

Abstract
7,8-Dihydro-8-oxoguanine (8-oxoG) is the most common form of oxidative DNA damage in human cells. Biochemical studies have shown that 8-oxoG decreases the DNA cleavage activity of human topoisomerase I, an enzyme vital to DNA metabolism and stability. We present the 3.1-Å crystal structure of human topoisomerase I in noncovalent complex with a DNA oligonucleotide containing 8-oxoG at the +1 position in the scissile strand. We find that 8-oxoG reorganizes the active site of human topoisomerase I into an inactive conformation relative to the structures of topoisomerase I–DNA complexes elucidated previously. The catalytic Tyr-723–Phe rotates away from the DNA cleavage site and packs into the body of the molecule. A second active-site residue, Arg-590, becomes disordered and is not observed in the structure. The docked, inactive conformation of Tyr-723–Phe is reminiscent of the related tyrosine recombinase family of integrases and recombinases, suggesting a common regulatory mechanism. We propose that human topoisomerase I binds to DNA first in an inactive conformation and then rearranges its active site for catalysis. 8-OxoG appears to impact topoisomerase I by stabilizing the inactive, DNA-bound state.
The most common type of oxidative damage identified in mammalian cells is 7,8-dihydro-8-oxoguanine (8-oxoG), with roughly 100,000 8-oxoG lesions formed per rat cell per day (1). DNA polymerases incorporate adenine bases opposite 8-oxoG, generating G:C → T:A transversion mutations, the second most common somatic mutation in human cancer. 8-OxoG:C base pairs are repaired by elaborate defense systems in nearly all organisms. In humans, the 8-oxoguanine DNA glycosylase enzyme (hOGG1; ref. 2) catalyzes the excision of the 8-oxoG base from a duplex and cleavage of the DNA backbone. hOGG1 recognizes 8-oxoG by forming a hydrogen bond between the protonated N7 atom in the damaged base and a main-chain carbonyl on the enzyme. The analogous enzyme in bacteria, MutM, is structurally unrelated to hOGG1 (3) and was shown to make base-specific contacts with the “estranged” base opposite the lesion (4). The impact of 8-oxoG lesions on DNA topoisomerases, enzymes necessary for the relaxation of DNA superhelical tension throughout the genome, is not expected to result from the specific recognition of damaged sites, but rather from random contacts made between such enzymes that act genomewide and sites of DNA damage.
Human topoisomerase I is vital for most cellular processes involving DNA, including replication, transcription, and recombination, and it is the sole target of the camptothecin anticancer drugs (5). Human topoisomerase I breaks one strand of duplex DNA and relaxes superhelical tension by a postulated “controlled rotation” mechanism (6). The 765-aa enzyme is composed of four major regions: a highly charged and putatively unstructured N-terminal domain (residues 1–200; removed for structural studies), a conserved core domain (residues 201–635), a flexible linker domain (residues 636–712), and a conserved C-terminal domain (residues 713–765) that contains the catalytic Tyr-723 residue (7). Several crystal structures of human topoisomerase I in both covalent and noncovalent complexes with DNA have been reported (6, 8–10). These structures revealed that the core domain is composed of CAP (residues 202–434) and CAT (residues 435–635) regions that wrap completely around the DNA duplex, positioning the active site around the scissile phosphate group. The active site contains the catalytic tyrosine (Tyr-723), two arginines (Arg-488, Arg-590), one histidine (His-632), and one lysine (Lys-532) (6, 8, 10).
The CAT region of human topoisomerase I is structurally similar to the tyrosine recombinase family of DNA integrases and recombinases from bacteria and phage (8). These recombinases and integrases also use an active site made up of a tyrosine, two arginines, a histidine (or tryptophan), and a lysine (8, 11–14). In the crystal structures of human topoisomerase I-DNA complexes (6, 8–10), the catalytic tyrosine (or inactive phenylalanine in noncovalent complexes) is positioned adjacent to the scissile DNA phosphate group, ready for catalysis. In the tyrosine recombinase family, however, the catalytic tyrosine has been observed docked up to 27 Å away from the remaining active-site residues (13). This alternative positioning of the active-site nucleophile has been interpreted as a regulatory mechanism used by these enzymes to prevent catalysis until the DNA substrate binds properly to the enzyme (13).
