogen). The NheI-HindIII fragment of the resulting vector containing the p300 stop codon was replaced with an NheI-HindIII DNA fragment having a tyrosine (TAC) codon instead of the stop codon and an in-frame Flag epitope sequence. The construct was verified by DNA sequencing. The pFastBac-Flag-Tip60α was created by cloning the Tip60α cDNA (28) obtained by PCR from the human HeLa QUICK-Clone cDNA (Clontech Laboratories, Inc.) by using the nested primers 5′-AGTGACGTCTCCCAGAGGGGCCGGAAGT and GGG TGGGCTATCAGCCCCAGTCCTGC (first PCR amplification) and then 5′-CGTCTCCCAGAGGGATCCGAAGTGGC and 5′-CCAGTCCTGCGGCCGCCTTGGC (that introduced BamHI and NotI sites during the second PCR amplification) between the BamHI and NotI sites of pFlagFast baculoviral vector, a derivative of pFastBac1 encoding a Flag epitope. The resulting pFastBac-Flag-Tip60α encodes a Tip60α protein having a Flag tag and a 16-amino-acid spacer sequence at its N terminus. The construct was verified by DNA sequencing.
Antibodies.
The antibodies used in immunoprecipitation and Western blot analyses were: hTBP (gift from R. G. Roeder), Gal4 DBD, Pol II N-20, p300 N-15, Max H2, Max C-17, Max C-124, Myc C-33, and Myc N-262 (Santa Cruz Biotechnology), Acetyl-K (Cell Signaling Technology), Flag M2, and Flag M2 affinity resin (Sigma).
Cell culture, transfection, immunoprecipitation, and Western blotting.
COS-7, HEK293, and HeLa cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum at 37°C with 5% CO2. For luciferase reporter assays, HeLa cells at 80% confluence were transfected in six-well plates by using Lipofectamine reagent (Invitrogen) and 0.5 μg of hTERT-Luc plasmid, 0.1 μg of pCbS-Flag-Myc, 1 μg of pcDNA3.1p300 or pRc/RSV-CBP, and 0.2 μg of pCMV-β-galactosidase (see Fig. 6) or 0.2 μg of pG5-E4-Luc, 0.1 μg of pSV-Gal4-Myc, 1 μg of pcDNA3.1p300 or pRc/RSV-CBP, and 0.2 μg of pCMV-β-galactosidase (see Fig. 7). Corresponding empty vectors were used to keep total DNA constant. Luciferase assays were performed as previously described (19). Since β-galactosidase activity was moderately increased by transfection of p300 and CBP, luciferase activity was normalized to protein concentration, and each result is the mean ± the standard deviation (SD) from at least three independent experiments, each performed in duplicate.
FIG. 6.

Synergistic activation of the hTERT promoter by Myc and p300/CBP in HeLa cells requires E-box elements, p300/CBP HAT activity, and the Myc TAD1-110 region. (A) Schematic of hTERT-luciferase reporters. Two E-box elements and five Sp1 sites are indicated. The bent arrow is the transcription start site. (B) HeLa cells were transiently transfected with hTERT wt (black), Del.E2 (gray) or Mut.E1 (white) constructs and expression vectors for Flag-Myc and p300 (lanes 4 to 6) or corresponding empty vectors (lanes 1 to 3). Luciferase activities are relative to the activity in lane 1 (set to 1, *). (C) Cells were transfected with hTERT wt (▪) (lanes 1 to 6) or Del.E2 ( ) (lanes 7 to 12) and with p300 and Flag-Myc wild-type (wt) and Δ1-110 (Δ110) expression vectors, as indicated. The “Fold Activation” is relative to the luciferase activities obtained with hTERT wt reporter alone (lane 1) or Del.E2 alone (lane 7), which were set to 1 (*). (D) Cells were transfected with hTERT wt (▪) (lanes 1 to 8) or Del.E2 () (lanes 9 to 16) and expression vectors for Flag-Myc proteins (as described above) and CBP or CBPΔ HAT, as indicated. (E) Cells were transfected with hTERT wt reporter and Flag-Myc wt or R6 mutant and either p300 (+) or the empty vectors (−), as indicated. Fold activation is relative to lane 1. (F) Extracts of cells transfected with either Flag-Myc wt or Flag-Myc Δ1-110 expression vectors and with (+) p300 (upper panel) or CBP or CBPΔHAT (bottom panels) or with the corresponding empty vectors (−) were analyzed by Western blotting with antibodies to Flag (wt and Δ110) and TATA-binding protein (TBP).
) (lanes 7 to 12) and with p300 and Flag-Myc wild-type (wt) and Δ1-110 (Δ110) expression vectors, as indicated. The “Fold Activation” is relative to the luciferase activities obtained with hTERT wt reporter alone (lane 1) or Del.E2 alone (lane 7), which were set to 1 (*). (D) Cells were transfected with hTERT wt (▪) (lanes 1 to 8) or Del.E2 () (lanes 9 to 16) and expression vectors for Flag-Myc proteins (as described above) and CBP or CBPΔ HAT, as indicated. (E) Cells were transfected with hTERT wt reporter and Flag-Myc wt or R6 mutant and either p300 (+) or the empty vectors (−), as indicated. Fold activation is relative to lane 1. (F) Extracts of cells transfected with either Flag-Myc wt or Flag-Myc Δ1-110 expression vectors and with (+) p300 (upper panel) or CBP or CBPΔHAT (bottom panels) or with the corresponding empty vectors (−) were analyzed by Western blotting with antibodies to Flag (wt and Δ110) and TATA-binding protein (TBP).
FIG. 7.
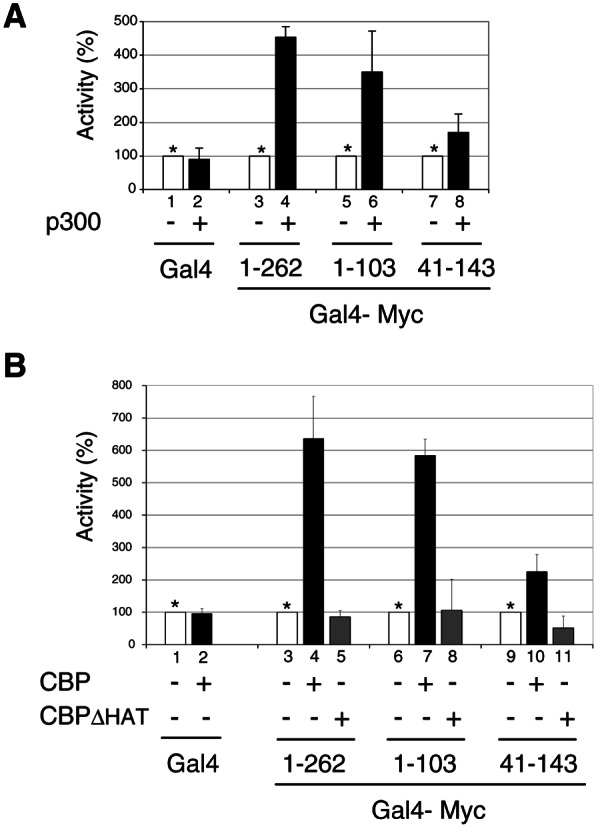
Myc TAD 1-103 region is sufficient for synergistic transcription activation with p300/CBP. (A) HeLa cells were transfected with pG5-E4-luciferase reporter and expression vectors for either Gal4 (lanes 1 and 2), Gal4-Myc(1-262) (lanes 3 and 4), Gal4-Myc(1-103) (lanes 5 and 6), or Gal4-Myc(41-143) (lanes 7 and 8) and with a p300 expression vector (+) or the corresponding empty vector (−). The activity of each Gal4 fusion activator in the presence of p300 (▪) is given as the percentage of its activity in the absence of p300, which was set arbitrarily to 100% (□, asterisks). (B) HeLa cells were transfected as described above but with (+) expression vectors for CBP, or CBPΔHAT or the corresponding empty vector (−), as indicated. The activity of each Gal4 fusion activator in the absence of CBP/CBPΔHAT was set to 100% as described above.
For immunoprecipitation of Flag-tagged proteins, HEK293 cells in 10-cm plates at ca. 95% confluence were transfected with Lipofectamine 2000 (Invitrogen) and pCI-p300-FLAG (10 μg) and pUHD-Myc (8 μg) as indicated in Fig. 1B or with pCbS-Flag-Myc (8 μg), pCbS-Flag-Max (3 μg), and pCDNA3.1p300 (10μg) as indicated in Fig. 1C. The corresponding empty vectors were included in control lanes, as indicated. At 48 h after transfection, whole-cell extracts were prepared in lysis buffer (50 mM HEPES [pH 7.9] at 4°C, 250 mM NaCl, 0.1% IGEPAL CA-630, 0.2 mM EDTA, 0.2 mM phenylmethylsulfonyl fluoride [PMSF], 2 mM 2-mercaptoethanol). Lysates were diluted with BC-0 (20 mM HEPES [pH 7.9] at 4°C, 20% glycerol, 0.2 mM EDTA, 0.05% IGEPAL CA-630, 0.2 mM PMSF, 5 mM 2-mercaptoethanol) to reach 179 mM NaCl final concentration (immunoprecipitation [IP] buffer), and cell extracts from one 10-cm plate were incubated with 10 μl of Flag M2 resin for 12 h at 4°C under constant rotation. Immunoprecipitates were washed three times with IP buffer, resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and analyzed by Western blotting with the indicated antibodies. For immunoprecipitation of endogenous Myc and Max proteins, HeLa cells were lysed as described above. Lysates in either IP buffer (Fig. 1A) or lysis buffer (see Fig. 3B) were precleared, respectively, with 15 μl of protein G-Sepharose resin (Amersham Biosciences) or with 25 μl of TrueBlot anti-rabbit immunoglobulin IP beads (eBioscience) for 30 min at 4°C. The indicated antibodies (5 μg) were incubated with the lysates (from one or two 10-cm plates) for 14 h at 4°C and precipitated after incubation with 10 μl of protein G-Sepharose for 4.5 h at 4°C (Fig. 1A) or 40 μl of TrueBlot anti-rabbit immunoglobulin IP beads (eBioscience) overnight at 4°C (Fig. 3B). Immunoprecipitates were washed four times with IP buffer or lysis buffer and analyzed by SDS-PAGE and Western blotting with the indicated antibodies and the enhanced chemiluminescence (ECL) kit (Amersham Biosciences). Rabbit immunoglobulin G TrueBlot-HRP reagent (eBioscience) was used in the Western blot of Fig. 3B (top panel) to lower the background signal of denatured immunoglobulin heavy chains.
