

Abstract
Chronic inflammation results in increased nitrogen monoxide (⋅NO) formation and the accumulation of nitrite (NO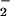 ). Neutrophils stimulated by various inflammatory mediators release myeloperoxidase to produce the cytotoxic agent hypochlorous acid (HOCl). Exposure of chondrocytic SW1353 cells to HOCl resulted in a concentration- and time-dependent loss in viability, ATP, and glutathione levels. Treatment of cells with NO
). Neutrophils stimulated by various inflammatory mediators release myeloperoxidase to produce the cytotoxic agent hypochlorous acid (HOCl). Exposure of chondrocytic SW1353 cells to HOCl resulted in a concentration- and time-dependent loss in viability, ATP, and glutathione levels. Treatment of cells with NO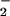 but not nitrate (NO
but not nitrate (NO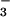 ) substantially decreased HOCl-dependent cellular toxicity even when NO
) substantially decreased HOCl-dependent cellular toxicity even when NO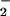 was added at low (μM) concentrations. In contrast, NO
was added at low (μM) concentrations. In contrast, NO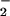 alone (even at 1 mM concentrations) did not affect cell viability or ATP and glutathione levels. These data suggest that NO
alone (even at 1 mM concentrations) did not affect cell viability or ATP and glutathione levels. These data suggest that NO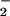 accumulation at chronic inflammatory sites, where both HOCl and ⋅NO are overproduced, may be cytoprotective against damage caused by HOCl. We propose that this is because HOCl is removed by reacting with NO
accumulation at chronic inflammatory sites, where both HOCl and ⋅NO are overproduced, may be cytoprotective against damage caused by HOCl. We propose that this is because HOCl is removed by reacting with NO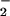 to give nitryl chloride (NO2Cl), which is less damaging in our cell system.
to give nitryl chloride (NO2Cl), which is less damaging in our cell system.
Keywords: inflammation, cell toxicity, nitryl chloride, nitric oxide, arthritis
At sites of chronic inflammation, activated neutrophils release the enzyme myeloperoxidase (MPO) and hydrogen peroxide (H2O2) to catalyze the formation of hypochlorous acid (HOCl; Eq. 1).
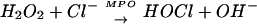 |
It is estimated that between 25 and 40% of the H2O2 generated by activated neutrophils is used to form HOCl (1, 2). Throughout this paper we use the term HOCl (pKa = 7.46) to refer to the ≈50% ionized mixture of HOCl and OCl− species that exists at pH 7.4 (3).
HOCl oxidizes many important biomolecules such as plasma membrane ATPases, collagen, ascorbate, proteins including α1-antiproteinase, nucleotides, sulfhydryls, thioethers, DNA, and DNA-repair enzymes (2, 4–11), depletes intracellular ATP and reduced glutathione (GSH), and causes cell death (12). HOCl is a reactive chlorine species (RCS), capable of chlorinating protein tyrosine residues to form the proposed biomarker for RCS, 3-chlorotyrosine (13–16). Levels of 3-chlorotyrosine are increased in the intima and circulating low-density lipoprotein of some atherosclerosis patients (17–19), bronchoalveolar lavage fluid of lung-transplant patients (20), and expectorated sputum specimens of cystic fibrosis patients (21). HOCl also chlorinates cholesterol in cell membranes (22) as well as the cytosine and adenine residues in DNA (23–26).
An additional reaction of HOCl is with nitrite (NO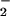 ) to form NO2Cl (Eq. 2; refs. 27–29).
) to form NO2Cl (Eq. 2; refs. 27–29).
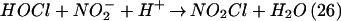 |
This reaction is favored with decreasing pH, such as may occur during chronic inflammation, and its second-order rate constant has been estimated at pH 7.2 and 25°C as 7.4 ± 1.3 × 103 M−1⋅s−1 (30).
Levels of NO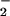 found in plasma taken from healthy human volunteers range between 0.5 and 21.0 μM (31, 32), and these levels are elevated significantly during inflammation, e.g., up to 36 μM in patients with HIV infection (33). Serum NO
found in plasma taken from healthy human volunteers range between 0.5 and 21.0 μM (31, 32), and these levels are elevated significantly during inflammation, e.g., up to 36 μM in patients with HIV infection (33). Serum NO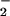 levels in patients with systemic sclerosis are reported to be much higher (34). In the synovial fluid of patients with rheumatoid arthritis, NO
levels in patients with systemic sclerosis are reported to be much higher (34). In the synovial fluid of patients with rheumatoid arthritis, NO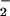 levels are reported to range from 0.3 to as high as 15 μM (26, 35–37). NO
levels are reported to range from 0.3 to as high as 15 μM (26, 35–37). NO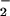 has been used for decades in the food industry as a preservative and for curing meat. It is also estimated that 5% of ingested nitrate (NO
has been used for decades in the food industry as a preservative and for curing meat. It is also estimated that 5% of ingested nitrate (NO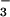 ) is reduced to NO
) is reduced to NO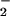 by oral microflora where it enters the gastrointestinal tract (reviewed in refs. 38 and 39). Furthermore, dietary NO
by oral microflora where it enters the gastrointestinal tract (reviewed in refs. 38 and 39). Furthermore, dietary NO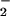 has been proposed as an oral and gut antimicrobial agent (38, 39). Salivary levels of NO
has been proposed as an oral and gut antimicrobial agent (38, 39). Salivary levels of NO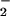 of up to 98 μM have been reported (40).
of up to 98 μM have been reported (40).
