

Abstract
Huntington's disease and several other neurological diseases are caused by expanded polyglutamine [poly(Gln)] tracts in different proteins. Mechanisms for expanded (>36 Gln residues) poly(Gln) toxicity include the formation of aggregates that recruit and sequester essential cellular proteins [Preisinger, E., Jordan, B. M., Kazantsev, A. & Housman, D. (1999) Phil. Trans. R. Soc. London B 354, 1029–1034; Chen, S., Berthelier, V., Yang, W. & Wetzel, R. (2001) J. Mol. Biol. 311, 173–182] and functional alterations, such as improper interactions with other proteins [Cummings, C. J. & Zoghbi, H. Y. (2000) Hum. Mol. Genet. 9, 909–916]. Expansion above the “pathologic threshold” (≈36 Gln) has been proposed to induce a conformational transition in poly(Gln) tracts, which has been suggested as a target for therapeutic intervention. Here we show that structural analyses of soluble huntingtin exon 1 fusion proteins with 16 to 46 glutamine residues reveal extended structures with random coil characteristics and no evidence for a global conformational change above 36 glutamines. An antibody (MW1) Fab fragment, which recognizes full-length huntingtin in mouse brain sections, binds specifically to exon 1 constructs containing normal and expanded poly(Gln) tracts, with affinity and stoichiometry that increase with poly(Gln) length. These data support a “linear lattice” model for poly(Gln), in which expanded poly(Gln) tracts have an increased number of ligand-binding sites as compared with normal poly(Gln). The linear lattice model provides a rationale for pathogenicity of expanded poly(Gln) tracts and a structural framework for drug design.
Experimental studies of polyglutamine [poly(Gln)] have historically been hampered by insolubility, and molecular modeling, biophysical characterization of solubilized peptides, and antibody-binding studies have provided conflicting results as to its structure. Theoretical work suggested either random coil or more ordered structures (e.g., a β-hairpin) (reviewed in ref. 1). Circular dichroism (CD) studies of normal poly(Gln) peptides (Q9 or Q17 flanked by α-helical peptides) (2) revealed random-coil structures, as did normal and expanded poly(Gln) peptides (K2QnK2; n = 5–49) in aqueous solution after a disaggregation procedure involving nonnative solvents (3). A recent NMR study of normal and expanded poly(Gln) fused to glutathione S-transferase also found random-coil structures (4). Several monoclonal antibodies preferentially bind expanded poly(Gln) on Western blots (1C2, refs. 5 and 6; 1F8, refs. 7 and 8; 3B5H10¶; and MW1–6, ref. 9) or in a biosensor-based assay (1C2, ref. 10), and these results were interpreted to indicate that expanded poly(Gln) adopts a pathologic conformation different from the conformation of normal poly(Gln), and that the antibodies specifically recognize this alternative conformation (8–10, ¶).
Here, we report the structural characterization of the N-terminal poly(Gln)-containing exon 1 domain of the Huntington's disease (HD) protein, huntingtin. N-terminal fragments of huntingtin are implicated in disease pathogenesis (11), and HD exon 1 is sufficient to cause a rapidly progressing HD-like phenotype in transgenic mice when it contains a large poly(Gln) tract (12). We have expressed and purified thioredoxin (TRX) fusion proteins with HD exon 1 containing 16, 25, 39, or 46 glutamines and have characterized these constructs by using a variety of biochemical and biophysical techniques, including antibody-binding studies.
Materials and Methods
Protein Preparation.
Huntingtin exon 1 was expressed as a fusion protein with Escherichia coli TRX. Inserts containing HD exon 1 with a C-terminal His6 tag produced by restriction enzyme digestions or PCR amplification (requiring the addition of 10% dimethyl sulfoxide or 10% glycerol) were subcloned into the NcoI and BamHI sites of a pET-32a vector (Novagen) that was modified to remove the N-terminal His6 tag. Constructs contained the TRX gene, a linker segment (GSGSGERQHMDSPDLGTDDDDK), the HD exon 1 insert, and a His6 tag (Fig. 1a). Constructs contain CAG or CAA/CAG repeats (13), the stabilities of which were verified by matrix-assisted laser desorption ionization (MALDI) mass spectrometry of purified proteins and postinduction DNA sequence analysis. TRX-tag is a control protein that contains the TRX gene, linker, and His6 tag. Proteins were overexpressed in BL21(DE3) cells (Novagen) and released by osmotic shock by using a modification of a published method (14). Briefly, cells were spun at 6,000 × g for 10 min at 4°C, and pellets were resuspended in 15 mM Tris⋅HCl, pH 8.0, and incubated 45 min with shaking at 4°C. Osmotically shocked cells were spun at 15,000 × g for 10 min at 4°C, and the supernatant containing the protein of interest was incubated with Ni nitrilotriacetate beads (Qiagen). Proteins were eluted in 250 mM imidazole, purified by gel filtration FPLC (Superdex-75; Amersham Pharmacia Biotech), and concentrated with an Amicon stirred ultrafiltration cell (Millipore). Proteins were stable for weeks at 4°C in 50 mM Tris⋅HCl, pH 8.0/150 mM NaCl/1 mM PMSF/1 mM EDTA.