FIG. 1.

Myc interacts with p300 in mammalian cells and in vitro via its TAD. (A) A HeLa whole-cell extract was immunoprecipitated (IP) with Flag (lane 2), Max H2 (lane 3), and Myc C-33 (lane 4) antibodies and analyzed by Western blotting (WB). Different stripes of the same blot were probed with p300 (N-15), Myc (N-262), and Max (C-17) antibodies. Lane 1 shows the input extract. (B) Extracts of HEK293 cells transfected with pUHD-Myc and/or pCI-p300-Flag, or the corresponding empty vectors as indicated with “+” and “−,” respectively (input, lanes 1 to 3), were immunoprecipitated with the Flag antibody (IP: Flag, lanes 4 to 6) and analyzed by Western blotting (WB) with p300 (N-15) and Myc (N-262) antibodies. (C) Extracts of HEK293 cells (input, lanes 1 to 7) transfected with Flag-Myc wild-type (wt), Flag-Myc deletion mutant Δ1-110 (Δ), Flag-Max, and p300, or corresponding empty vectors as indicated (+ and −), were immunoprecipitated with the Flag antibody (IP: Flag, lanes 8 to 14) and analyzed by Western blotting with p300, Flag, and Max antibodies, as indicated (WB). The positions of Myc wt and Δ1-110 are indicated. A nonspecific band was detected in lanes 13 and 14 (*). (D) GST pull-down assays. Purified recombinant p300 (lanes 1 and 8; see also Fig. 2A) was incubated with immobilized GST (lanes 2 and 9) and GST-Myc fusion proteins with different Myc regions as indicated (lanes 3 to 7, 10, and 11, see also panel E). Specific binding of p300 was analyzed by Western blotting with p300 (N-15) antibodies. (E) Summary of the above p300-Myc interaction results obtained by coimmunoprecipitation assays (in cells) and by GST pull-down assays (in vitro). MB1 and MB2 are Myc boxes 1 and 2; bHLHZip is the basic helix-loop-helix leucine zipper domain. Strong interaction with p300, i.e., comparable to wild-type Myc (+++), medium (++), weak (+), and no detectable (−) interactions are indicated.
FIG. 3.

Myc is acetylated by p300 in mammalian cells. (A) Acetyl-K antibody recognizes p300-acetylated Myc in vitro. The Myc:Max complex (0.3 pmol) was acetylated in vitro in the absence (−) and presence (+) of p300 (50 ng) and cold acetyl-CoA (2 μM, lane 3, and 10 μM, lanes 4 and 5). The reactions were analyzed by Western blotting (WB) with p300 (N-15), Myc (C-33), and acetyl-K antibodies. The Myc portion of the filter was probed first with acetyl-K antibody and then stripped and reprobed with Myc (C-33) antibody. Acetylated Myc is indicated with an arrowhead. (B) Endogenous Myc-Max complexes were immunoprecipitated with Myc (N-262) and Max (C-124) antibodies (lanes 2 and 3, respectively) from HeLa cells transfected with p300 and treated for 4.5 h with HDAC inhibitors and MG-132. A control immunoprecipitation was performed with a Gal4 (DBD) antibody (lane 1). Western blot was performed first with the acetyl-K antibody and, after stripping the same filter, was reprobed with Myc (C-33) antibody. TrueBlot reagents were used for IP and Western blotting (see Materials and Methods). Arrowheads indicate the position of Myc. (C) COS-7 cells and (D) HEK293 cells were transfected as described in Materials and Methods with either Flag-Myc wt or Flag-MycΔ1-110 mutant (Δ1-110) and p300 (+) or the corresponding empty vectors (−), as indicated. Cells were treated (+) or mock-treated (−) with HDAC inhibitors (HDAC inh.), as indicated. Immunoprecipitations were performed with the Flag antibody and Flag-Myc proteins were analyzed by Western blotting (WB) with the acetyl-K antibody first (top panel) and then, after stripping the filter, with the Flag antibody (bottom panel). Flag-Myc wt and Δ1-110 are indicated with filled and open arrowheads, respectively. An asterisk indicates the immunoglobulin H chain.
Expression and purification of recombinant proteins and glutathione S-transferase (GST) pull-down assays.
Recombinant human Myc-Max and Max-Max complexes and recombinant human GCN5-S were expressed in bacteria and purified as previously described (7). The STAGA complex was purified from a HeLa cell line expressing Flag-tagged SPT3, as described previously (22). The recombinant baculoviruses for Flag-Tip60 and p300-Flag were prepared by using the Bac-to-Bac baculovirus expression system (Life Technologies). Flag-tagged Tip60 and p300 were expressed in insect Sf9 cells and purified from whole-cell lysates by anti-Flag affinity chromatography as described before (15). GST pull-down assays were performed essentially as previously described (22).
In vitro acetylation assays.
Acetylation reactions were performed in a 20-μl final volume by incubating about 2.5 pmol of recombinant Myc-Max or Max-Max complex or a 2-μg mixture of calf thymus histones (Roche) with one of the following HATs: 6 to 13 ng of GCN5-S (or equivalent HAT units of STAGA complex), 36 ng of Tip60α, and 4 to 8 ng of p300 in the presence of 2 μM [3H]acetyl-coenzyme A (CoA; 27.5 Ci/mmol, 1.0 mCi/ml; Sigma) in HAT buffer (50 mM Tris-HCl [pH 8.0], 14% glycerol, 70 mM KCl, 0.18 mg of bovine serum albumin/ml, 0.1% IGEPAL CA-630, 10 mM sodium butyrate, 8 mM 2-mercaptoethanol, and 0.3 mM PMSF) for 1 h at 30°C. Proteins were resolved by SDS-PAGE and stained with Coomassie blue. For fluorographic detection of 3H-acetylated proteins, the gels were further soaked in “Amplify” solution (Amersham Biosciences), dried, and exposed to autoradiography films with intensifying screens at −80°C. Cold acetylation reactions were performed essentially as described above but with 50 ng of p300 and the indicated amount of unlabeled acetyl-CoA, and the acetylated proteins were identified by Western blotting with the acetyl-K antibody.
Analysis of Myc acetylation in cultured cells and stability assays.
HEK293 cells in a 10-cm plate were transfected as described above, with 7 μg of pCbS-Flag-Myc wt or Δ1-110, 1 μg of pCbS-Flag-Max, and 10 μg of pcDNA3.1p300, or the corresponding empty vectors, as indicated (Fig. 3D). COS-7 cells were transfected twice with a 6- to 8-h gap between transfections, with 4.5 μg of pCbS-Flag-Myc wt or mutants, 0.5 μg of pCbS-Flag-Max, and 4 μg of pCDNA3.1p300 (or pCMVβ-p300-CHA). After 36-h expression, the cells were incubated with histone deacetylase (HDAC) inhibitors (10 mM sodium butyrate, 10 mM nicotinamide, 2 μM trichostatin A) or the corresponding solvents for an additional 12 h before extract preparation in lysis buffer containing 10 mM sodium butyrate. Immunoprecipitations and Western blots were as described above. For the experiment of Fig. 4C, HEK293 cells were transfected, three times with a 4-h gap between transfections, with 3 μg of pCbS-Flag-Myc wt or the indicated mutants, 5 μg of pCMVβ-p300-CHA, or the corresponding empty vectors. After 40-h expression the cells were incubated for 8 h with the HDAC inhibitors before extract preparation, immunoprecipitation, and Western blot analysis with the indicated antibodies. Mass spectrometry mapping of the Myc acetylated residues is described in the supplemental material.
FIG. 4.

Mapping p300-acetylated Myc residues in mammalian cells by mass spectrometry and site-directed mutagenesis. (A) Diagram of mouse c-Myc wt indicating seven lysine residues acetylated by p300 in vitro (arrowheads; see reference 37 and additional data in the supplemental material). Filled arrowheads indicate those lysine residues acetylated by p300 in HEK293 cells and mapped by MS/MS (see text and mass spectra in the supplemental material). The lysine-to-arginine (K→R) substitution mutants used below are indicated. Boxes 1 and 2 are MB1 and MB2. “N” is a nuclear localization signal. (B) Analysis of Flag-Myc wt and K→R mutants in transfected COS-7 cells treated (+) or mock treated (−) with HDAC inhibitors (HDAC inh.). Equal amounts of immunoprecipitated Flag-Myc wt and mutants were analyzed by Western blot as in Fig. 3C. Acetylated (Ac-Myc) and total Flag-Myc (Total Myc) is indicated. (C) HEK293 cells transfected with Flag-Myc wt or the indicated K→R and Δ1-110 (ΔN) mutants, and with p300 (+) or the empty vector (−) were treated with HDAC inhibitors and equal amounts of immunoprecipitated (IP: Flag) Flag-Myc proteins were analyzed by Western blot as described above. Endogenous Max coimmunoprecipitated with Flag-Myc proteins was analyzed on the same blot with the acetyl-K antibody (Ac-Max) and with Max (C-124) antibody (Total Max).