NO2Cl formation by activated human neutrophils in the presence of added NO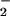 has been demonstrated (29). NO2Cl is capable of nitrating, chlorinating, and dimerizing phenolic compounds such as tyrosine (27–29), and exposure of isolated human low-density lipoprotein to NO2Cl results in the depletion of β-carotene and α-tocopherol as well as protein modification (30). The addition of HOCl to isolated DNA or cells in the presence of NO
has been demonstrated (29). NO2Cl is capable of nitrating, chlorinating, and dimerizing phenolic compounds such as tyrosine (27–29), and exposure of isolated human low-density lipoprotein to NO2Cl results in the depletion of β-carotene and α-tocopherol as well as protein modification (30). The addition of HOCl to isolated DNA or cells in the presence of NO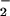 results in increased cytosine chlorination and DNA oxidation compared with DNA or cells in the absence of added NO
results in increased cytosine chlorination and DNA oxidation compared with DNA or cells in the absence of added NO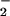 (26, 41). Bacteria phagocytosed by polymorphonuclear cells contain nitrated and chlorinated protein tyrosine residues, suggesting an important host–defense mechanism for reactive nitrogen and chlorine species such as HOCl, NO2Cl, and peroxynitrite (42, 43). These studies have emphasized the potential deleterious effects of NO
(26, 41). Bacteria phagocytosed by polymorphonuclear cells contain nitrated and chlorinated protein tyrosine residues, suggesting an important host–defense mechanism for reactive nitrogen and chlorine species such as HOCl, NO2Cl, and peroxynitrite (42, 43). These studies have emphasized the potential deleterious effects of NO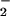 /HOCl-reaction products. However, NO
/HOCl-reaction products. However, NO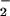 has been reported also to inhibit the antimicrobial activity of HOCl (44–46), isolated MPO (47), and MPO-mediated low-density lipoprotein oxidation (48).
has been reported also to inhibit the antimicrobial activity of HOCl (44–46), isolated MPO (47), and MPO-mediated low-density lipoprotein oxidation (48).
Although NO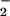 , NO
, NO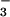 , and HOCl are formed in substantial amounts during inflammation and are present normally in saliva and the gut at high micromolar concentrations, there are few data on the effects of HOCl on human cells in the presence of these nitrogen monoxide (⋅NO) metabolites (24). Therefore, in this paper we describe the effects of physiologically relevant concentrations of NO
, and HOCl are formed in substantial amounts during inflammation and are present normally in saliva and the gut at high micromolar concentrations, there are few data on the effects of HOCl on human cells in the presence of these nitrogen monoxide (⋅NO) metabolites (24). Therefore, in this paper we describe the effects of physiologically relevant concentrations of NO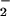 on HOCl-induced cell toxicity in chondrocytic SW1353 cells as a model of cartilage cells exposed to HOCl/NO
on HOCl-induced cell toxicity in chondrocytic SW1353 cells as a model of cartilage cells exposed to HOCl/NO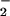 during inflammatory joint disease (49, 50).
during inflammatory joint disease (49, 50).
Experimental Procedures
Materials.
DMEM, Earle's balanced salt solution (EBSS), trypsin-EDTA solution, GSH, oxidized glutathione, Aspergillus nitrate reductase (EC 1.6.6.2), firefly lantern extract, sodium nitrite (NaNO2), sodium nitrate (NaNO3), and all other reagents were purchased from Sigma–Aldrich (Poole, Dorset, U.K.). Sodium arsenite was obtained from BDH (Poole). All cell-culture flasks and microplates were obtained from Greiner (Greiner, Nurtingen, Germany).
Cell Culture.
Human chondrosarcoma cells were obtained from American Type Culture Collection (SW1353) and cultured in DMEM with 1% (vol/vol) penicillin, 10% (wt/vol) FCS, and 5% CO2/95% O2 with ≈95% humidity to 90% confluency before use (49, 50).
Measurement of HOCl.
HOCl concentration was quantified immediately before use spectrophotometrically at 290 nm (pH 12.0, ɛ = 350 M−1⋅cm−1) (3). HOCl was diluted in ice-cold EBSS to a concentration of 10 mM and stored on ice for no longer than 1 min. After this time, the solution was discarded, and fresh HOCl (10 mM) was prepared. Monitoring HOCl concentration (as OCl−) spectrophotometrically at 290 nm showed that there was no significant loss of HOCl over this time.
Exposure of SW1353 Cells to HOCl.
Cells were washed twice with PBS and once with EBSS warmed to 37°C. Fresh EBSS then was added followed by HOCl addition. Cells then were incubated at 37°C, and the reaction was terminated by the addition of 10 mM oxidized glutathione. Adding HOCl did not alter the pH of the reaction mixture significantly.
Exposure of Cells to NO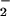 /NO
/NO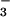 .
.
Cells were washed as described above and incubated in EBSS. NaNO2 or NaNO3 solutions were prepared (10 mM) in EBSS and added to the cells for 10 min. HOCl then was added, and the cells were incubated further at 37°C for up to 30 min when the reaction was terminated by the addition of 10 mM oxidized glutathione. The addition of NaNO2 or NaNO3 did not alter the pH of the reaction mixture significantly. NO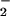 and NO
and NO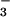 remaining in the culture medium were measured by the Griess assay after reduction of NO
remaining in the culture medium were measured by the Griess assay after reduction of NO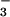 with NO
with NO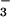 reductase as described (51).
reductase as described (51).
Assessment of Cell Viability.