Figure 1.

Expression and purification of HD exon 1 constructs. (a) HD exon 1 and TRX-tag constructs are represented schematically. (b) SDS/PAGE analysis of purified proteins (0.1 nmol; ≈2.5 μg of each protein) under reducing conditions.
Antibody Reagents.
MW1 (IgG2b/κ) (9) was purified from ascites fluid by protein G affinity chromatography. Western blots were probed with 1C2 ascites fluid (Chemicon), anti-His6 antibody (Qiagen) (data not shown), or purified MW1, and detected with horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibody. MW1 Fab was prepared by papain cleavage (15) with a ratio of 1:3000 (papain:MW1 by weight) for 30 min at 35°C, purified by using protein A affinity chromatography and gel filtration chromatography (Superdex-75; Amersham Pharmacia Biotech), and concentrated in 100 mM sodium acetate, pH 5.5/150 mM NaCl.
Analytical Ultracentrifugation.
Protein concentrations were determined spectrophotometrically at 280 nm by using the following extinction coefficients: HD-16Q, HD-25Q, HD-39Q, HD-46Q, and TRX-tag, 13,940 M−1⋅cm−1 [calculated (16) from the amino acid sequences of HD exon 1 fusion proteins or TRX-tag]; MW1 Fab, 76,620 M−1⋅cm−1 [mean value calculated (16) from IgG2b/κ Fab sequences in the Protein Data Bank: PDB codes 1IBG, 1FAI, and 1CGS] and verified by comparing side-by-side dilutions of HD-25Q or MW1 Fab in 6 M guanidine⋅HCl and aqueous solution. Proteins were dialyzed into 50 mM Tris⋅HCl, pH 8.0/150 mM NaCl and analyzed using a Beckman–Coulter XL-I Ultima analytical ultracentrifuge with absorption optics. Sedimentation equilibrium experiments were conducted at 16,000, 25,000, and 30,000 rpm at 25°C. Data files were processed and analyzed as described (17). Sedimentation velocity experiments were performed at 36,000, 42,000, or 48,000 rpm (at 20° or 25°C), and the data were analyzed with svedberg (18). Each sample was spun at three concentrations and the apparent sedimentation coefficients were corrected for dilution and buffer effects to give s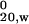 .
.
CD.
Samples were dialyzed into 10 mM sodium phosphate, pH 7.0, and filtered (0.2 μm). In this buffer, proteins with expanded poly(Gln) were not aggregated as assessed by gel filtration FPLC chromatography or analytical ultracentrifugation. CD data were collected from samples (6–14 μM) at 25°C by using an Aviv 62A DS spectropolarimeter (Aviv Instruments, Lakewood, NJ) (1-mm path-length cuvette). Three wavelength scans (250 to 190 nm) were collected for each HD exon 1 fusion protein and TRX-tag with a 1-sec averaging time per data point. Data were processed with KaleidaGraph (Abelbeck Software, Reading, PA). The CD spectrum of TRX-tag is very similar to that of E. coli TRX (Promega).
NMR Spectroscopy.
15N-labeled HD-46Q or TRX-tag was expressed in minimal M9 medium prepared with 15N-labeled ammonium chloride (Aldrich) and purified. Samples were concentrated to 100 μM in 50 mM sodium phosphate, pH 7.0/50 mM NaCl/5% D2O. Under these conditions, HD-46Q was not aggregated as shown by the elution of the post-NMR sample at the same time point as freshly prepared HD-46Q in gel filtration FPLC. Shigemi tubes were used to minimize the amount of sample required. One-dimensional 1H and two-dimensional gradient-enhanced 1H,15N heteronuclear single quantum coherence (HSQC) NMR spectra were acquired at 4°C on a Varian INOVA 600-MHz spectrometer equipped with a three-axis gradient HCN probe. A 2-sec relaxation delay and 8,610-Hz proton spectral width were used in all experiments. The one-dimensional spectra were acquired with 5,024 complex points and 2,048 transients; the solvent signal was attenuated by presaturation. The two-dimensional spectra were acquired with 128 complex increments, 2,500-Hz nitrogen spectral width and 64 (TRX-tag) or 256 (HD-46Q) transients. Proton chemical shifts were referenced relative to TSP [3-(trimethylsilyl)propionic-2,2,3,3-d4 acid, sodium salt] by means of the residual solvent resonance at 4.9846 ppm; nitrogen chemical shifts were indirectly referenced to the proton resonance frequency.