For the stability assays (see Fig. 5), HEK293 cells were transfected in a 6-cm plate at 80 to 90% confluence with 3 μg of pCbS-Flag-Myc wt or R6 mutant and 5 μg of pCMVβ-p300-CHA, pRc/RSV-CBP, or pRc/RSV-CBP(F1541/ΔHAT) or their empty vectors. After 12 h, the transfected cells from one 6-cm plate were treated with trypsin and distributed into six new plates (one for each cycloheximide time point). At 12 h after the replating, HDAC inhibitors were added to the medium, and the cells were incubated for 8 h before addition of 100 μg of cycloheximide/ml (time zero). MG-132 (20 μM) was added to the culture medium 90 min before cycloheximide, where indicated. Whole-cell extracts were prepared after the indicated times of cycloheximide treatment and Flag-Myc content was analyzed by Western blotting directly or after immunoprecipitation with the Flag antibody. The relative amount of Flag-Myc in cell extracts at different times after cycloheximide addition was determined by densitometry scanning of Western blot signals on X-ray films by using the NIH Image software. Signals obtained on the same blot with an antibody to vinculin were used for normalization. For each time course analysis a serial dilution of the extract at time zero min (100% Flag-Myc) was analyzed on the same Western blot. Standard curves were generated for each blot from the Flag-Myc signals in the serial dilution. Film exposures were selected for linearity of film response and to avoid signal saturation. The amount of Flag Myc remaining at each time point and its half-life (i.e., time at which 50% Flag-Myc remains) were determined from the standard curves and linear regression of the logarithmic values of Flag-Myc signals as a function of time. Flag-Myc half-life under each condition was determined from at least three independent experiments. The effect of p300/CBP on Flag-Myc half-life within each independent experiment (i.e., relative half-life) was obtained by dividing the half-life of Flag-Myc in the presence of p300/CBP by that in the absence of cotransfected p300/CBP. The average relative half-lives (±SD) are shown in Fig. 5F.
FIG. 5.

Regulation of Myc turnover by p300/CBP in HEK293 cells. (A) Cells were transfected with Flag-Myc wt and, as indicated, with expression vectors for p300, CBP, CBPΔHAT, or the empty vector (−). Cycloheximide (CHX) was added at time zero and whole-cell extracts were prepared at the indicated times (in minutes), and Flag-Myc (arrowhead) was detected by Western blotting with the Flag antibody. An asterisk indicates vinculin detected on the same blot with a specific antibody, as a loading reference. (B and C) Same as in panel A, but cells were incubated with HDAC inhibitors for 8 h prior to (and during) treatment with cycloheximide. In panel C, Flag-Myc wt and mutant R6 were analyzed with or without cotransfected p300. (D) As above, but Flag-Myc wt was immunoprecipitated with the Flag antibody, and approximately equal amounts of immunoprecipitated Flag-Myc were loaded on the gel (equal. load) and analyzed by Western blotting by successively probing with the Acetyl-K antibody (Ac-Myc) and then with the Flag antibody (Total Myc). (E) Same as in panel D except that MG-132 was present during cycloheximide treatment. (F) The half-lives (T1/2) of Flag-Myc wt and R6 mutant without and with cotransfected p300, CBP, and CBPΔHAT were determined in the absence (−) and presence of HDAC inhibitors and normalized to the half-life of Flag Myc wt without cotransfected HAT which was set to 1* (average Myc wt T1/2 ± the SD indicated in minutes). The relative T1/2 is the average (±the SD) from at least three independent experiments. The value in parentheses indicated by a superscript “a” is from one experiment. n.a., not analyzed.
RNA interference (RNAi), stable cell lines, and reverse transcription-PCR (RT-PCR).
The vector pSUPER.retro.puro (OligoEngine) was used for transient (see Fig. 8A) and stable (see below) expression of human c-Myc short hairpin RNAs (shRNAs) in HeLa cells. Oligonucleotides encoding shRNAs were cloned between the BglII and HindIII sites, and expression vectors were verified by DNA sequencing. The pSUPER-Myc827 vector was obtained by cloning the human c-Myc-si827 annealed oligonucleotides: 5′-GATCCCCGGTCAGAGTCTGGATCAACTTCAAGAGAGGTGATCCAGACTCTGACCTTTTTA (sense) and 5′-AGCTTAAAAAGGTCAGAGTCTGGATCACCTCTCTTGAAGTTG ATCCAGACTCTGACCGGG (antisense). The control pSUPER-GL2 vector encoding an shRNA against luciferase was obtained by inserting the annealed Luc-siGL2 oligonucleotides 5′-GATCCCCCGTACGCGGAATACTTCGATTCAAGAGATCGAAGTATTCCGCGTACGTTTTTA (sense) and 5′-AGCTTAAAAACGTACGCGGAATACTTCGATCTCTTGAATCGAAGTATTCCGCGTACG GGG (antisense). The specific targeting sequences are in boldface and underlined.
FIG. 8.

Myc is required for activation of the endogenous hTERT gene and for recruitment of p300 to the hTERT promoter in HeLa cells. (A) HeLa cells were transiently transfected with pSUPER vectors expressing shRNAs for luciferase (GL2) or human c-Myc (h-Myc) and either the mouse Myc (m-Myc) expression vector pCbS-Flag-Myc (lane 3) or its empty vector pCbS (lanes 1 and 2), and transcripts for hTERT and β-actin were analyzed by RT-PCR. Several hTERT splicing variants (α+ β+ and β−) are indicated. (B) HeLa cells were transfected as described above with vectors expressing GL2 or h-Myc shRNAs alone (lanes 1 and 2) or together with expression vectors for human Myc (h-Myc) (lanes 3 and 4) or mouse Flag-Myc (m-Myc) (lanes 5 and 6). After 72 h endogenous and ectopic human Myc (h-Myc) and ectopic mouse Myc (m-Myc) were analyzed by Western blotting with a Myc antibody and with the hTBP antibody. (C) HeLa.Myc-shRNA cells stably expressing the h-Myc shRNA (+) and HeLa.pSuper control cells (−) were analyzed by Western blotting (WB) with p300, Myc, SPT3, and TBP antibodies (upper panels) and by RT-PCR (bottom panels). The hTERT splicing variants and β-actin transcripts are shown. (D) Equivalent amounts of chromatin from HeLa.Myc-shRNA and HeLa.pSuper control cells, which express (+) or not (−) Myc shRNA were immunoprecipitated (ChIP) with either rabbit serum (C, lanes 7 and 8), or specific antibodies to Myc (lanes 9 and 10) or p300 (lanes 11 and 12). The position of the hTERT promoter PCR fragment is indicated with an arrowhead. PCRs with dilutions of input chromatin from both cell lines are shown (lanes 1 to 6). A region in the third intron of the β-globin gene was amplified as background control. Chromatin precipitated with hTBP and RNA polymerase II antibodies and PCR amplified with GAPDH promoter primers is shown in lanes 19 to 22.
For transient knockdown of human Myc, HeLa cells (at 90% confluence in 35-mm plates) were transfected with 2 μg of pSUPER-Myc827 or the control pSUPER-GL2 vector. At 24 h after the first transfection, cells were transfected again with the same amount of pSUPER-shRNA vector with Lipofectamine 2000. To verify the specificity of the knockdown, 1.5 μg of the mouse Myc expression vector (pCbS-Flag-Myc) or the human Myc expression vector (pUHD-Myc) was also included during the second transfection. Six hours after the second transfection, the medium was changed to fresh medium containing puromycin (1.5 μg/ml). After 48 to 72 h, total RNA was isolated for RT-PCR and Western blot analyses (Fig. 8A and B).
For stable knockdown of Myc, HeLa.Myc-shRNA and HeLa.pSuper control cells were generated by transfecting HeLa S3 cells in 35-mm plates with 2 μg of pSUPER-Myc827 or pSUPER.retro.puro plasmids, respectively, with Lipofectamine 2000 (Invitrogen). At 24 h after transfection, cells were transferred to 150-mm dishes in normal growth medium. After another 24 h, the medium was replaced with fresh medium containing puromycin (500 ng/ml). Single colonies were analyzed by Western blotting with Myc, p300, and TBP antibodies to select Myc-specific knockdown cells. The pool of resistant colonies was used as HeLa.pSuper-control cells. Experiments were performed with low-passage-number HeLa.Myc-shRNA cells as they eventually stop growing after 1 to 2 months in culture.
For RT-PCR, 3 μg of total RNA was isolated by using RNeasy minikit (QIAGEN) and reverse-transcribed by using the SuperScript First-Strand Synthesis System (Gibco) with oligo(dT) according to the manufacturer's instructions. Alternative splice variants of hTERT transcripts were detected by PCR with oligonucleotides TERT 2109 (5′-GCCTGAGCTGTACTTTGTCAA-3′) and TERT 2531R (5′-AGGCTGCAGAGCAGCGTGGAGAGG-3′) (36). Human β-actin was amplified with primers β-actin-forward (5′-TGACGGGGTCACCCACACTGTGCCCA-3′) and β-actin-reverse (5′-CTAGAAGCATTTGCGGTGGACGATGGAGGG-3′). PCR conditions within the exponential phase of product accumulation were chosen for each primer set and determined by serial dilution of template cDNA.