Cells were seeded overnight at a density of 3.5 × 104 cells per well in 96-well plates. After exposure to HOCl/NO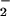 /NO
/NO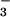 , cells were washed gently twice in warm (37°C) EBSS, and then 200 μl of warm DMEM plus 50 μl of 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide (0.5 mg/ml dissolved in DMEM) was added. Cells were incubated at 37°C in the dark for 1 hr and then washed twice with PBS, and 200 μl of DMSO was added to solubilize the formazan dye. Absorbance at 550 nm then was read on a Molecular Devices Spectramax190 plate reader after gentle shaking in the dark for 30 min. Where required, NO
, cells were washed gently twice in warm (37°C) EBSS, and then 200 μl of warm DMEM plus 50 μl of 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide (0.5 mg/ml dissolved in DMEM) was added. Cells were incubated at 37°C in the dark for 1 hr and then washed twice with PBS, and 200 μl of DMSO was added to solubilize the formazan dye. Absorbance at 550 nm then was read on a Molecular Devices Spectramax190 plate reader after gentle shaking in the dark for 30 min. Where required, NO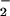 or NO
or NO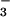 was premixed for 5 min with HOCl in warm EBSS before addition to cells. Control experiments showed that there was no significant HOCl loss over this time.
was premixed for 5 min with HOCl in warm EBSS before addition to cells. Control experiments showed that there was no significant HOCl loss over this time.
Measurement of Cellular Glutathione and ATP.
Cells were seeded overnight at a density of 0.25 × 106 cells per well in 24-well plates. After washing twice with EBSS, 2.0 ml of fresh EBSS was added and HOCl added. The reaction was terminated by the addition of 10 mM oxidized glutathione. Cells then were washed twice in ice-cold PBS to remove any residual NO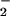 or NO
or NO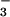 . This was followed by the addition of 250 μl of ice-cold trichloroacetic acid 6.5% wt/vol) and left on ice for 10 min. The trichloroacetic acid extract then was removed and either stored at −80°C or used immediately for analysis. NaOH (200 μl of 1M solution) then was added to solubilize cellular protein. Protein concentration then was measured by the Bradford method. The trichloroacetic acid extract then was used to assess cellular GSH and ATP.
. This was followed by the addition of 250 μl of ice-cold trichloroacetic acid 6.5% wt/vol) and left on ice for 10 min. The trichloroacetic acid extract then was removed and either stored at −80°C or used immediately for analysis. NaOH (200 μl of 1M solution) then was added to solubilize cellular protein. Protein concentration then was measured by the Bradford method. The trichloroacetic acid extract then was used to assess cellular GSH and ATP.
Analysis of cellular GSH was performed as described (52). Briefly, 7.5 μl of trichloroacetic acid extract was added to 96-well fluorescence plates followed by the addition of 227.5 μl of 100 mM KH2PO4-KOH buffer, pH 10.0, and 15 μl of o-phthaldialdehyde (10 mg/ml freshly prepared in methanol). Samples were stored in the dark at room temperature for 25 min and measured by fluorescence (excitation = 350 nm, emission = 420 nm) using a Gemini Fluorescence plate reader (Molecular Devices). Concentrations of GSH then were determined by comparing the values obtained with a freshly prepared standard curve of GSH.
Loss of cellular ATP was assessed by using firefly lantern extract as described (53). Briefly, 3 μl of sample was incubated with 200 μl of sodium arsenite buffer (comprising 26.67 mM MgSO4⋅7H2O/3.33 mM KH2PO4/33.33 mM Na2HASO4⋅7H2O, pH 7.4). After the addition of 10 μl of filtered firefly lantern extract per sample, light emission then was measured for 10 sec per sample by using a LUMI-ONE portable luminometer (Trans Orchid Enterprises, Tampa, FL). Concentrations of ATP then were determined by comparing the values obtained with a freshly prepared standard curve of ATP. Where required, NO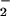 or NO
or NO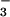 was premixed for 5 min with HOCl in warm EBSS before addition to cells.
was premixed for 5 min with HOCl in warm EBSS before addition to cells.
Assessment of Lactate Dehydrogenase (LDH) Release.
Cells were seeded overnight at a density of 0.25 × 106 per well in 24-well plates, washed, and exposed to NO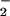 , NO
, NO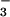 , and HOCl as described above. After 10 min of exposure, the cells were washed gently in warm PBS, and DMEM was added. Preliminary experiments showed that HOCl inactivated LDH; therefore, cells were washed and HOCl was removed before measuring LDH leakage after 24 hrs of incubation in DMEM at 37°C. LDH activity was measured by using a commercially available kit (Sigma, LD-50) and a Molecular Devices SpectraMax 190 microplate reader and compared as a percentage of LDH activity from cells lysed with 0.1% Triton X for 20 min at 37°C.
, and HOCl as described above. After 10 min of exposure, the cells were washed gently in warm PBS, and DMEM was added. Preliminary experiments showed that HOCl inactivated LDH; therefore, cells were washed and HOCl was removed before measuring LDH leakage after 24 hrs of incubation in DMEM at 37°C. LDH activity was measured by using a commercially available kit (Sigma, LD-50) and a Molecular Devices SpectraMax 190 microplate reader and compared as a percentage of LDH activity from cells lysed with 0.1% Triton X for 20 min at 37°C.
Where required, NO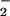 or NO
or NO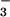 was premixed for 5 min with HOCl in warm EBSS before addition to cells.
was premixed for 5 min with HOCl in warm EBSS before addition to cells.
Data Analysis.
All graphs are plotted with mean ± standard deviation of the mean. In all cases the mean values were calculated from data taken from at least six separate experiments performed on separate days by using freshly prepared reagents. Where significance testing was performed, an independent Student's t test (two populations) was used (*, P < 0.1; **, P < 0.05; ***, P < 0.01).
Results
Effect of HOCl, NO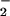 , and NO
, and NO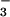 on Cell Viability.
on Cell Viability.