Biosensor Binding Studies.
A BIAcore 2000 biosensor system (Pharmacia LKB Biotechnology) was used to assay interactions between HD exon 1 fusion proteins and MW1 Fab. Binding between a molecule coupled to a biosensor chip (ligand) and a second molecule injected over the chip (analyte) results in surface plasmon resonance (SPR) changes that are read out in real time as resonance units (RU). MW1 Fab (0.02 mg/ml) in 5 mM sodium acetate, pH 4.6, was immobilized by standard amine coupling chemistry on a CM5 chip (Pharmacia LKB Biotechnology) at low (380 RU), medium (970 RU), or high (2080 RU) density. HD exon 1 fusion proteins or TRX-tag was injected at 100 μl/min at 25°C in 50 mM Tris⋅HCl, pH 8.0/150 mM NaCl/0.005% BIAcore surfactant P20. Each injection was preceded by an identical injection over a mock-coupled flow-cell to subtract out nonspecific responses. HD exon 1 fusion proteins do not bind an IgG2b/κ isotype control antibody (PharMingen) (data not shown). Biosensor response curves were processed and the equilibrium binding responses (Req values) were obtained with Scrubber (developed by T. Morton and D. G. Myzska; www.cores.utah.edu/interaction). Dissociation constants (KDs) were determined by linear regression analysis of plots of Req versus the log of the concentration of injected protein. For each HD exon 1 fusion protein, data for three coupling densities were globally fit in Scientist (MicroMath Scientific Software, Salt Lake City) with monovalent, bivalent (17, 19), or trivalent analyte (see analysis in supporting information on the PNAS web site, www.pnas.org) models and the best-fit curve was used to derive KD1, KD2, and KD3.
Results
Solution Structure of HD Exon 1 with Normal or Expanded Poly(Gln).
We expressed TRX fusion proteins containing HD exon 1 with 16, 25, 39, or 46 glutamines (Fig. 1a). The C-terminally tagged constructs reported here yield exclusively full-length products that are purified under native conditions in milligram quantities (Fig. 1b), in contrast to previous HD exon 1 fusion proteins with N-terminal affinity tags produced in our laboratory and by others (20), which copurify with truncated products. Thus, we have a system for characterizing poly(Gln) in solution within the context of a naturally occurring sequence. In this system, HD exon 1 is soluble and remains monomeric with up to 46 Gln at micromolar concentrations as assessed by analytical ultracentrifugation (Table 1). In contrast to TRX-tag alone, the exon 1 fusion proteins with normal or expanded poly(Gln) are nonglobular by several measures, including anomalous migration in size exclusion chromatography and high frictional ratios (f/f0) in sedimentation velocity ultracentrifugation (Table 1).
Table 1.
Masses and sedimentation coefficients of HD exon 1, TRX-tag, and MW1 Fab
| Protein | Mass, kDa
|
s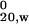 ,‡ S ,‡ S |
f/f0§ | |||
|---|---|---|---|---|---|---|
| Calculated* | MS† | FPLC† | AUC† | |||
| HD-25Q | 25.2 | 25.2 | 56.4 | 26.2 | 1.58 | 1.9 |
| HD-46Q | 27.9 | 27.9 | 61.0 | 27.2 | 1.66 | 2.0 |
| TRX-tag | 14.8 | 14.8 | 19.8 | 15.2 | 1.56 | 1.4 |
| MW1 Fab | 47‖ | 47.5 | 43.8 | 37.2 | 3.30 | 1.4 |
| Typical values | ||||||
| Elongated | >1.6 | |||||
| Globular | ≈1.2–1.3 | |||||
Calculated mass for protein expressed in bacteria, assuming cleavage of N-terminal Met.
Masses determined by mass spectrometry (MS) or sedimentation velocity analytical ultracentrifugation (AUC), or estimated from gel filtration chromatography (FPLC). Although fusion proteins with normal or expanded poly(Gln) migrate in gel filtration with high apparent molecular weights, they are monomeric as shown by AUC.