Several types of DNA damage impact the catalytic activity of human topoisomerase I (15). In particular, 8-oxoG lesions at the +1 position (just downstream) relative to the site of cleavage distinctly affect the action of human topoisomerase I (16). Single-turnover DNA cleavage assays with suicide substrates revealed that strand cleavage activity by human topoisomerase I is decreased in the presence of an 8-oxoG lesion whereas topoisomerase I binding to DNA is enhanced (16). To unravel the structural basis of the impact of 8-oxoG on human topoisomerase I, we determined the crystal structure of a noncovalent complex of the enzyme with a 22-bp DNA oligonucleotide containing an 8-oxoG base at the +1 position on the scissile strand. We find the active site Tyr-723–Phe residue is docked into the interior of the protein, 7.4 Å away from the scissile phosphate group. This positioning of the catalytic residue is reminiscent of the inactive conformations of the tyrosine recombinases and integrases and may represent a central intermediate on the path toward DNA cleavage and relaxation by human topoisomerase I.
Materials and Methods
Protein Purification and DNA Oligonucleotides.
N-terminally truncated human topoisomerase I (70 kDa, residues 175–765) containing a Tyr-723–Phe mutation was expressed by using baculovirus in Spodoptera frugiperda (Sf9) insect cells (7). The inactive Tyr-723–Phe mutant form of the enzyme was used to produce a noncovalent complex with DNA. Single-stranded DNA oligonucleotides were purchased in reverse-phase HPLC purity from Oligos Etc. (Midlands, TX) and annealed as described (6) to create the following 22-bp DNA duplex (16):
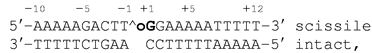 |
where ∧ indicates the weakly preferred cleavage site (17), and the bold oG indicates the position of the 8-oxoG base. This sequence has proven useful for previous studies of human topoisomerase I (6–10, 18–22).
Crystallization and Data Collection.
Crystals of N-terminally truncated Tyr-723–Phe mutant form of human topoisomerase I in noncovalent complex with duplex DNA were grown by sitting drop vapor diffusion at 22°C as described (6). Crystals were cryoprotected at 22°C in crystallant containing 20% ethylene glycol before flash-cooling in liquid nitrogen and belong to space group P21 with one protein–DNA complex in the asymmetric unit. Diffraction data were collected at 100 K at Brookhaven National Laboratory, Upton, NY (beamline X12B) from a single crystal and were processed and reduced by using denzo and scalepack (23).
Structure Determination and Refinement.
The structure was determined by molecular replacement (amore; ref. 24) using the structure of a human topoisomerase I in complex with DNA as a search model (Protein Data Bank ID code ) (6). Model adjustments were performed by using o, and the structure was refined by using torsion angle dynamics in cns (25). Residues 202–633 and 641–765 of the enzyme and all 22 bp of the DNA duplex are present in the final model. The 8-oxoG base and the novel positioning of the active site Tyr-723–Phe residue were placed by using composite annealed omit maps. The Tyr-723–Phe residue exhibits thermal displacement parameters of 63 Å2, similar to that exhibited by the protein region of the structure (Table 1). Late in refinement, bulk solvent and overall anisotropic B-factor corrections were added, as well as 26 well-ordered waters. The final model exhibits good geometry (Table 1) and no Ramachandran outliers (26). Figures were generated with molscript (28), RASTER3D (29), and bobscript (27).
Table 1.
Crystallographic refinement statistics
| Resolution, Å (highest shell) | 20–3.14 (3.19–3.14) |
| Space group | P21 |
| Unit cell dimensions | a = 57.2, b = 122.5, c = 72.0, β = 97.4° |
| No. of total reflections | 38,120 |
| No. of unique reflections | 17,165 |
| Mean redundancy | 2.22 |
| Rsym, % (highest shell) | 5.9 (30.0) |
| Completeness, % (highest shell) | 95.6 (90.5) |
| Mean I/σ (highest shell) | 11.9 (2.7) |
| Rcryst | 0.258 |
| Rfree | 0.300 |
| rms deviation | |
| Bond length, Å | 0.00821 |
| Bond angles, degree | 1.41 |
| Dihedral angles, degree | 21.5 |
| Improper angles, degree | 1.02 |
| No. of protein atoms | 4,457 |
| Mean B value, protein atoms, Å2 | 59.0 |
| No. of DNA atoms | 896 |
| Mean B value, DNA atoms, Å2 | 45.3 |
| No. of solvent atoms | 26 |
| Mean B value, solvent atoms, Å2 | 27.5 |
Results
Overall Structure.