Incubation of 100 μM NO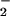 with increasing concentrations of HOCl in EBSS resulted in the oxidation of 1 mol of NO
with increasing concentrations of HOCl in EBSS resulted in the oxidation of 1 mol of NO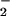 to NO
to NO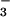 per mole of HOCl added (Fig. 1). The addition of HOCl to SW1353 cells substantially reduced cell viability as measured by 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide in a time- and concentration-dependent manner. Preliminary experiments (data not shown) established that low concentrations of HOCl (30 μM) were sufficient to significantly reduce cell viability after a 10-min incubation time at 37°C, pH 7.4. From these data, an incubation time of 10 min was chosen for subsequent studies. However, the presence of NaNO2 before HOCl addition substantially inhibited HOCl-induced cytotoxicity with marked protection observed with 30 μM NO
per mole of HOCl added (Fig. 1). The addition of HOCl to SW1353 cells substantially reduced cell viability as measured by 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide in a time- and concentration-dependent manner. Preliminary experiments (data not shown) established that low concentrations of HOCl (30 μM) were sufficient to significantly reduce cell viability after a 10-min incubation time at 37°C, pH 7.4. From these data, an incubation time of 10 min was chosen for subsequent studies. However, the presence of NaNO2 before HOCl addition substantially inhibited HOCl-induced cytotoxicity with marked protection observed with 30 μM NO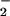 (Fig. 2A). The same degree of inhibition was observed if HOCl (125 μM) was premixed with NO
(Fig. 2A). The same degree of inhibition was observed if HOCl (125 μM) was premixed with NO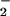 for 5 min before its addition to cells (Fig. 2B). This effect was not observed with NO
for 5 min before its addition to cells (Fig. 2B). This effect was not observed with NO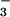 even when tested at 1 mM (data not shown). However, if the cells were incubated first with HOCl for 10 min followed by NO
even when tested at 1 mM (data not shown). However, if the cells were incubated first with HOCl for 10 min followed by NO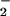 addition, NO
addition, NO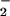 was unable to prevent HOCl-mediated loss of viability (data not shown), which indicates that NO
was unable to prevent HOCl-mediated loss of viability (data not shown), which indicates that NO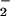 cannot reverse damage that has been caused by HOCl. Control experiments showed that incubating SW1353 cells with up to 1 mM NaNO2 or NaNO3 for 1 hr did not cause significant loss of cell viability (0.98 ± 0.5 and 1.02 ± 0.6% reduction in viability, respectively).
cannot reverse damage that has been caused by HOCl. Control experiments showed that incubating SW1353 cells with up to 1 mM NaNO2 or NaNO3 for 1 hr did not cause significant loss of cell viability (0.98 ± 0.5 and 1.02 ± 0.6% reduction in viability, respectively).
Fig 1.

Reaction of NO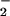 with HOCl. EBSS was incubated with NaNO2, and HOCl was subsequently added. Residual NO
with HOCl. EBSS was incubated with NaNO2, and HOCl was subsequently added. Residual NO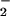 was measured by the Griess assay as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments.
was measured by the Griess assay as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments.
Fig 2.

HOCl-mediated loss of cellular viability: Effect of NO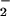 . NO
. NO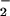 was added to cells in EBSS 10 min before HOCl (A) or NO
was added to cells in EBSS 10 min before HOCl (A) or NO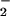 premixed with HOCl (125 μM) for 5 min and then added to cells for 10 min (B). Cells were washed in DMEM, and cellular viability was measured by using the 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide assay as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments. *, P < 0.1; **, P < 0.05; and ***, P < 0.01 compared with HOCl addition alone.
premixed with HOCl (125 μM) for 5 min and then added to cells for 10 min (B). Cells were washed in DMEM, and cellular viability was measured by using the 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide assay as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments. *, P < 0.1; **, P < 0.05; and ***, P < 0.01 compared with HOCl addition alone.
Effect of HOCl, NO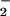 , and NO
, and NO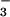 on Intracellular ATP Depletion.
on Intracellular ATP Depletion.
To characterize further the effects of HOCl and NO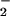 on cellular metabolism, levels of ATP were examined (Fig. 3 A and B). HOCl caused a rapid depletion of ATP when added to SW1353 cells at low (<30 μM) concentrations. The presence of NO
on cellular metabolism, levels of ATP were examined (Fig. 3 A and B). HOCl caused a rapid depletion of ATP when added to SW1353 cells at low (<30 μM) concentrations. The presence of NO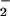 (but not NO
(but not NO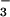 , tested up to 1 mM) before HOCl addition significantly inhibited HOCl-dependent ATP loss even in the presence of high HOCl concentrations (250 μM; Fig. 3A). The same degree of inhibition was observed if HOCl (125 μM) was premixed with NO
, tested up to 1 mM) before HOCl addition significantly inhibited HOCl-dependent ATP loss even in the presence of high HOCl concentrations (250 μM; Fig. 3A). The same degree of inhibition was observed if HOCl (125 μM) was premixed with NO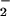 for 5 min before the addition to cells (Fig. 3B).
for 5 min before the addition to cells (Fig. 3B).
Fig 3.

HOCl-mediated loss of cellular ATP: Effect of NO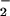 . NO
. NO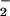 was added to cells in EBSS 10 min before HOCl (A) or NO
was added to cells in EBSS 10 min before HOCl (A) or NO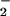 premixed with HOCl (125 μM) for 5 min and then added to cells for 10 min (B). Cells were washed in DMEM, and cellular ATP was measured by using firefly lantern extract as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments. *, P < 0.1; **, P < 0.05; and ***, P < 0.01 compared with HOCl addition alone.
premixed with HOCl (125 μM) for 5 min and then added to cells for 10 min (B). Cells were washed in DMEM, and cellular ATP was measured by using firefly lantern extract as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments. *, P < 0.1; **, P < 0.05; and ***, P < 0.01 compared with HOCl addition alone.
However, if the cells were incubated first with HOCl for 10 min followed by NO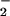 addition, NO
addition, NO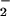 was unable to prevent HOCl-mediated ATP (data not shown). Control experiments showed that incubating SW1353 cells with up to 1 mM NaNO2 or NaNO3 for 1 hr did not affect cellular ATP levels significantly.
was unable to prevent HOCl-mediated ATP (data not shown). Control experiments showed that incubating SW1353 cells with up to 1 mM NaNO2 or NaNO3 for 1 hr did not affect cellular ATP levels significantly.