Three concentrations each of HD-25Q, HD-46Q, or TRX-tag were analyzed by sedimentation velocity and apparent sedimentation coefficients (s*) were extrapolated to infinite dilution and adjusted to standard conditions of 20°C in water (s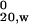 ). A single concentration of MW1 Fab was analyzed and correction of s* to infinite dilution was estimated with the program sednterp (33).
). A single concentration of MW1 Fab was analyzed and correction of s* to infinite dilution was estimated with the program sednterp (33).
Ratio of the observed frictional coefficient (f) and that calculated for an anhydrous sphere of equivalent radius (f0), calculated according to the Teller method with sednterp (33).
Mean of calculated masses from IgG2b/κ Fab sequences in the Protein Data Bank, PDB codes 1IBG, 1FAI, and 1CGS.
To determine whether the nonglobular structures of HD exon 1 contain regular secondary structures, we analyzed the fusion proteins by CD. The CD spectra of HD exon 1 with different lengths of poly(Gln) (Fig. 2a) resemble the spectra of disordered proteins. In particular, the spectra are reminiscent of denatured collagen (21), which has a similar high imino acid (e.g., proline) content. If the poly(Gln) tract in HD exon 1 underwent a global folding transition above the pathologic threshold of 36 Gln, we would expect to observe differences between the CD spectra of HD-25Q and HD-46Q because the poly(Gln) tract accounts for ≈40% of the amino acid sequence in the latter. However, the CD spectra are very similar (Fig. 2a), with no significant differences in the wavelength regions characteristic of regular α or β secondary structures (21). These results are consistent with a random-coil structure for both normal and expanded poly(Gln) tracts within HD exon 1. Moreover, the data suggest that flanking sequences in HD exon 1 (17 residues at the N terminus and 49 residues at the C terminus) do not adopt stable α or β secondary structures and that their presence does not induce a global transition in the poly(Gln) tract above the pathologic threshold.
Figure 2.

Structural characterization of HD exon 1. (a) CD spectra. Each curve represents the difference between the spectrum of an HD exon 1 fusion protein and the spectrum of the TRX-tag control protein (i.e., the CD signal attributable to HD exon 1). (b) Western blots using 1C2 and MW1. Equimolar amounts of HD exon 1 fusion proteins or TRX-tag were loaded and probed with a 1:2000 dilution of ascites fluid containing 1C2 (Chemicon) or 200 nM purified MW1. (c) Comparison between 1H NMR spectra of TRX-tag and HD-46Q. Only the amide and aromatic proton region and the upfield-shifted methyl region are shown. (Insets) Expanded views.
Because random-coil-like CD spectra have been observed for some folded proteins (21, 22), we also performed NMR spectroscopy to assess the structure of HD exon 1 by means of its chemical shift dispersion. Well dispersed resonances are a hallmark of NMR spectra of folded proteins. The one-dimensional 1H NMR spectra of TRX-tag and HD-46Q (Fig. 2c) have identical peaks in the spectral regions outside the extensively overlapped envelope of resonances (Fig. 2c Insets), indicating that TRX is folded in both proteins. The absence of additional peaks in these regions of the HD-46Q spectrum (Fig. 2c Insets) suggests that the N-terminal portion of HD exon 1, which contains two Phe residues (Fig. 1a) capable of producing ring current shifts in the context of a folded protein, does not participate in stable tertiary structure. Furthermore, in the one-dimensional spectrum (Fig. 2c) and in the 1H,15N-HSQC spectrum (data not shown) of HD-46Q, we see little dispersion of amide proton chemical shifts. The majority of backbone amide proton resonances not due to TRX-tag fall in the region characteristic of unfolded proteins (7.7–8.7 ppm). Terminal amide proton resonances of Gln side chains are also narrowly dispersed about their random-coil chemical shifts (6.9 and 7.6 ppm) (23). These regions of the spectra do not provide insight into the structure of the proline residues of HD exon 1, which lack amide protons and may form polyproline II helices. Such helices are inherently flexible and have been considered a subset of random coil, being common in unfolded proteins, even those that are not Pro-rich (21). The data also do not exclude the possible presence of transiently folded species, in which chemical shift perturbations would be diminished by conformational averaging. Our conclusion from the NMR and CD data taken together is that HD exon 1 proteins containing normal and expanded poly(Gln) tracts are random-coil structures that do not adopt a stable conformation involving extensive α or β secondary structures.
Western Blot Analysis of HD Exon 1.