Chromatin immunoprecipitation (ChIP).
HeLa.pSuper and HeLa.Myc-shRNA cells at 90% confluence in 15-cm plates were cross-linked with 1% formaldehyde, lysed, and sonicated to obtain 0.4- to 1.0-kbp genomic DNA, essentially as previously described (8). The normalized and precleared chromatin lysates were incubated with 2 to 3 μg of anti-Myc (N-262), anti-p300 (N-15), and anti-Pol II (N-20) antibodies or 15 μl of anti-TBP or preimmune (as negative control) rabbit serum at 4°C overnight. The immune complexes were recovered with 40 μl of preblocked protein A beads. Beads were washed sequentially with 1 ml each of ChIP buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 0.06% SDS, 1% Triton X-100), ChIP-500 buffer (25 mM Tris-HCl [pH 8.0], 500 mM NaCl, 2 mM EDTA, 0.06% SDS, 1% Triton X-100), and LiCl buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate, 0.25M LiCl) and three times with TE buffer, and protein-DNA complexes were eluted twice with 1% SDS-0.1 M NaHCO3. Eluates adjusted to 0.3 M NaCl were heated at 65°C overnight, and DNA was purified with the PCR purification kit (QIAGEN). PCR was performed with hTERT promoter-specific primers CH3 (5′-AGTGGATTCGCGGGCACAGA-3′) and CH4 (5′-AGCACCTCGCGGTAGTGGCT-3′) (24). As a negative control and as a means to normalize samples to their nonspecific DNA content, the third intron region of the human β-globin gene was PCR-amplified by using the following primers: β-globin/5′ (5′-ATCTTCCTCCCACAGCTCCT-3′) and β-globin/3′ (5′-TTTGCAGCCTCACCTTCTTT-3′) (24). The GAPDH promoter fragment encompassing nucleotides −147 to +48 was amplified with the primers GAPDH/5′ (5′-GCTACTAGCGGTTTTACGGG-3′) and GAPDH/3′ (5′-CGACGCAAAAGAAGATGCA-3′). PCR products were analyzed on 2% agarose gels stained with ethidium bromide.
RESULTS
Myc interacts with p300 in vivo and in vitro through its TAD domain.
To test whether endogenous Myc-Max complexes and p300 interact in human cells, Myc and Max proteins were separately immunoprecipitated from HeLa cell lysates with specific antibodies, and immune precipitates were analyzed for the presence of p300 by Western blotting (Fig. 1A). Both the anti-Myc and the anti-Max antibodies coimmunoprecipitated p300 from HeLa cells, although the anti-Myc antibody was more efficient (lanes 3 versus 4). As a control, the anti-Flag antibody did not precipitate endogenous p300 or the Myc/Max proteins (lane 2). Consistent with an association of Myc with p300, Flag-tagged p300 and untagged Myc overexpressed in HEK293 cells by transient transfection were also both specifically coimmunoprecipitated with the anti-Flag antibody (Fig. 1B, lane 6). To further address whether p300 interacts specifically with Myc and/or Max and to test the role of the Myc TAD domain in this interaction, Flag-tagged Myc wild type (wt) and a mutant deleted of the first N-terminal 110 amino acids (Δ1-110), as well as Flag-tagged Max, were separately transfected in HEK293 cells and immunoprecipitated with the anti-Flag antibody (Fig. 1C). Although p300 coimmunoprecipitated with Flag-Myc wt (lane 11), no p300 interaction with Flag-Max could be detected (lane 14). Notably, the amount of p300 coimmunoprecipitated with Flag-MycΔ1-110 was significantly lower than with Flag-Myc wt, whereas similar amounts of Max coimmunoprecipitated in each case (lane 12 versus lane 11). This indicated an important role of Myc N-terminal TAD residues 1 to 110 in p300 binding in vivo.
To further test whether p300 directly interacts with Myc and to address the role of the N-terminal Myc TAD region, we performed GST pull-down experiments in vitro with purified recombinant p300 and GST-Myc fusion proteins containing various domains of Myc. Consistent with the above results p300 strongly interacted with Myc N-terminal TAD-containing region 1-263 (Fig. 1D, lane 3) but not with the C-terminal 92-amino-acid region comprising the bHLHZip domain (C-92, lane 11). Myc residues 2 to 108 were sufficient for efficient p300 binding (lanes 4 and 10), while the 21-108 region still bound p300 but less efficiently (lane 5) and the 47-108 and 1-48 regions did not bind p300 at all (lanes 6 and 7). Altogether, these results indicate that p300 interacts preferentially with the N-terminal Myc region 1-263 and that an N-terminal TAD domain comprising residues 1 to 110 is necessary and sufficient for efficient and direct p300 binding (Fig. 1E).
Direct and differential acetylation of the Myc-Max complex by p300, GCN5, and the GCN5-containing STAGA complex but not by Tip60 in vitro.
Since Myc interacts with p300, GCN5, and Tip60 HATs, we tested whether Myc is a direct substrate for acetylation by these HATs in vitro. Recombinant full-length p300, Tip60, GCN5-S (short form), and the native GCN5-containing STAGA complex were purified (Fig. 2A), and their HAT activities were normalized in acetylation assays in vitro with purified histones as substrates (Fig. 2B). We first tested whether a purified recombinant Myc-Max complex (Fig. 2A), which is functional in specific DNA binding (7), can be acetylated by full-length p300. As shown in Fig. 2C, p300 acetylated both Myc and Max subunits (lane 2). Notably, p300 auto-acetylation was specifically stimulated in the presence of the Myc-Max complex (compare lanes 2 and 3, top) but not in the presence of histones (lane 4) or the Max-Max homodimeric complex (data not shown). In accord with this, we have shown recently that the isolated p300-HAT domain also acetylates Myc-Max in vitro; however, autoacetylation of the HAT domain is not stimulated by Myc-Max (37). These results establish that both Myc and Max within a functional Myc-Max complex are direct targets for acetylation by p300 in vitro and suggest the intriguing possibility that the Myc-Max complex might stimulate autoacetylation of p300 and perhaps its acetyltransferase activity.
FIG. 2.

Acetylation of the Myc-Max complex by p300, GCN5, and STAGA but not Tip60 in vitro. (A) Purified recombinant proteins analyzed by SDS-PAGE and stained with Coomassie blue: p300 (1 μg), Tip60α (1 μg), GCN5-S (0.2 μg), Max (1 μg), and Myc-Max heterodimer (1 pmol). The STAGA complex (8 μl) was stained with silver, and asterisks indicate nonspecific proteins. Positions of molecular mass standards are on the left. (B) In vitro acetylation of histones with the indicated HATs and [3H]acetyl-CoA was analyzed by SDS-PAGE and fluorography (3H-Acetyl). Acetylated histones H3 and H4 are indicated. (C) In vitro acetylation of the Myc-Max complex (2 pmol, lanes 1 and 2) and histones (25 pmol, lane 4) without (−) and with (+) 4 ng of purified recombinant p300 as indicated, was analyzed by SDS-PAGE and fluorography. Lanes 1 to 3 and lane4 are from distinct regions of the same gel (spliced together) and same autoradiographic exposure (33 h). Acetylated Myc, Max, p300, and histones are indicated. (D) In vitro acetylation (as described above) with Myc-Max complex and the indicated HATs normalized for their HAT activity (see panel B). Myc-Max was omitted in lanes 4 to 6. The left panel (lanes 1′ to 3′) is the Coomassie blue-stained gel of the middle fluorographic image (lanes 1 to 3). Coomassie blue-stained BSA, Myc and Max are indicated on the left. The positions of acetylated Myc and Max are indicated on the right and with dots next to the respective bands in lanes 1 and 3. Autoacetylated p300 and Tip60 are indicated on the right (open arrowhead and arrow, respectively). Autoacetylated Tip60 is indicated with open arrowheads in lanes 2 and 5. (E) In vitro acetylation of the Myc-Max complex, as described above, but with normalized GCN5-S and STAGA. (F) In vitro acetylation of Max-Max homodimer (lanes 1 to 3), as in panel D. Max-Max was omitted in lanes 4 to 6. The top panels show an autoradiography of the portion of the Coomassie blue-stained gel shown in the bottom panels.
To determine whether the other Myc-interacting HATs can also directly acetylate the Myc-Max complex and to compare their activities to that of p300, similar HAT units of each GCN5-S, Tip60 and p300 (see Fig. 2B) were used in acetylation assays as described above. Interestingly, whereas p300 efficiently acetylated both Myc and Max (Fig. 2D, lane 3, dots), GCN5-S only acetylated Myc (lane 1), and Tip60 did not acetylate Myc or Max (lane 2), although it was efficiently autoacetylated (open arrowheads in lanes 2 and 5) and efficiently acetylated histones (Fig. 2B, lane 4). The native GCN5-L (long form)-containing STAGA complex also acetylated Myc (Fig. 2E, lane 2) but not Max (data not shown). Similarly, the homodimeric Max-Max complex was specifically acetylated by p300 (Fig. 2F, lane 3), although acetylation of Max within the Myc-Max heterodimer was more efficient (data not shown). As expected from the results presented above, neither GCN5-S, STAGA or Tip60 acetylated Max-Max homodimers (lanes 1 and 2, and data not shown). These results demonstrate that p300, GCN5, and the GCN5-containing STAGA complex can all directly acetylate Myc within a Myc-Max heterodimer, but only p300 acetylates both subunits of the Myc-Max complex and the Max homodimer. In contrast, neither Myc-Max nor Max:Max dimers are acetylation substrates for purified Tip60.