Effect of HOCl, NO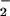 , and NO
, and NO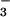 on Intracellular GSH.
on Intracellular GSH.
Preliminary experiments also showed that HOCl caused a rapid depletion of cellular GSH. Increasing the time of incubation increased the extent of GSH loss, and a 10-min incubation time was selected for subsequent studies. The addition of NO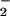 (but not NO
(but not NO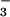 , tested up to 1 mM) to the cells before HOCl addition significantly prevented HOCl-dependent GSH loss even in the presence of high HOCl concentrations (250 μM; Fig. 4A). A similar degree of inhibition was observed if HOCl (125 μM) was premixed with NO
, tested up to 1 mM) to the cells before HOCl addition significantly prevented HOCl-dependent GSH loss even in the presence of high HOCl concentrations (250 μM; Fig. 4A). A similar degree of inhibition was observed if HOCl (125 μM) was premixed with NO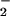 for 5 min before the addition to cells (Fig. 4B). Again, if the cells were incubated first with HOCl for 10 min followed by NO
for 5 min before the addition to cells (Fig. 4B). Again, if the cells were incubated first with HOCl for 10 min followed by NO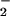 addition, NO
addition, NO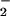 was unable to prevent HOCl-mediated GSH loss (data not shown). Control experiments showed that incubating SW1353 cells with up to 1 mM NaNO2 or NaNO3 for 1 hr did not affect cellular GSH levels significantly.
was unable to prevent HOCl-mediated GSH loss (data not shown). Control experiments showed that incubating SW1353 cells with up to 1 mM NaNO2 or NaNO3 for 1 hr did not affect cellular GSH levels significantly.
Fig 4.

HOCl-mediated loss of cellular GSH: Effect of NO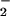 . NO
. NO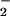 was added to cells in EBSS 10 min before HOCl (A) or NO
was added to cells in EBSS 10 min before HOCl (A) or NO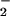 premixed with HOCl (125 μM) for 5 min and then added to cells for 10 min (B). Cells were washed in DMEM, and cellular GSH was measured as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments. *, P < 0.1; **, P < 0.05; and ***, P < 0.01 compared with HOCl addition alone.
premixed with HOCl (125 μM) for 5 min and then added to cells for 10 min (B). Cells were washed in DMEM, and cellular GSH was measured as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments. *, P < 0.1; **, P < 0.05; and ***, P < 0.01 compared with HOCl addition alone.
Effect of HOCl, NO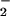 , and NO
, and NO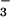 on LDH Release.
on LDH Release.
Fig. 5 shows the effect of high concentrations of HOCl (200 μM) on LDH release from SW1353 cells. HOCl induced substantial cellular LDH release 24 hr after exposure. This effect was inhibited significantly when cells were incubated with NO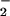 (>30 μM), but not NO
(>30 μM), but not NO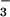 (up to 1 mM), for 10 min before HOCl addition. A similar degree of inhibition of LDH release was observed if HOCl (200 μM) was premixed with NO
(up to 1 mM), for 10 min before HOCl addition. A similar degree of inhibition of LDH release was observed if HOCl (200 μM) was premixed with NO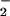 for 5 min before the addition to cells (Fig. 5). However, treatment of cells with HOCl followed by NO
for 5 min before the addition to cells (Fig. 5). However, treatment of cells with HOCl followed by NO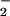 or NO
or NO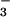 addition did not inhibit LDH release significantly (data not shown).
addition did not inhibit LDH release significantly (data not shown).
Fig 5.

HOCl-mediated LDH leakage: Effect of NO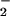 . NO
. NO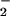 was added to cells in EBSS 10 min before HOCl (▪) or NO
was added to cells in EBSS 10 min before HOCl (▪) or NO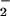 premixed with HOCl (200 μM) for 5 min and then added to cells for 10 min (□). After treatment cells were washed in DMEM, and LDH leakage was measured in the medium after 24 hrs as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments. *, P < 0.1; **, P < 0.05; and ***, P < 0.01 compared with HOCl addition alone.
premixed with HOCl (200 μM) for 5 min and then added to cells for 10 min (□). After treatment cells were washed in DMEM, and LDH leakage was measured in the medium after 24 hrs as described in Experimental Procedures. Data are expressed as mean ± standard deviation of six or more separate experiments. *, P < 0.1; **, P < 0.05; and ***, P < 0.01 compared with HOCl addition alone.
Discussion
There is considerable evidence that HOCl and the end products of ⋅NO metabolism (NO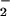 and NO
and NO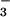 ) accumulate at sites of chronic inflammation (1, 2, 33–36). HOCl is known to oxidize many important classes of biomolecules (1–12) as well as chlorinate protein, DNA, and cholesterol (13–26) and cause cell death (12). Although there is a wealth of information on HOCl-mediated processes at sites of chronic inflammation, HOCl is coproduced with ⋅NO, and relatively little information is available on the consequences to cells of HOCl generated in the presence of NO
) accumulate at sites of chronic inflammation (1, 2, 33–36). HOCl is known to oxidize many important classes of biomolecules (1–12) as well as chlorinate protein, DNA, and cholesterol (13–26) and cause cell death (12). Although there is a wealth of information on HOCl-mediated processes at sites of chronic inflammation, HOCl is coproduced with ⋅NO, and relatively little information is available on the consequences to cells of HOCl generated in the presence of NO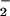 (24). The relatively fast second-order rate constant of reaction for NO
(24). The relatively fast second-order rate constant of reaction for NO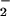 and HOCl (pH 7.2, 25°C, 7.4 ± 1.3 × 103 M−1⋅s−1; ref. 30), the high concentrations of HOCl, and the high concentration of NO
and HOCl (pH 7.2, 25°C, 7.4 ± 1.3 × 103 M−1⋅s−1; ref. 30), the high concentrations of HOCl, and the high concentration of NO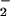 at sites of chronic inflammation (36–39) or present in saliva (40) and the gut (38) make this reaction plausible in vivo. Therefore, it was pertinent to examine the effects of HOCl on cell function in the presence of physiologically relevant concentrations of NO
at sites of chronic inflammation (36–39) or present in saliva (40) and the gut (38) make this reaction plausible in vivo. Therefore, it was pertinent to examine the effects of HOCl on cell function in the presence of physiologically relevant concentrations of NO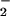 .