We also assessed the structure of HD exon 1 by evaluating its binding to anti-poly(Gln) monoclonal antibodies in Western blots. In contrast to previous results in Western blots (6, 9, 10), we observe binding of 1C2 and MW1 to both normal and expanded poly(Gln) (Fig. 2b). Thus, the epitope recognized by these antibodies contains 16 or fewer Gln residues. However, the signal is more intense in long poly(Gln) as compared with short poly(Gln). To understand the molecular basis for this pattern of binding and its implications for poly(Gln) structure, we studied the interaction between HD exon 1 and the Fab fragment derived from MW1 (MW1 Fab) by using analytical ultracentrifugation and a biosensor binding assay.
Stoichiometry of MW1 Fab–HD Exon 1 Complexes.
To determine the number of antibody-binding sites in normal versus expanded poly(Gln), we investigated the stoichiometries of complexes between the Fab of MW1 and HD exon 1 with 25 or 46 Gln residues by sedimentation equilibrium analytical ultracentrifugation. Mixtures were analyzed with input molar ratios of MW1 Fab to HD exon 1 that ranged from less than 1:1 to more than 3:1. The weight-average buoyant molecular weight (σw), which corresponds to an average of all species in solution, was greater than that of free Fab or free HD-25Q, but less than that of a 1:1 complex for all of the input mixtures of MW1 Fab:HD-25Q, indicating that the predominant complex formed was 1:1 at the concentrations used in these experiments (1–4 μM). Input mixtures of MW1 Fab:HD-46Q with greater than 1:1 stoichiometry, however, had a σw value significantly higher than that of a 1:1 complex when measured at the same concentrations as MW1 Fab:HD-25Q. This increase in σw indicates the presence of higher-order complexes. Thus, the poly(Gln) tract in HD-46Q is a multivalent antigen that has more than one binding site for MW1 Fab.
Binding Affinity of MW1 Fab for HD Exon 1.
We next used biosensor binding studies to determine binding affinities between HD exon 1 and MW1 Fab. Injected HD exon 1 fusion proteins with normal or expanded poly(Gln) bind immobilized MW1 Fab (Fig. 3a), and a plot of the equilibrium response (Req) versus concentration demonstrates specific responses over a large concentration range (10−10 to 10−5 M) (Fig. 3b). None of the response curves can be fit well to a 1:1 (monovalent analyte) binding model, suggesting that each poly(Gln) tract interacts with two or more MW1 Fab molecules on the chip. This hypothesis is supported by analytical ultracentrifugation results showing HD-46Q is a multivalent antigen for MW1 Fab (discussed above). Because a 1:1 model does not fit the data even at the lowest MW1 Fab coupling density that is experimentally feasible, we used multivalent analyte models to analyze the binding data.
Figure 3.

Biosensor analyses of HD exon 1–MW1 Fab interactions. (a) Representative sensorgrams in which MW1 Fab is coupled at a density of 2,080 RU and HD-25Q (Left) or HD-46Q (Right) is injected at indicated concentrations. (b) Req versus the concentration (logarithmic scale) of injected analyte. Multivalent interactions are favored at low concentrations of each injected protein (low RU response region) and monovalent interactions are favored at high concentrations (high RU response region). The greatest differentiation between expanded poly(Gln) (e.g., HD-46Q) and normal poly(Gln) (e.g., HD-16Q) occurs in the low RU response region, because of multivalent binding of HD-46Q to MW1 Fab. (c–f) Global fits of biosensor data for HD exon 1 fusion proteins injected over MW1 Fab coupled to the chip at low, medium, or high density. Best-fit binding curves to the experimental data points are shown as continuous lines. Alternative models that do not adequately fit the data are shown as dashed lines.
Microscopic KDs [i.e., the affinity for binding one Fab arm to one epitope on the poly(Gln) tract] can be determined from multivalent analyte models, as shown by our previous work involving homodimeric transferrin receptor (TfR) and its ligand, HFE (17, 19). In that system, a bivalent analyte model was developed to extract the KD1 and KD2 affinity constants from equilibrium-based data, which describe the binding of the first and second HFE molecules to TfR (17). In the present analysis the bivalent analyte model fits the HD-16Q data (Fig. 3c, solid lines) but does not adequately describe the HD-25Q, -39Q, and -46Q data (Fig. 3 d–f, dashed lines). However, a trivalent analyte model (see supporting information) does fit the latter data (Fig. 3 d–f, solid lines). Thus, we derived microscopic KD values for the sequential binding of one, two, or three MW1 Fab molecules to a single HD exon 1 molecule. The affinities determined (Table 2) are micromolar, and they differ modestly as a function of poly(Gln) length (e.g., a 17-fold difference in KD1 for HD-16Q vs. HD-46Q). This modest affinity difference is in contrast to the >100-fold differences in avidity (macroscopic KD) at low injected protein concentrations, where multivalent binding is favored (Fig. 3b).