Reversible acetylation of Myc in mammalian cells by endogenous HATs and p300 requires the Myc TAD1-110 region.
To analyze Myc acetylation in mammalian cells, we used an antibody directed against acetyl-lysine residues (acetyl-K antibody hereafter) that specifically recognizes Myc acetylated by p300 in vitro (Fig. 3A, bottom panel, lanes 3 and 4) but not unacetylated Myc (lanes 1, 2, and 5) in Western blots. Endogenous cellular Myc-Max complexes were immunoprecipitated with specific antibodies to Myc and Max from HeLa cells that were transfected with a p300 expression vector and treated with proteasome and HDAC inhibitors to prevent the rapid turnover of acetylated Myc (see below). A control immunoprecipitation was performed with an antibody to yeast Gal4. Western blot analysis indicated that immunoprecipitated Myc was specifically recognized by the acetyl-K antibody (Fig. 3B, top panel, lanes 2 and 3) and, as expected, no Myc was immunoprecipitated with the Gal4 antibody (lane 1). Interestingly, the proportion of acetylated (versus total) Myc was higher in complexes immunoprecipitated with antibodies to Max (lane 3) compared to anti-Myc immunoprecipitated complexes (lane 2). This might suggest that acetylated Myc is preferentially found in association with Max or alternatively that Myc acetylation hinders its direct recognition by Myc antibodies. Acetylation of endogenous Myc in HeLa cells was further confirmed by tandem mass spectrometry analyses of complexes immunoprecipitated with Max antibodies, which identified K148 as one of the residues acetylated in human c-Myc (see Fig. S6 in the supplemental material and further below). These results demonstrate acetylation of endogenous Myc and that acetylated Myc can associate with Max in HeLa cells.
To further analyze Myc acetylation by endogenous cellular HATs and p300 in mammalian cells and to test the role of the TAD1-110 region of Myc, both Flag-tagged Myc wt and the Δ1-110 Myc deletion mutant were transiently transfected in COS-7 cells with or without a p300 expression vector. The cells were either incubated with HDAC inhibitors or mock incubated and Flag-tagged Myc proteins were immunoprecipitated and analyzed by Western blotting with the acetyl-K and Flag antibodies. As shown in Fig. 3C, in the absence of transfected p300, increased levels of Myc acetylation were observed in cells treated with HDAC inhibitors (lane 5, top panel) compared to untreated cells (lane 3). This indicates that Myc is a target for both endogenous HATs and HDACs in COS-7 cells. Significantly, Myc acetylation levels were further increased by coexpression of p300 (lane 6), although p300 alone had basically no effect (lane 4), indicating a synergistic action of HDAC inhibitors and p300 in acetylation of Myc in COS-7 cells. In stark contrast, the Myc Δ1-110 mutant protein was not detectably acetylated under the same conditions, either in the absence or presence of p300 (lanes 7 and 8). A similar synergistic action of HDAC inhibitors and p300 on acetylation of Myc wt but not Myc Δ1-110 was observed in HEK293 cells (Fig. 3D). These results demonstrate that Myc can be acetylated in mammalian cells by p300 and that Myc acetylation in COS-7 and HEK293 cells by both ectopic p300 and endogenous cellular HATs requires the TAD1-110 region and is actively counteracted by cellular HDACs.
Myc is stabilized by p300/CBP independently of acetylation and destabilized by acetylation of lysine residues located between the TAD and bHLHZip domains.
To address the role of p300 and Myc acetylation we mapped the location of p300-acetylated Myc residues in vitro and in mammalian cells (COS-7 and HEK293 cells). We have recently identified by mass spectrometry six Myc lysine residues that are direct substrates for acetylation by p300 in vitro (37), and we report here the identification by tandem mass spectrometry (MS/MS) of a novel p300-targeted site in vitro (see data in the supplemental material). Thus, p300 directly acetylates a total of seven lysine residues in vitro, which are in mouse (and human) c-Myc: K144 (143), K149 (148), K158 (157), K275, K317, K323, and K371 (mouse and human c-Myc coordinates differ only at the N terminus). We have further found that four of these residues, K149 (148), K158 (157), K317, and K323 are also acetylated in HEK293 cells upon cotransfection of Flag-Myc and p300 (see MS/MS data in the supplemental material). Interestingly, these residues are not located within the 1-110 region that is essential for p300 binding and Myc acetylation in vivo, but reside between the TAD and the HLHZip domains (Fig. 4A). Notably, the TAD1-110 region contains only two lysine residues at position 51 and 52 (K51 and K52), whereas the bHLHZip domain contains 10 conserved lysine residues (Fig. 4A).
To determine whether the p300-acetylated sites identified by mass spectrometry are also acetylated by endogenous HATs in COS-7 cells and include the major acetylation sites in vivo, we expressed Flag-tagged Myc mutant proteins containing various combinations of lysine-to-arginine substitutions targeting a total of 17 of the 27 lysine residues present in mouse Myc and analyzed their acetylation by Western blot, as described above. A Myc mutant (NRC, Fig. 4A) having each of these 17 lysine residues substituted to arginine was not acetylated (Fig. 4B, top panel, lane 9). In contrast, a Myc mutant having only K51 and K52 substituted to arginine (NR, Fig. 4A) and a mutant Myc protein (CR) having all of the 10 conserved lysine residues within the bHLHZip domain substituted to arginines were both as efficiently acetylated as the wild-type Myc protein (Fig. 4B, lanes 5 and 8 versus lane 4). Significantly, the substitution of 6 (mutant R6) or only 5 (mutant R5) lysine residues drastically reduced Myc acetylation (Fig. 4B, lanes 6 and 7). Notably, Myc acetylation was not significantly affected by substituting these lysine residues individually; however, a significant but partial reduction (ca. 60% inhibition) in acetylation was observed with a mutant having both residues K158 and K323 substituted to arginine, indicating that these two residues are major acetylation sites in vivo but also that other residues substituted in R5 and R6 mutants contribute significantly to the overall Myc acetylation level (data not shown).
To determine whether these residues comprise the major acetylation targets for p300, Flag-Myc wt and mutants were expressed in HEK293 cells in the presence or absence of p300 and the cells were treated with HDAC inhibitors. Flag-Myc proteins were immunoprecipitated and analyzed by Western blot as described above. As shown in Fig. 4C (top panel) p300 induced acetylation of Myc wt (lane 3 versus 4), as well as the Myc mutants with lysine-to-arginine substitutions in the N terminus (NR, lane 5) and C terminus (CR, lane 8). However, consistent with the above results, p300-mediated acetylation of Myc was drastically reduced (but apparently not completely eliminated) in mutants R6 and R5 and in the Δ1-110 deletion mutant (ΔN, lane 10). Consistent with the fact that Max is directly acetylated by p300 in vitro and in vivo (see above; F.Faiola and E. Martinez, submitted for publication) endogenous Max coprecipitated with Flag-Myc and was also acetylated by p300 in these assays (two bottom panels, lane 3 versus 4). Interestingly, p300-mediated Max acetylation was independent of Myc acetylation (lanes 6, 7, and 9) but required the N-terminal 1-110 region of Myc (lane 10). These results suggest that there are two main regions within Myc that are targeted for acetylation by p300 in vivo: i.e., a region immediately flanking MB2 in which lysine residues K144, K149, and K158 are acetylated and a region overlapping with a nuclear localization signal containing the acetylated residues K317 and K323 (Fig. 4A). Thus, we conclude that efficient p300-mediated acetylation of both Myc and Max within Myc-Max complexes in mammalian cells requires the Myc N-terminal (i.e., 1-110) p300-binding region.
Since acetylation often regulates protein stability we analyzed the role of p300/CBP in regulation of Myc protein turnover in vivo under both nonacetylating and acetylating conditions. To first address a possible role of p300/CBP in regulating Myc protein turnover independently of Myc acetylation, Flag-tagged Myc and p300/CBP expression vectors were cotransfected in HEK293 cells cultured in the absence of HDAC inhibitors. Under these conditions Myc is not detectably acetylated (Fig. 3). Protein synthesis was then inhibited with cycloheximide and the relative amounts of Flag-Myc were determined by semiquantitative Western blot analyses (see Materials and Methods) at different times after cycloheximide addition. As shown in the representative experiment of Fig. 5A (top 3 panels) and the relative half-lives obtained from several independent experiments (Fig. 5F), both p300 and CBP stabilized Myc by increasing its half-life by ∼3-fold. Significantly, a mutant CBPΔHAT that lacks acetyltransferase activity due to an amino acid substitution in its HAT domain (20) also stabilized Myc to a similar extent (Fig. 5A, bottom panel, and Fig. 5F). Thus, these results demonstrate that p300/CBP can stabilize Myc independently of acetylation (see also below).