.
HOCl caused rapid depletion of cellular ATP and GSH as well as reduced cell viability. However, when HOCl was added in the presence of physiological concentrations of NO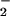 , the latter was oxidized to NO
, the latter was oxidized to NO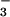 , and HOCl-induced cytotoxicity was inhibited substantially (Figs. 2–5), presumably because of removal of the HOCl. This was observed even at high concentrations of HOCl (>200 μM). Such high concentrations of HOCl are unlikely to occur in vivo, but the fact that NO
, and HOCl-induced cytotoxicity was inhibited substantially (Figs. 2–5), presumably because of removal of the HOCl. This was observed even at high concentrations of HOCl (>200 μM). Such high concentrations of HOCl are unlikely to occur in vivo, but the fact that NO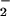 can protect cells from toxicity at low concentrations even when HOCl is in molar excess is notable. We speculate that the NO2Cl formed from the reaction of HOCl with NO
can protect cells from toxicity at low concentrations even when HOCl is in molar excess is notable. We speculate that the NO2Cl formed from the reaction of HOCl with NO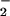 is less cytotoxic than HOCl in our cell system. Perhaps it does not penetrate the cell as readily as HOCl. Indeed, preincubating HOCl with NO
is less cytotoxic than HOCl in our cell system. Perhaps it does not penetrate the cell as readily as HOCl. Indeed, preincubating HOCl with NO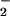 also removed the cytotoxic activity of HOCl (Figs. 2B, 3B, 4B, and 5).
also removed the cytotoxic activity of HOCl (Figs. 2B, 3B, 4B, and 5).
Thus under our reaction conditions at physiologically attainable concentrations, NO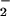 but not NO
but not NO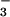 seems to act as an antioxidant in that it reacts preferentially with HOCl by a two-electron process to form NO
seems to act as an antioxidant in that it reacts preferentially with HOCl by a two-electron process to form NO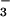 in a 1:1 molar ratio (Fig. 1; ref. 29) to prevent HOCl-mediated cytotoxicity. Indeed, NO
in a 1:1 molar ratio (Fig. 1; ref. 29) to prevent HOCl-mediated cytotoxicity. Indeed, NO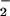 substantially inhibits the bactericidal activity of HOCl toward Escherichia coli (44–46) and prevents HOCl-mediated inactivation of α1-antiproteinase, the major inhibitor of serine proteases in human body fluids (25), as well as HOCl-mediated loss of 8-hydroxyguanine and FAPy guanine in isolated calf thymus DNA (41). NO
substantially inhibits the bactericidal activity of HOCl toward Escherichia coli (44–46) and prevents HOCl-mediated inactivation of α1-antiproteinase, the major inhibitor of serine proteases in human body fluids (25), as well as HOCl-mediated loss of 8-hydroxyguanine and FAPy guanine in isolated calf thymus DNA (41). NO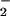 also inhibits MPO to limit tyrosine nitration and chlorination as well as oxidation reactions (47, 48).
also inhibits MPO to limit tyrosine nitration and chlorination as well as oxidation reactions (47, 48).
Based on studies of nitration of phenolics and proteins, oxidation of lipids, depletion of α-tocopherol, and chlorination of DNA and proteins, previous reports have suggested that NO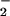 might enhance HOCl-mediated toxicity at sites of inflammation (13–26). The current findings indicate that this is unlikely to be the case, because NO
might enhance HOCl-mediated toxicity at sites of inflammation (13–26). The current findings indicate that this is unlikely to be the case, because NO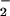 decreases the toxicity of HOCl for cells in vitro. Nevertheless, NO2Cl, the product of HOCl and NO
decreases the toxicity of HOCl for cells in vitro. Nevertheless, NO2Cl, the product of HOCl and NO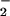 , is likely to cause some cell damage (12–23). The present studies indicate that, in our cell system, NO2Cl is far less damaging than HOCl. The removal of HOCl by NO
, is likely to cause some cell damage (12–23). The present studies indicate that, in our cell system, NO2Cl is far less damaging than HOCl. The removal of HOCl by NO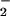 could represent an additional endogenous defense mechanism to limit HOCl-mediated oxidative damage of cellular targets. Therefore it is plausible that some of the antiinflammatory and cytoprotective actions of ⋅NO could involve NO
could represent an additional endogenous defense mechanism to limit HOCl-mediated oxidative damage of cellular targets. Therefore it is plausible that some of the antiinflammatory and cytoprotective actions of ⋅NO could involve NO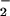 (54, 55).
(54, 55).
In summary, we have shown that low and physiologically relevant concentrations of NO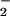 substantially inhibit HOCl-mediated cellular toxicity to human SW1353 chondrosarcoma cells. This may be of great importance in diseases that are associated with oxidative stress such as rheumatoid arthritis (56) and inflammatory bowel disease (57), whereby endogenous HOCl-removing antioxidants such as GSH and ascorbate are depleted substantially and ⋅NO, NO
substantially inhibit HOCl-mediated cellular toxicity to human SW1353 chondrosarcoma cells. This may be of great importance in diseases that are associated with oxidative stress such as rheumatoid arthritis (56) and inflammatory bowel disease (57), whereby endogenous HOCl-removing antioxidants such as GSH and ascorbate are depleted substantially and ⋅NO, NO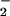 , and HOCl formation are elevated substantially. Further work is needed to address these issues.