Table 2.
Affinity constants for binding of HD exon 1 to MW1 Fab
| Protein | Affinity constant,* μM
|
||
|---|---|---|---|
| KD1 | KD2 | KD3 | |
| HD-16Q | 25.3 | 73.9 | Not applicable |
| HD-25Q | 14.0 | 78.7 | ≈29† |
| HD-39Q | 2.2 | 65.2 | ≈7† |
| HD-46Q | 1.5 | 37.5 | ≈5† |
KDs determined from global fits of equilibrium-based biosensor data (at low, medium, and high ligand coupling density) to the best-fit bivalent or trivalent binding models (Fig. 3 c–f).
KD3 values are not well determined because they are sensitive to possible tetravalent (or higher valency) interactions that are not accounted for in our binding models, which lead to artefactually low values for KD3. Lower values of KD3 relative to KD2 are not predicted by the linear lattice model unless there is positive cooperativity.
Discussion
The deviation of biosensor binding data from a monovalent analyte model (Fig. 3 c–f) and the trends in KD values determined by using bivalent and trivalent analyte models (Table 2) are consistent with MW1 Fab binding to a “linear lattice” of poly(Gln) (Fig. 4). The linear lattice model has been described in theoretical and experimental studies of ligand interactions with biopolymers such as DNA (24, 25) or oligosaccharides (26). In these systems, binding affinities increase with the length of the lattice, because of entropic (statistical) factors (24, 26) [lower KD values for longer poly(Gln) in Table 2]. Similarly, in the absence of positive cooperativity, affinity for binding a second molecule is reduced (24) (larger KD2 values relative to KD1 in Table 2). The linear lattice model is also consistent with our other data, including the evidence from analytical ultracentrifugation of more than one Fab binding site in HD-46Q and the evidence from CD and NMR that HD exon 1 with both normal and expanded poly(Gln) is a random coil. The linear lattice model is also supported by data in the literature showing that MW1 binds to short peptide epitopes (9 Gln) in dot-blot assays (9). Taking all results into account, we conclude that the pathologic threshold observed in CAG-expansion diseases does not correlate with a global transition of soluble, monomeric poly(Gln) to a folded “pathologic conformation.” Instead, the data support a model of poly(Gln) as a linear lattice, in which the number of binding epitopes increases with length, which accounts for the interaction of poly(Gln) with MW1, and by extension, with other anti-poly(Gln) antibodies [e.g., MW2–5 (ref. 9), 1F8 (ref. 8), 3B5H10,¶ and 1C2 (refs. 5, 6, and 10)].
Figure 4.

Linear lattice model for poly(Gln). Poly(Gln) is a linear array of n repeating units (red dots). A monovalent ligand binds the lattice by interacting with several consecutive units. Binding of a first monovalent ligand occurs with an affinity that increases modestly with lattice length (e.g., 14 μM for HD-25Q and 1.5 μM for HD-46Q binding MW1 Fab, Table 2). A second ligand molecule can bind to a poly(Gln) tract that contains more than one binding site, but binds with weaker affinity than the first ligand. A multivalent ligand can bind with high avidity to a longer poly(Gln) tract by interacting with two binding sites simultaneously (the macroscopic KD is the product of the microscopic KD1 and KD2 values). Thus, normal and pathologic poly(Gln) can be distinguished if a ligand binds multivalently only to pathologic poly(Gln). The existence of such a multivalent ligand in vivo at concentrations below the KD for binding normal (≤36) poly(Gln), but above the KD for binding expanded (>36) poly(Gln) could explain the correlation between poly(Gln) length and disease incidence.