To address the role of acetylation, the assays described above were repeated in HEK293 cells treated with HDAC inhibitors since this allows p300-mediated Myc acetylation (see Fig. 4C). Under these conditions and without cotransfected p300/CBP, Myc half-life was only slightly reduced from 97 to 88 min on average (Fig. 5F, Myc wt column). In stark contrast to the results presented above, in the presence of HDAC inhibitors neither p300 nor CBP stabilized Myc, and p300 overexpression led to a small but reproducible decrease in Myc half-life (Fig. 5B and C, compare two top panels, and Fig. 5F, columns wt+p300 and wt+CBP). Interestingly, the CBPΔHAT mutant increased Myc half-life more than twofold under the same conditions (Fig. 5B, bottom panel, and Fig. 5F, column wt+CBPΔHAT). Thus, a functional HAT domain interferes with the Myc stabilizing functions of p300/CBP in the presence of HDAC inhibitors. These results suggested specifically the possibility that the Myc stabilizing function of p300/CBP was counteracted by acetylation of the specific lysine residues identified above. To test this, the acetylation-defective Myc R6 mutant was analyzed similarly. In the presence (but not in the absence) of HDAC inhibitors Myc R6 had a 30% longer half-life than Myc wt (Fig. 5F, Myc R6+HDACinh.). Significantly, cotransfection of p300 further increased the half-life of Myc R6 mutant to more than the double that of Myc wt under the same conditions (Fig. 5C and Fig. 5F, +HDAC inh., compare Myc wt and wt+p300 with R6+p300). This result further supports the notion that p300-mediated Myc acetylation increases Myc protein turnover. However, since only a fraction of total Myc was acetylated under these conditions (see below), we also analyzed the turnover of acetylated Myc more directly. Flag-Myc proteins were immunoprecipitated at different times after cycloheximide addition and then analyzed by Western blotting with the acetyl-K antibody (for acetylated Myc) and with the Flag antibody (for total acetylated and unacetylated Myc). If the fraction of acetylated Myc was more stable than unacetylated Myc, a time-dependent increase in therelative amount of acetylated Myc versus total Myc should be observed after cycloheximide addition. Furthermore, since we inhibited cellular HDACs but not HATs, ongoing acetylation during cycloheximide treatment (see below) should also lead to accumulation of acetylated Myc even if acetylation had no effect on Myc stability. If, however, acetylated Myc was less stable than unacetylated Myc, a time-dependent decrease in the ratio of acetylated versus total Myc (or minimally a constant ratio) should be expected. Since the total cellular amount of Myc decreases during the cycloheximide time course, approximately equal amounts of immunoprecipitated Myc were analyzed for each time point to facilitate this comparison. As shown in Fig. 5D, the ratio of acetylated to total Myc did not increase but rather decreased from 30 to 210 min of cycloheximide treatment. Thus, these results confirm that p300-mediated acetylation does not increase Myc stability but instead further support an increased turnover of p300-acetylated Myc (see also below).
To determine whether the turnover of acetylated Myc is proteasome dependent, the same assay (as in Fig. 5D) was performed in the presence of the proteasome inhibitor MG-132. As shown in Fig. 5E, MG-132 increased the relative amount of acetylated versus total Myc during cycloheximide treatment. This specific increase in the amount of acetylated Myc, while total Myc remained constant in cells treated with MG-132 (data not shown), confirmed that only a fraction of total Myc was acetylated and that acetylation continues during cycloheximide treatment in the presence of HDAC inhibitors, a finding consistent with the fact that p300 levels remained unchanged during the time course analysis (data not shown). These results therefore indicate a proteasome-dependent turnover of acetylated Myc. The fact that Myc acetylation continues after inhibition of protein synthesis, but the fraction of acetylated Myc versus total Myc does not increase over time (Fig. 5D), strongly supports the notion that p300-mediated acetylation increases Myc turnover.
Altogether the above results suggest that p300/CBP both stabilizes Myc independently of acetylation and increases proteasome-dependent Myc turnover via acetylation of one or several of the acetylated lysine residues located between the TAD and bHLHZip domains. We propose that under acetylating conditions (e.g., cellular HDAC inhibition) the Myc stabilization effect of p300/CBP is counteracted by its acetylation-dependent Myc destabilizing function.
Synergistic transcription activation by Myc and p300/CBP from the hTERT promoter requires Myc TAD1-110 region and p300/CBP HAT activity but not p300/CBP-mediated Myc acetylation.
To determine whether p300 can function as a transcription coactivator for Myc, we analyzed activation of the natural hTERT promoter (an established Myc-regulated promoter [12]) by transient transfection of Myc and p300 in HeLa cells. All experiments described hereafter were performed in the absence of HDAC inhibitors. Under these conditions we did not detect Myc acetylation either in the presence or absence of coexpressed p300 (data not shown and see below). The activity of a chimerical hTERT promoter-luciferase reporter gene construct having both Myc E-box binding sites (E1 and E2, hTERT wt, Fig. 6A) was stimulated eight- to ninefold by coexpression of Myc and p300 (Fig. 6B, lane 1 versus lane 4). Deletion of hTERT promoter sequences (+6 to +40) downstream of the transcription start site, which include the downstream E-box (E2), increased promoter activity in the absence of transfected Myc and p300 (lane 2) and resulted in a reduced activation by Myc and p300 (lane 5), suggesting that the downstream E-box element (E2) has both activating and repressive functions, as previously proposed (12). A single nucleotide substitution in the upstream E-box (CACGTG → CACCTG), which was shown previously to strongly inhibit Myc activation of the hTERT promoter (34), reduced hTERT promoter activity by ca. 50% (lane 3 versus lane 1) and was not (or only marginally) activated by coexpression of Myc and p300 (compare lanes 6 and 3). Thus, both E box elements are required for maximal activation of the hTERT promoter by the combined action of Myc and p300 (see also below).
We further analyzed the mechanisms of hTERT promoter activation by Myc and p300 and also addressed the role of CBP. Interestingly, whereas the combined action of Myc and p300 led to an eight- to ninefold activation of the hTERT wt promoter, Myc and p300 separately only activated about threefold and less than twofold, respectively (Fig. 6C, lanes 2 to 4). Similarly, cotransfection of Myc and CBP activated the hTERT promoter nine- to tenfold (Fig. 6D, lane 5), whereas each separately activated only about threefold (Fig. 6D, lanes 2 and 4). The synergistic activation by Myc and p300/CBP was abolished by deletion of the Myc TAD region 1-110 (Fig. 6C, lane 6, and Fig. 6D, lane 8). The failure of the Δ1-110 Myc mutant to synergize with p300/CBP did not result from a complete loss of transactivating potential of this truncated Myc protein since under these conditions the Δ1-110 Myc mutant still activated the hTERT promoter about two- to threefold (Fig. 6C, lane 5, and Fig. 6D, lane 7). Consistent with the fact that both E-box elements were important for Myc-dependent activation, the synergistic activation by Myc and p300/CBP was also abolished by mutation of the upstream E-box element (Fig. 6B, lane 6) or by deletion of the downstream E-box sequence (Fig. 6C, lane 10, and Fig. 6D, lane 13). Thus, the Myc1-110 region and both E-box elements are important for synergistic activation of the hTERT promoter by Myc and p300/CBP.
To address the role of p300/CBP HAT activity in this synergism, we analyzed Myc-dependent activation of the hTERT promoter in the presence of the CBPΔHAT mutant (20). As shown in Fig. 6D, CBPΔHAT did not activate hTERT transcription by itself or in combination with cotransfected Myc (lanes 3 and 6, respectively). Western blot analyses further indicated that, under these conditions, Myc protein levels (both Flag-Myc wt and Δ1-110) were not significantly affected by cotransfection of p300/CBP (Fig. 6F) and Myc was not detectably acetylated (data not shown). These results suggest that stimulation of Myc transactivation by p300/CBP was not simply the result of increased Myc protein levels or Myc acetylation. Consistent with this, the acetylation-defective Myc R6 mutant was as active as wt Myc (Fig. 6E, lane 5), and its transactivating function was also stimulated by p300 (lane 6).
Altogether these results demonstrate a synergistic activation of the hTERT promoter by Myc and p300/CBP, which requires both E-box elements, p300/CBP HAT activity and the Myc TAD1-110 region and strongly suggest that activation of Myc-dependent transcription by p300/CBP is not simply mediated through Myc acetylation and/or indirectly through alterations in Myc protein levels.
Myc TAD1-103 region is sufficient for synergistic transcription activation with p300/CBP.
To further determine whether the synergistic transactivation by Myc and p300/CBP is mediated through interactions with the N-terminal Myc TAD and does not require the rest of the Myc protein, Max, or other gene-specific promoter-binding transcription factors, transient transfections were performed in HeLa cells with a luciferase reporter gene under the control of an artificial promoter composed of five Gal4 binding sites upstream of the adenovirus E4 TATA box and expression vectors for p300/CBP and Gal4-Myc fusion proteins containing different regions of Myc TAD fused the Gal4 DNA-binding domain. As shown in Fig. 7A, the Gal4 reporter gene was activated four- to fivefold by p300 in the presence a Gal4-Myc1-262 fusion protein containing the N-terminal 262 amino acids of Myc (Fig. 7A, lane 4) but not in the presence of the Gal4 DNA-binding domain alone (lane 2). Significantly, a minimal Myc TAD1-103 region was sufficient to confer a three- to fourfold induction by p300, whereas the Myc TAD region 41-143, which lacks residues 1 to 40 that are important for interaction with p300 (see Fig. 1D), conferred less than twofold activation by p300 (lane 8). The fact that the MB2-containing Myc 41-143 region did not significantly synergize with p300 was not due to a lack of transactivating potential of this domain as Gal4-Myc41-143 activated this reporter better than Gal4-Myc1-262 and Gal4-Myc1-103, as reported previously (16; data not shown).
Similarly, CBP also synergized (6-7-fold) with Gal4-Myc1-262 and Gal4-Myc1-103 (Fig. 7B, lanes 4 and 7) but much less efficiently (two- to threefold) with Gal4-Myc41-143 (lane 10). Furthermore, the CBPΔHAT mutant did not stimulate Myc TAD-dependent transcription (lanes 5 and 8), suggesting that acetylation of histones or other targets distinct from Myc and Max is important for the synergistic action of Myc TAD and p300/CBP. These results demonstrate a functional synergy between p300/CBP and an isolated p300-interacting Myc TAD region that requires the HAT activity of p300/CBP but not Myc acetylation and is independent of other gene-specific activators.