, and HOCl formation are elevated substantially. Further work is needed to address these issues.
Acknowledgments
We thank Dr. Jeremy P. E. Spencer (King's College, London) for his critical discussion of this manuscript. We are grateful also to the National Medical Research Council of Singapore (NMRC/0474/2000 and NMRC/0481/2000) and the National University of Singapore Academic Research Fund (R183000053214) for their generous research support.
Abbreviations
MPO, myeloperoxidase
HOCl, hypochlorous acid
GSH, reduced glutathione
NO
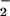 , nitrite
, nitriteNO
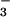 , nitrate
, nitrateLDH, lactate dehydrogenase
NO2Cl, nitryl chloride
References
- 1.Foote C. S., Goyne, T. E. & Lehrer, T. (1983) Nature (London) 301, 715-716. [DOI] [PubMed] [Google Scholar]
- 2.Weiss S. J., Klein, P., Slivka, A. & Wei, M. (1992) J. Clin. Invest. 70, 1341-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris J. C. (1966) J. Phys. Chem. 70, 3798-3805. [Google Scholar]
- 4.Schrauffstatter I. U., Browne, K., Harris, A., Hyslop, P. A., Jackson, J. H., Quehenberger, O. & Cochrane, C. G. (1989) J. Clin. Invest. 85, 554-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aruoma O. I., Halliwell, B., Hoey, B. M. & Butler, J. (1989) Free Radical Biol. Med. 6, 593-597. [DOI] [PubMed] [Google Scholar]
- 6.Halliwell B., Wasil, M. & Grootveld, M. (1987) FEBS Lett. 213, 15-18. [DOI] [PubMed] [Google Scholar]
- 7.Folkes L. K., Candeias, L. P. & Wardman, P. (1995) Arch. Biochem. Biophys. 323, 120-126. [DOI] [PubMed] [Google Scholar]
- 8.Prutz W. A. (1996) Arch. Biochem. Biophys. 332, 110-120. [DOI] [PubMed] [Google Scholar]
- 9.Van Rensberg C. E. J., Van Staden, A. M. & Anderson, R. (1991) Free Radical Biol. Med. 1, 285-291. [DOI] [PubMed] [Google Scholar]
- 10.Van Rensberg C. E. J., Van Staden, A. M., Anderson, R. & Van Rensberg, E. J. (1992) Mutat. Res. 265, 255-261. [DOI] [PubMed] [Google Scholar]
- 11.Pero R. W., Sheng, Y., Olsson, A., Bryngelsson, C. & Lund-Pero, M. (1996) Carcinogenesis 17, 13-18. [DOI] [PubMed] [Google Scholar]
- 12.Jenner A. M., Ruiz, E. J., Dunster, C., Halliwell, B., Mann, G. E. & Siow, R. C. M. (2002) Arterioscler. Thromb. Vasc. Biol. 22, 574-580. [DOI] [PubMed] [Google Scholar]
- 13.Domigan N. M., Charlton, T. S., Duncan, M. W., Winterbourn, C. C. & Kettle, A. J. (1995) J. Biol. Chem. 270, 16542-16848. [DOI] [PubMed] [Google Scholar]
- 14.Kettle A. J. (1995) FEBS Lett. 379, 103-106. [DOI] [PubMed] [Google Scholar]
- 15.Olszowski S., Olszowska, E., Stelmaszynska, T., Krawczyk, A., Marcinkiewicz, J. & Baczek, N. (1996) Acta Biochim. Pol. 43, 661-672. [PubMed] [Google Scholar]
- 16.Hazen S. L., Crowley, J. R., Mueller, D. M. & Heinecke, J. W. (1997) Free Radical Biol. Med. 23, 909-916. [DOI] [PubMed] [Google Scholar]
- 17.Daugherty A., Dunn, J. L., Rateri, D. L. & Heinecke, J. W. (1994) J. Clin. Invest. 94, 437-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hazen S. J. & Heinecke, J. W. (1997) J. Clin. Invest. 99, 2075-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hazell L. J., Arnold, L., Flowers, D., Waeg, G., Malle, E. & Stocker, R. (1996) J. Clin. Invest. 97, 1535-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van der Vliet A., Nguyen, M. N., Shigenaga, M. K., Eiserich, J. P., Marleich, G. P. & Cross, C. E. (2001) Am. J. Physiol. 279, L537-L546. [DOI] [PubMed] [Google Scholar]
- 21.de Andrade J. A., Crow, J. P., Viera, L., Alexander, C. B., Yong, K. R., McGiffin, D. C., Zorn, G. L., Zhu, G., Matalon, S. & Jackson, R. M. (2000) Am. J. Respir. Crit. Care Med. 161, 2035-2042. [DOI] [PubMed] [Google Scholar]
- 22.Carr A., Vanderberg, J. J. M. & Winterbourn, C. (1996) Arch. Biochem. Biophys. 332, 63-69. [DOI] [PubMed] [Google Scholar]
- 23.Whiteman M., Jenner, A. & Halliwell, B. (1997) Chem. Res. Toxicol. 10, 1240-1246. [DOI] [PubMed] [Google Scholar]
- 24.Spencer J. P. E., Whiteman, M., Jenner, A. & Halliwell, B. (2000) Free Radical Biol. Med. 28, 1039-1050. [DOI] [PubMed] [Google Scholar]
- 25.Whiteman M., Jenner, A. & Halliwell, B. (1999) Biomarkers 4, 303-310. [Google Scholar]
- 26.Whiteman M., (1998) Ph.D. thesis (Univ. of London, London).