The linear lattice model provides a structural framework for poly(Gln) in vivo because the ligand whose binding we have characterized (MW1 Fab) is derived from an antibody that recognizes full-length huntingtin or HD exon 1 in mouse brain (9). In particular, the linear lattice model suggests a structural basis for “aberrant interaction” mechanisms of cytotoxicity in which soluble expanded poly(Gln) interacts abnormally with other proteins, leading to their functional dysregulation (27). A number of proteins have been identified that interact with normal and expanded poly(Gln) in N-terminal huntingtin fragments (27), and in several cases binding is enhanced for expanded poly(Gln) tracts (28–30). The linear lattice model provides a plausible mechanism for these observations, in that higher-affinity binding is predicted as the poly(Gln) tract is lengthened (Table 2). Furthermore, the model suggests that at low (i.e., ≤ nM) concentrations in vivo, some multivalent ligands (e.g., multimers or monomers with more than one binding site) may bind significantly only to expanded poly(Gln) (Fig. 4), thereby rationalizing the pathogenicity of expanded poly(Gln) tracts. The linear lattice model does not preclude aggregation of expanded poly(Gln), which may contribute to toxicity by recruiting and/or sequestering itself or other proteins (13). Although the linear lattice model does not address the structure of aggregated poly(Gln), it can account for the structural and ligand-binding properties of soluble poly(Gln) species that exist as random coils during a lag period preceding aggregation in vitro (3). Our model predicts that during this lag period, soluble expanded poly(Gln) binds ligands with higher affinity than does normal poly(Gln). Thus in either aberrant interaction or recruitment/sequestration models of expanded poly(Gln) toxicity, the linear lattice model provides a structural framework for rational drug design.
Previous drug design efforts focused mainly on targeting the proposed pathologic conformation of expanded poly(Gln) with small molecules or single-chain Fv (scFv) molecules (9, 31). However, our results suggest that such molecules are unlikely to discriminate sufficiently between normal and expanded poly(Gln). For example, an scFv version of MW1 would be expected to bind normal and expanded poly(Gln) with similar affinities (≈10-fold differences in KD1 for MW1 Fab binding HD-25Q or HD-46Q in Table 2). Use of such molecules therapeutically could lead to low specificity and potential disruption of other proteins containing poly(Gln) [e.g., transcription factors (13)]. An alternative approach is suggested by our demonstration that multivalent interactions with expanded poly(Gln) lead to high-avidity binding (Fig. 3b). The covalent linkage of monovalent compounds with micromolar affinities can produce a new bivalent compound for which the binding affinity is nanomolar (32). Thus, by covalently linking two or more scFvs or small molecule compounds, it should be possible to obtain therapeutic agents with high avidity and specificity for pathologic soluble poly(Gln) tracts (Fig. 4).
Supplementary Material
Acknowledgments
We thank J. Ko and P. H. Patterson for providing MW1 hybridoma cell lines, Susan Ou in the Caltech Monoclonal Antibody Facility for ascites production, R. Myers and A. Kazantsev for proving HD exon 1 DNA templates, the California Institute of Technology Protein/Peptide MicroAnalytical Laboratory (PPMAL) for peptide and protein analyses, and A. M. Giannetti and W. L. Martin for assistance with data processing and helpful discussions. M.J.B. was supported by a fellowship from the Wills Foundation, K.E.H.-T. was supported by a grant from the Hereditary Disease Foundation Cure HD Initiative to M.J.B. and by the Howard Hughes Medical Institute, A.B.H. was supported by a fellowship from the Damon Runyon Cancer Research Foundation (DRG-1658), and A.P.W. was supported by a Burroughs Wellcome Fund Career Award in the Biomedical Sciences.
Abbreviations
- poly(Gln)
polyglutamine
- HD
Huntington's disease
- TRX
thioredoxin
- RU
resonance unit
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on May 1, 2001.
Foley, S., Curtis, J., Saudou, F. & Finkbeiner, S. (2000) Soc. Neurosci. 26, 1294 (abstr.).