Myc is essential for p300 recruitment and transcription activation of the endogenous hTERT promoter in HeLa cells.
To address the role of endogenous Myc in p300 recruitment and activation of the natural hTERT gene in HeLa cells, we used RNAi to inhibit Myc expression. Expression vectors for shRNAs directed against human Myc (h-Myc) and against luciferase (GL2, as control) were transiently transfected in HeLa cells. Expression of the h-Myc shRNA but not the control GL2 shRNA decreased the mRNA levels for several established splicing variants of hTERT (36) as determined by RT-PCR (Fig. 8A, lane 1 versus lane 2). To confirm that the reduction in hTERT mRNA levels is due specifically to the knockdown of Myc, we overexpressed mouse Myc (m-Myc), which in contrast to the endogenous and overexpressed human Myc cannot be silenced by the h-Myc shRNA sequence used here (see Fig. 8B). Mouse Myc restored hTERT mRNA levels in cells expressing the h-Myc shRNA (Fig. 8A, compare lanes 2 and 3). These results establish an essential role of endogenous Myc in activation of the hTERT gene in HeLa cells.
To address whether Myc and p300 are recruited to the endogenous hTERT promoter and whether Myc is required for p300 recruitment, we generated HeLa S3 cells that stably express the h-Myc shRNA described above. As shown in Fig. 8C, cells stably transfected with the h-Myc shRNA expression vector had a reduced expression of endogenous Myc compared to cells stably transfected with the control vector, whereas p300 protein levels were not affected (lane 1 versus lane 2, top panels [WB]), and this correlated with reduced levels of hTERT transcripts (lane 2, bottom RT-PCR panels). We then analyzed the recruitment of Myc and p300 to the endogenous hTERT promoter in these cells by ChIP assay. As shown in Fig. 8D, control cells had both Myc and p300 bound to the endogenous hTERT promoter (lanes 9 and 11). In contrast, the interaction of both Myc and p300 with the hTERT promoter was inhibited in Myc shRNA-expressing cells (lanes 10 and 12). The association of Myc and p300 with the hTERT promoter was specific since a region of the β-globin gene was not immunoprecipitated with Myc and p300 antibodies (lanes 9 to 12, bottom panel). In addition, the inhibition of Myc and p300 binding in Myc shRNA-expressing cells was not due to a generalized nonspecific inhibition of gene expression, since the β-actin and GAPDH genes were not inhibited (Fig. 8C, bottom panel, and data not shown) and the binding of TBP and Pol II to the GAPDH promoter was not affected (Fig. 8D, lanes 19 to 22). These results establish an essential role of endogenous Myc in p300 recruitment and transcription activation of the hTERT gene in HeLa cervical cancer cells, which is consistent with the synergistic activation of the hTERT promoter by Myc and p300 presented above.
DISCUSSION
In this study we have uncovered a complex regulation of Myc by p300 involving p300 interaction with Myc N-terminal TAD1-110 region, a region that is essential for Myc-dependent oncogenic transformation. We provide evidence that p300 controls Myc protein turnover via acetylation-dependent and -independent mechanisms and functions as a Myc coactivator that stimulates Myc-dependent activation of the hTERT promoter in human cancer cells.
We have shown that Myc (but not Max) recruits p300 in vivo and in vitro through direct physical interactions with its N-terminal TAD1-110 region. Interestingly, the fact that Max does not interact directly with p300 but that Myc TAD is essential for efficient acetylation of both Myc and Max within Myc:Max heterodimers in mammalian cells suggests that Myc TAD might stimulate acetylation by functioning as a docking site that tethers p300 in close proximity to its substrates. In contrast, GCN5 (or STAGA) only acetylates Myc and Tip60 does not acetylate either Myc or Max in vitro. Thus, our analyses demonstrate important differences and specificities in the acetylation of the Myc-Max and Max-Max complexes by p300 and by the other Myc-interacting HATs and suggest that Tip60-induced Myc acetylation in transfected cells (26) is either indirect or requires additional Tip60-associated proteins.
We have identified 5 Myc lysine residues (K144, K149, K158, K317, and K323) located between the TAD and bHLHZip domains that are direct targets for acetylation by p300 in vitro and in mammalian cells. The fact that individual and combinations of two lysine substitutions do not abolish Myc acetylation in vivo (data not shown) suggests that most (if not all) of theses five lysine residues contribute to the net acetylation of Myc by p300 in cultured cells. Although it is possible that additional Myc lysine residues might be acetylated under specific conditions (and/or by other HATs [see below]) in vivo, their contribution to the overall acetylation level of Myc under the conditions used here is clearly negligible.
Interestingly, we show that p300 targets Myc residue K149, which was previously found acetylated in vivo by an unknown HAT different from GCN5 (26). Thus, K149 appears to be a specific substrate for p300. Notably, one of the lysine residues acetylated by p300 (K323) is also acetylated by GCN5 in transfected cells and GCN5 in addition acetylates K417, a residue that is not phylogenetically conserved (26) and does not appear from our results to be a major acetylation site for p300. Although the complete set of Myc lysine residues acetylated by GCN5 (and Tip60) remains to be determined, it is clear from these combined observations and from the fact that only p300 acetylates Max that GCN5 and p300 have overlapping but also distinct acetylation target specificities within the Myc-Max complex. This suggests the possibility that acetylation by p300/CBP and GCN5 might play distinct roles in the regulation of Myc functions.
Our results indicate a complex regulation of Myc protein turnover by p300/CBP involving both stabilizing and destabilizing functions. Myc is stabilized by p300/CBP under nonacetylating conditions, including in the absence of a functional HAT domain and of Myc residues that are substrates for p300-mediated acetylation. This is partly consistent with a previous report that indicated Myc stabilization and reduced Myc ubiquitination by overexpressed CBP in HEK293 cells (cultured apparently in the absence of HDAC inhibitors). That study concluded that Myc stabilization results from its acetylation by CBP (31). However, as shown here, Myc is not detectably acetylated in HEK293 cells in the absence of HDAC inhibitors (Fig. 3D). Moreover, we have found no evidence for a Myc-stabilizing role of p300/CBP involving acetylation but rather our data indicate that p300-acetylated Myc has a faster turnover than the bulk of unacetylated Myc and is disposed by a proteasome-dependent mechanism in HEK293 cells. Thus, our results strongly suggest that Myc is stabilized by p300/CBP in HEK293 cells independently of its acetylation and destabilized by p300/CBP-mediated acetylation at one or several of the lysine residues identified here. In contrast, GCN5 has been reported to stabilize Myc in H1299 lung cancer cells in a manner that is dependent on GCN5-acetylated lysines 323 and/or 417 (26). Thus, Myc acetylation by p300/CBP and GCN5 appears to have opposite effects on Myc stability. It remains, however, to be shown whether this difference is cell type or growth condition dependent or whether it reflects a more general difference in Myc regulation by these two types of HATs.
The mechanisms involved in regulation of Myc protein turnover by either GCN5 or p300/CBP remain to be determined. While acetylation of specific Myc lysine residues by GCN5 could prevent their ubiquitination and ubiquitin-mediated proteasomal degradation (26), the interaction of p300/CBP with Myc could potentially interfere with the recruitment of specific Myc ubiquitin ligases and/or could mask potential ubiquitination sites, resulting in acetylation-independent Myc stabilization. This would be consistent with the direct interaction of Myc TAD with p300 and the role of Myc TAD in recruitment of ubiquitin ligases and in regulation of Myc turnover (reviewed in reference 1). Although much less frequently observed, acetylation of lysine residues has also been associated with increased recruitment of ubiquitin ligases and ubiquitin-dependent proteasomal degradation of proteins (reviewed in reference 35). Such a mechanism could potentially explain the increased turnover of p300-acetylated Myc.
In addition to its role in acetylating Myc and regulating its turnover, we provide evidence that p300/CBP also functions as a coactivator for Myc via its interaction with the N-terminal Myc TAD domain. We demonstrate that endogenous Myc in HeLa cells is essential for transcription of the chromosomal hTERT gene and for recruitment of p300 to the hTERT promoter. We show that p300/CBP stimulates Myc-dependent transcription from the hTERT promoter and that this requires both E-box elements, the Myc TAD1-110 region, and p300/CBP HAT activity. Although these results do not exclude additional functions of p300/CBP through the C terminus of Myc at certain promoters as suggested previously (31), our data strongly suggest that Myc directly recruits p300/CBP to promoters in vivo via interactions with its TAD domain. Indeed, p300 directly binds to Myc TAD in vitro and p300/CBP also stimulates activation by Gal4-MycTAD fusion proteins. The p300/CBP HAT activity-dependent stimulation of Myc transactivation described here is not merely the result of increased Myc stability since a functional p300/CBP HAT domain is not required for Myc stabilization and most likely involves acetylation of targets other than Myc since p300 can stimulate transcription activation of the hTERT promoter by an acetylation-defective Myc mutant. The mechanism underlying the synergy between Myc and p300/CBP remains, however, to be elucidated. The fact that synergistic transactivation by Myc and p300/CBP from the hTERT promoter requires the downstream E-box sequence, although an hTERT promoter containing only the upstream E-box can support a significant level of activation by Myc and p300/CBP separately, suggests a possible role of p300/CBP HAT activity in antagonizing a repressor, perhaps including nucleosomal histones, which might interfere with Myc binding or function at the downstream E-box element. To our knowledge, these results provide the first evidence for a role of p300/CBP in Myc-dependent transcription activation of the hTERT promoter in cancer cells.