The crystal structure of a 70-kDa form of human topoisomerase I in noncovalent complex with a 22-bp duplex DNA oligonucleotide containing 8-oxoG at the +1 position on the scissile strand (the 8-oxoG structure) was refined to 3.1 Å and crystallographic R and cross-validating Rfree values of 0.258 and 0.300, respectively (Table 1). While this structure of topoisomerase I is in complex with a site of DNA damage, it is similar overall to the human topoisomerase I–DNA complexes reported previously (Fig. 1, Table 2) (6, 9, 10). Maximum shifts between protein domains of 1.3 Å for the CAP, 3.0 Å for the linker domain, and 3.6 Å for the C-terminal domain are observed between the 8-oxoG structure and the complex of human topoisomerase I with nondamaged DNA (Fig. 1). These values are within the limits of flexibility described for human topoisomerase I protein–DNA complexes (9).
Fig 1.

Crystal structure of human topoisomerase I in complex with DNA containing an 8-oxoG lesion. Superimposed (using 544 Cα atoms) in gray is the structure of human topoisomerase I in complex with nondamaged DNA (Protein Data Bank ID code ; ref. 6) The DNA duplexes from three other human topoisomerase I structures are also shown.
Table 2.
rms and distance deviations between the 8-oxoG structure and the structure of the 70-kDa form of human topoisomerase I in noncovalent complex with DNA
| Deviation | Residue(s) | rmsd/distance, Å |
|---|---|---|
| rmsd | ||
| All nonsolvent atoms | 1.7 | |
| All protein atoms | 1.8 | |
| CAP protein atoms | 202–434 | 1.8 |
| CAT protein atoms | 435–633 | 1.3 |
| Linker protein atoms | 641–712 | 3.1 |
| C-term protein atoms | 713–765 | 1.4 |
| All DNA atoms | 0.87 | |
| P positions | 0.86 | |
| Distance | ||
| Active site residues | Tyr-723F (CZ) | 7.4 |
| Arg-488 (NH1) | 1.2 | |
| Arg-590 | Not observed | |
| Lys-532 (NZ) | 1.3 | |
| His-632 (CE1) | 1.3 |
rmsd, rms deviation. Protein Data Bank ID code (6).
The 8-oxoG base in this structure was built into clear electron density at the +1 position in the scissile strand by using composite annealed omit maps. A small number of changes are observed in the overall protein–DNA contacts in the 8-oxoG structure relative to previous topoisomerase I-DNA complexes (7–10): interactions between DNA phosphates and His-367, Lys-374, Thr-411, Ala-489, and Asn-491 are lost, but new interactions are formed with Gln-578 and Lys-493. Each change is at least 15 Å from the 8-oxoG base and thus are likely to reflect simple variability in enzyme–DNA contacts and not structural distortions caused by the DNA lesion. The O8 atom on the 8-oxoG base forms no polar or nonpolar contacts with the protein. The closest protein side chain is Asn-722, which places its Nδ2 atom 3.9 Å from O8 on the 8-oxoG base. We find that the Asn-722 side chain can be modeled such that its Nδ2 atom is 3.4 Å from the O8 atom. Thus, during the DNA binding phase of catalysis, Asn-722 could form a hydrogen bond with the 8-oxoG base. The Asn-722 residue does not make this interaction in our current structure, however. Instead, its Nδ2 atom is involved in a 3.2-Å hydrogen bond with the main-chain carbonyl oxygen of Thr-718.
Impact of 8-OxoG on the Topoisomerase I Active Site.
Although the overall structure of the 8-oxoG complex is similar to previous topoisomerase I–DNA structures, the active-site region deviates significantly. When CAT regions of the 8-oxoG structure and the structure of topoisomerase I in complex with nondamaged DNA (6) are superimposed, several shifts in the active site of the 8-oxoG structure are evident (Fig. 2, Table 2). Moderate changes in side-chain positions (1.2–1.3 Å) occur for the active-site residues Arg-488, Lys-532, and His-632. The catalytic Tyr-723–Phe, however, undergoes a rotamer shift (described in detail below) and the Arg-590 side chain becomes disordered and is not observed. Arg-590 helps to neutralize the charge on the scissile DNA phosphate group, likely facilitating nucleophilic attack by the catalytic Tyr-723 residue (6, 10). These observations suggest that the positioning of the Tyr-723 side chain adjacent to the scissile phosphate is required for proper Arg-590 ordering at the active site.
Fig 2.