- 27.Johnson D. W. & Margerum, D. W. (1991) Inorg. Chem. 30, 4845-4851. [Google Scholar]
- 28.Eiserich J. P., Cross, C. E., Jones, A. D., Halliwell, B. & Van Der Vliet, A. (1996) J. Biol. Chem. 271, 19199-19208. [DOI] [PubMed] [Google Scholar]
- 29.Eiserich J. P., Hristova, M., Cross, C. E., Jones, A. D., Freeman, B. A., Halliwell, B. & Van Der Vliet, A. (1998) Nature (London) 391, 393-397. [DOI] [PubMed] [Google Scholar]
- 30.Panasenko O. M., Briviba, J., Klotz, L. O. & Sies, H. (1997) Arch. Biochem. Biophys. 343, 254-259. [DOI] [PubMed] [Google Scholar]
- 31.Leone A. M., Francis, P. L., Rhodes, P. & Moncada, S. (1994) Biochem. Biophys. Res. Commun. 200, 951-957. [DOI] [PubMed] [Google Scholar]
- 32.Ueda T., Maekawa, T., Sadamitsu, D., Ohshita, S., Ogino, K. & Nakamura, K. (1995) Electrophoresis 16, 1002-1004. [DOI] [PubMed] [Google Scholar]
- 33.Torre D., Ferrario, G., Speranza, F., Fiori, G. P. & Zeroli, P. (1996) J. Clin. Pathol. 49, 574-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sud A., Khullar, M., Wanchu, A. & Bambert, P. (2000) Nitric Oxide 4, 615-619. [DOI] [PubMed] [Google Scholar]
- 35.Davies C. A., Perrett, D., Zhang, Z., Nielsen, B. R., Blake, D. R. & Winyard, P. G. (1999) Electrophoresis 20, 2111-2117. [DOI] [PubMed] [Google Scholar]
- 36.Zuber M. & Miesle, R. (1994) in Biology of Nitric Oxide, eds. Moncada, S., Feelisch, M., Busse, R. & Higgs, E. A. (Portland Press, London).
- 37.Ueki Y., Tominaga, Y. & Eguchi, K. (1996) J. Rheumatol. 23, 230-235. [PubMed] [Google Scholar]
- 38.McKnight G. M., Duncan, C. W., Leifert, C. & Golden, M. H. (1999) Br. J. Nutr. 81, 349-358. [DOI] [PubMed] [Google Scholar]
- 39.Weitzberg E. & Lundberg, J. O. N. (1998) Nitric Oxide 2, 1-7. [DOI] [PubMed] [Google Scholar]
- 40.Helaleh M. I. H. & Korenaga, T. (2000) J. Chromatogr. B Biomed. Sci. Appl. 744, 433-437. [DOI] [PubMed] [Google Scholar]
- 41.Whiteman M., Spencer, J. P. E., Jenner, A. & Halliwell, B. (1999) Biochem. Biophys. Res. Commun. 257, 572-576. [DOI] [PubMed] [Google Scholar]
- 42.Evans T. J., Buttery, L. D. K., Carpenter, A., Springall, D. R., Polak, J. M. & Cohen, J. (1996) Proc. Natl. Acad. Sci. USA 93, 9553-9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hazen S. L., Hsu, F. F., Mueller, D. M., Crowley, J. R. & Heinecke, J. W. (1996) J. Clin. Invest. 98, 1283-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klebanoff S. J. (1993) Free Radical Biol. Med. 14, 351-360. [DOI] [PubMed] [Google Scholar]
- 45.Kono Y. (1995) Biochem. Mol. Biol. Int. 36, 275-283. [PubMed] [Google Scholar]
- 46.Marcinkiewicz J., Chain, B., Nowak, B., Grabowska, A., Bryniarski, K. & Baran, J. (2000) Inflamm. Res. 49, 280-289. [DOI] [PubMed] [Google Scholar]
- 47.Van Dalen C. J., Winterbourn, C. C., Senthilmohan, R. & Kettle, A. J. (2000) J. Biol. Chem. 275, 11638-11644. [DOI] [PubMed] [Google Scholar]
- 48.Carr A. C. & Frei, B. Z. (2001) J. Biol. Chem. 276, 1822-1828. [DOI] [PubMed] [Google Scholar]
- 49.Kerkela E., Bohling, T., Herva, R., Uria, J. A. & Saarialho-Kere, U. (2001) Bone (NY) 29, 487-493. [DOI] [PubMed] [Google Scholar]
- 50.Vincenti M. P. & Brinckerhoff, C. E. (2001) Arthritis Res. 3, 381-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nims M R. W., Cook, J. C., Krishna, M. C., Christodoulu, D., Poore, C. M. B., Miles, A. M., Grisham, M. B. & Wink, D. A. (1996) Methods Enzymol. 268, 93-105. [DOI] [PubMed] [Google Scholar]
- 52.Hissin P. J. & Hilf, R. (1976) Anal. Biochem. 74, 214-226. [DOI] [PubMed] [Google Scholar]
- 53.Kalbhem D. D. & Koch, H. J. (1967) Z. Klin. Chem. Klin. Biochem. 5, 299-394. [PubMed] [Google Scholar]
- 54.Khattab M. M., Gad, M. Z. & Abdallah, D. (2001) Pharmacol. Res. 43, 463-467. [DOI] [PubMed] [Google Scholar]
- 55.Lauer T., Preik, M., Rassaf, T., Strauer, B. E., Deussen, A., Feelisch, M. & Kelm, M. (2001) Proc. Natl. Acad. Sci. USA 98, 12814-12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Halliwell B. (1995) Ann. Rheum. Dis. 54, 505-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grisham M. B. (2000) Trends Pharmacol. 21, 119-120. [DOI] [PubMed] [Google Scholar]