In conclusion, the analyses reported here have uncovered a dual role of p300/CBP in Myc regulation: a “positive” role as a coactivator-HAT that stabilizes Myc and mediates Myc transactivating functions on natural target genes such as hTERT and a “negative” role as an inducer of Myc instability via direct Myc acetylation. These apparently antagonistic activities of p300/CBP in Myc regulation are reminiscent of the roles of p300/CBP in cell proliferation and as a tumor suppressor and inducer of cell differentiation (4, 11). This “double-edged sword” activity of p300/CBP in Myc regulation trough the TAD domain appears also strikingly similar to the role of Skp2 in ubiquitin/proteasome-mediated degradation of Myc and in stimulation of Myc transactivation (17, 32; for a review, see reference 1) and suggests a potential cross talk between these two Myc regulatory pathways that warrants further investigation.
Supplementary Material
Acknowledgments
We thank J. Carl Barrett, M. D. Cole, R. A. Currie, C. V. Dang, M. Eilers, R. Goodman, W. C. Greene, X. Liu, Y. Nakatani, R. G. Roeder, and H. T. M. Timmers for generous gifts or reagents.
This study was supported by a NIH grant CA100464 from the National Cancer Institute and by a grant from the UCR Genomics Institute Core Instrumentation Facility.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Amati, B. 2004. Myc degradation: dancing with ubiquitin ligases. Proc. Natl. Acad. Sci. USA 101:8843-8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouchard, C., O. Dittrich, A. Kiermaier, K. Dohmann, A. Menkel, M. Eilers, and B. Lüscher. 2001. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 15:2042-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brand, M., K. Yamamoto, A. Staub, and L. Tora. 1999. Identification of TATA-binding protein-free TAFII-containing complex subunits suggests a role in nucleosome acetylation and signal transduction. J. Biol. Chem. 274:18285-18289. [DOI] [PubMed] [Google Scholar]
- 4.Chan, H. M., and N. B. La Thangue. 2001. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 114:2363-2373. [DOI] [PubMed] [Google Scholar]
- 5.Chen, L. F., Y. Mu, and W. C. Greene. 2002. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J. 21:6539-6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Currie, R. A. 1998. NF-Y is associated with the histone acetyltransferases GCN5 and P/CAF. J. Biol. Chem. 273:1430-1434. [DOI] [PubMed] [Google Scholar]
- 7.Farina, A., F. Faiola, and E. Martinez. 2004. Reconstitution of an E box-binding Myc:Max complex with recombinant full-length proteins expressed in Escherichia coli. Protein Expr. Purif. 34:215-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank, S. R., M. Schroeder, P. Fernandez, S. Taubert, and B. Amati. 2001. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 15:2069-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frank, S. R., T. Parisi, S. Taubert, P. Fernandez, M. Fuchs, H. M. Chan, D. M. Livingston, and B. Amati. 2003. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 4:575-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grandori, C., S. M. Cowley, L. P. James, and R. N. Eisenman. 2000. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 16:653-699. [DOI] [PubMed] [Google Scholar]
- 11.Grossman, S. R., M. E. Deato, C. Brignone, H. M. Chan, A. L. Kung, H. Tagami, Y. Nakatani, and D. M. Livingston. 2003. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 300:342-344. [DOI] [PubMed] [Google Scholar]
- 12.Horikawa, I., and J. C. Barrett. 2003. Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis 24:1167-1176. [DOI] [PubMed] [Google Scholar]
- 13.Horikawa, I., P. L. Cable, C. Afshari, and J. C. Barrett. 1999. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res. 59:826-830. [PubMed] [Google Scholar]
- 14.Ikura, T., V. V. Ogryzko, M. Grigoriev, R. Groisman, J. Wang, M. Horikoshi, R. Scully, J. Qin, and Y. Nakatani. 2000. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102:463-473. [DOI] [PubMed] [Google Scholar]
- 15.Ito, T., M. E. Levenstein, D. V. Fyodorov, A. K. Kutach, R. Kobayashi, and J. T. Kadonaga. 1999. ACF consists of two subunits, Acf1 and ISWI, that function cooperatively in the ATP-dependent catalysis of chromatin assembly. Genes Dev. 13:1529-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato, G. J., J. Barrett, M. Villa-Garcia, and C. V. Dang. 1990. An amino-terminal c-myc domain required for neoplastic transformation activates transcription. Mol. Cell. Biol. 10:5914-5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim, S. Y., A. Herbst, K. A. Tworkowski, S. E. Salghetti, and W. P. Tansey. 2003. Skp2 regulates Myc protein stability and activity. Mol. Cell 11:1177-1188. [DOI] [PubMed] [Google Scholar]
- 18.Kwok, R. P., J. R. Lundblad, J. C. Chrivia, J. P. Richards, H. P. Bachinger, R. G. Brennan, S. G. Roberts, M. R. Green, and R. H. Goodman. 1994. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature 370:223-226. [DOI] [PubMed] [Google Scholar]
- 19.Liu, X., J. Tesfai, Y. A. Evrard, S. Y. Dent, and E. Martinez. 2003. c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. J. Biol. Chem. 278:20405-20412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Balbas, M. A., A. J. Bannister, K. Martin, P. Haus-Seuffert, M. Meisterernst, and T. Kouzarides. 1998. The acetyltransferase activity of CBP stimulates transcription. EMBO J. 17:2886-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez, E., T. K. Kundu, J. Fu, and R. G. Roeder. 1998. A human SPT3-TAFII31-GCN5-L acetylase complex distinct from transcription factor IID. J. Biol. Chem. 273:23781-23785. [DOI] [PubMed] [Google Scholar]
- 22.Martinez, E., V. P. Palhan, A. Tjernberg, E. S. Lymar, A. M. Gamper, T. K. Kundu, B. T. Chait, and R. G. Roeder. 2001. Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo. Mol. Cell. Biol. 21:6782-6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMahon, S. B., H. A. Van Buskirk, K. A. Dugan, T. D. Copeland, and M. D. Cole. 1998. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell 94:363-374. [DOI] [PubMed] [Google Scholar]
- 24.Nikiforov, M. A., S. Chandriani, J. Park, I. Kotenko, D. Matheos, A. Johnsson, S. B. McMahon, and M. D. Cole. 2002. TRRAP-dependent and TRRAP-independent transcriptional activation by Myc family oncoproteins. Mol. Cell. Biol. 22:5054-5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogryzko, V. V., T. Kotani, X. Zhang, R. L. Schiltz, T. Howard, X. J. Yang, B. H. Howard, J. Qin, and Y. Nakatani. 1998. Histone-like TAFs within the PCAF histone acetylase complex. Cell 94:35-44. [DOI] [PubMed] [Google Scholar]
- 26.Patel, J. H., Y. Du, P. G. Ard, C. Phillips, B. Carella, C. J. Chen, C. Rakowski, C. Chatterjee, P. M. Lieberman, W. S. Lane, G. A. Blobel, and S. B. McMahon. 2004. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 24:10826-10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Philipp, A., A. Schneider, I. Vasrik, K. Finke, Y. Xiong, D. Beach, K. Alitalo, and M. Eilers. 1994. Repression of cyclin D1: a novel function of MYC. Mol. Cell. Biol. 14:4032-4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ran, Q., and O. M. Pereira-Smith. 2000. Identification of an alternatively spliced form of the Tat interactive protein (Tip60), Tip60β. Gene 258:141-146. [DOI] [PubMed] [Google Scholar]
- 29.Roth, S. Y., J. M. Denu, and C. D. Allis. 2001. Histone acetyltransferases. Annu. Rev. Biochem. 70:81-120. [DOI] [PubMed] [Google Scholar]
- 30.Sakamuro, D., and G. C. Prendergast. 1999. New Myc-interacting proteins: a second Myc network emerges. Oncogene 18:2942-2954. [DOI] [PubMed] [Google Scholar]
- 31.Vervoorts, J., J. M. Luscher-Firzlaff, S. Rottmann, R. Lilischkis, G. Walsemann, K. Dohmann, M. Austen, and B. Lüscher. 2003. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 4:484-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von der Lehr, N., S. Johansson, S. Wu, F. Bahram, A. Castell, C. Cetinkaya, P. Hydbring, I. Weidung, K. Nakayama, K. I. Nakayama, O. Soderberg, T. K. Kerppola, and L. G. Larsson. 2003. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol. Cell 11:1189-1200. [DOI] [PubMed] [Google Scholar]
- 33.Walhout, A. J., J. M. Gubbels, R. Bernards, P. C. van der Vliet, and H. T. M. Timmers. 1997. c-Myc/Max heterodimers bind cooperatively to the E-box sequences located in the first intron of the rat ornithine decarboxylase (ODC) gene. Nucleic Acids Res. 25:1493-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu, K. J., C. Grandori, M. Amacker, N. Simon-Vermot, A. Polack, J. Lingner, and R. Dalla-Favera. 1999. Direct activation of TERT transcription by c-MYC. Nat. Genet. 21:220-224. [DOI] [PubMed] [Google Scholar]
- 35.Yang, X. J. 2004. Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays 26:1076-1087. [DOI] [PubMed] [Google Scholar]
- 36.Yi, X., J. W. Shay, and W. E. Wright. 2001. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res. 29:4818-4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang, K., F. Faiola, and E. Martinez. 2005. Six lysine residues on c-Myc are direct substrates for acetylation by p300. Biochem. Biophys. Res. Commun. 336:274-280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.