References
- 1.Masino L, Pastore A. Brain Res Bull. 2001;56:183–189. doi: 10.1016/s0361-9230(01)00582-2. [DOI] [PubMed] [Google Scholar]
- 2.Altschuler E, Hud N V, Mazrimas J A, Rupp B. J Peptide Res. 1997;50:73–75. doi: 10.1111/j.1399-3011.1997.tb00622.x. [DOI] [PubMed] [Google Scholar]
- 3.Chen S, Berthelier V, Yang W, Wetzel R. J Mol Biol. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 4.Masino L, Kelly G, Leonard K, Trottier Y, Pastore A. FEBS Lett. 2002;513:267–272. doi: 10.1016/s0014-5793(02)02335-9. [DOI] [PubMed] [Google Scholar]
- 5.Lescure A, Lutz Y, Eberhard D, Jacq X, Krol A, Grummt I, Davidson I, Chambon P, Tora L. EMBO J. 1994;13:1166–1175. doi: 10.1002/j.1460-2075.1994.tb06366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, et al. Nature (London) 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 7.Huang C C, Faber P W, Persichetti F, Mittal V, Vonsattel J-P, MacDonald M E, Gusella J F. Somatic Cell Mol Genet. 1998;24:217–233. doi: 10.1023/b:scam.0000007124.19463.e5. [DOI] [PubMed] [Google Scholar]
- 8.Persichetti F, Trettel F, Huang C C, Fraefel C, Timmers H T M, Gusella J F, MacDonald M E. Neurobiol Dis. 1999;6:364–375. doi: 10.1006/nbdi.1999.0260. [DOI] [PubMed] [Google Scholar]
- 9.Ko J, Ou S, Patterson P H. Brain Res Bull. 2001;56:319–329. doi: 10.1016/s0361-9230(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 10.Trottier Y, Zeder-Lutz G, Mandel J-L. In: Genetic Instabilities and Hereditary Neurological Diseases. Wells R D, Warren S T, editors. San Diego: Academic; 1998. pp. 447–453. [Google Scholar]
- 11.Cummings C J, Zoghbi H Y. Hum Mol Genet. 2000;9:909–916. doi: 10.1093/hmg/9.6.909. [DOI] [PubMed] [Google Scholar]
- 12.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies S W, Bates G P. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 13.Preisinger E, Jordan B M, Kazantsev A, Housman D. Philos Trans R Soc London B. 1999;354:1029–1034. doi: 10.1098/rstb.1999.0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LaVallie E R, DiBlasio E A, Kovacic S, Grant K L, Schendel P F, McCoy J M. Bio/Technology. 1993;11:187–193. doi: 10.1038/nbt0293-187. [DOI] [PubMed] [Google Scholar]
- 15.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 16.Gill S C, von Hippel P H. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 17.West A P, Giannetti A M, Herr A B, Bennett M J, Nangiana J S, Pierce J R, Weiner L P, Snow P M, Bjorkman P J. J Mol Biol. 2001;313:385–397. doi: 10.1006/jmbi.2001.5048. [DOI] [PubMed] [Google Scholar]
- 18.Philo J. Biophys J. 1997;72:435–444. doi: 10.1016/S0006-3495(97)78684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.West A P, Bjorkman P J. Biochemistry. 2000;39:9698–9708. doi: 10.1021/bi000749m. [DOI] [PubMed] [Google Scholar]
- 20.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates G P, Davies S W, Lehrach H, Wanker E E. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 21.Fasman G D. Circular Dichroism and the Conformational Analysis of Biomolecules. New York: Plenum; 1996. [Google Scholar]
- 22.Wu J, Yang J T, Wu C-S C. Anal Biochem. 1992;200:359–364. doi: 10.1016/0003-2697(92)90479-q. [DOI] [PubMed] [Google Scholar]
- 23.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley Interscience; 1986. [Google Scholar]
- 24.Kelly R C, Jensen D E, von Hippel P H. J Biol Chem. 1976;251:7240–7250. [PubMed] [Google Scholar]
- 25.McGhee J D, von Hippel P H. J Mol Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 26.Olson S T, Halvorson H R, Bjork I. J Biol Chem. 1991;266:6342–6352. [PubMed] [Google Scholar]
- 27.Gusella J F, MacDonald M E. Curr Opin Neurobiol. 1998;8:425–430. doi: 10.1016/s0959-4388(98)80071-8. [DOI] [PubMed] [Google Scholar]
- 28.Li X-J, Li S-H, Sharp A H, Nucifora F C, Schillling G, Lanahan A, Worley P, Snyder S H, Ross C A. Nature (London) 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- 29.Faber P W, Barnes G T, Srinidhi J, Chen J, Gusella J G, MacDonald M E. Hum Mol Genet. 1998;7:1463–1474. doi: 10.1093/hmg/7.9.1463. [DOI] [PubMed] [Google Scholar]
- 30.Boutell J M, Thomas P, Neal J W, Weston V J, Duce J, Harper P S, Jones A L. Hum Mol Genet. 1999;8:1647–1655. doi: 10.1093/hmg/8.9.1647. [DOI] [PubMed] [Google Scholar]
- 31.Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N, Lehrach H, Wanker E E. Proc Natl Acad Sci USA. 2000;97:6739–6744. doi: 10.1073/pnas.110138997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shuker S B, Hadjuk P J, Meadows R P, Fesik S W. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 33.Laue T M, Shah B D, Ridgeway T M, Pelletier S M. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding S E, Rowe A J, Horton J C, editors. London: R. Soc. Chem.; 1992. pp. 90–125. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.