(A) Human topoisomerase I active-site residues in the 8-oxoG structure with the 8-oxoG base (8OG) shown. Superimposed in transparent gray is the Protein Data Bank ID code structure (6). (B) Stereoview of the active site of the 8-oxoG structure in the same orientation as A, showing 3.1-Å resolution 2Fobs − Fcalc electron density (contoured at 1σ).
The Tyr-723–Phe side chain in the 8-oxoG structure undergoes a 7.4-Å shift in position relative to previous structures and packs by using a preferred rotamer into a novel cavity within the body of the enzyme (Table 2; Fig. 3). Difference density in composite annealed Fobs − Fcalc omit maps, as well as density in initial 2Fobs − Fcalc maps, provided clear evidence for placement of this side chain in its new conformation (Fig. 3A). Tyr-723–Phe in this orientation packs well within a novel cavity formed between the CAT and C-terminal domains and makes van der Waals contacts with several residues, including Ala-594, Thr-718, Ser-719, and Leu-724 (Fig. 3B). As a tyrosine, this side chain could also form two hydrogen bonds within this pocket. First, a 2.8-Å hydrogen bond could form between the phenolic oxygen of the tyrosine and the main-chain carbonyl oxygen of Val-626. Second, if the side chain of Ser-719 is shifted by 120° to a favored rotamer, its hydroxyl oxygen is 2.2 Å from the phenolic oxygen of Tyr-723. Both Val-626 and Ser-719 are completely conserved in the type IB topoisomerases of known sequence (30). Thus, given small structural shifts to accommodate the tyrosyl hydroxyl group, it is likely that the catalytic side chain of human topoisomerase I can pack into the interior of the enzyme and be stabilized by hydrogen bonds. This structure is distinct from the topoisomerase I–DNA complexes determined to date; in all previous structures, the Tyr-723–Phe residue is positioned ready for catalysis (e.g., Fig. 2A).
Fig 3.

(A) Catalytic Tyr-723–Phe residue before (white; from Protein Data Bank ID code ) and after (red) repositioning, with composite annealed omit Fobs − Fcalc density (3.1-Å resolution). (B) The buried Tyr-723-Phe in the 8-oxoG structure. The tyrosyl hydroxyl, if it were present, could form two hydrogen bonds (with Val-626 and Ser-719, if this residue were modeled in the preferred rotamer position shown).
Creating the Tyr-723–Phe Binding Pocket.
The CAT region and C-terminal domain of human topoisomerase I undergo subtle, but important, structural changes to create a binding pocket in the interior of the enzyme (Fig. 4A). The largest alterations occur in helices α16, α17 from the CAT, and α21 from the C-terminal domain. Thr-597 and Leu-602 in α16 shift by 2.4 and 1.5 Å (Cα-Cα), respectively; Val-626, Leu-629 and Cys-630 in α17 shift by 2.2, 1.8, and 1.7 Å, respectively; and Trp-732 in α21 shifts by 1.9 Å. The combined effect of these concerted shifts is to create the binding pocket for the Tyr-723–Phe side chain (Fig. 4A). The presence of the O8 atom on the 8-oxoG base appears to facilitate these conformational shifts. If the CAT and C-terminal regions of topoisomerase I remained unchanged, the CG atom of Thr-718 would clash sterically with the O8 atom (at 2.9 Å; Fig. 4A). In the 8-oxoG structure, however, the position of Thr-718 has shifted by 2.6 Å, safely distancing itself from the 8-oxoG base (now 4.3 Å away). The electron density is clear for this positioning and rotamer orientation of Thr-718, and we find that it cannot be modeled to form a hydrogen bond with the O8 atom on the 8-oxoG base.
Fig 4.

(A) Stereoview of changes required to form the interior binding pocket for Tyr-723–Phe (green, active; magenta, inactive). The 8-oxoG structure is in red and blue (+1 8-oxoG:C base pair in yellow), and the Protein Data Bank ID code structure (and A:T base pair) is in gray (6). (B) Stereoview of the superposition of the 8-oxoG structure (yellow) on that of λ-integrase (green; 171 equivalent Cα positions, rms deviation = 3.6 Å) revealing the shift in the catalytic residues for each structure.
The related tyrosine recombinase family of bacterial and phage integrases and recombinases has also been found to dock its catalytic tyrosines away from its remaining active-site residues (11–14). The λ-integrase places its catalytic residue 17.9 Å from the active site (Tyr-342 CZ—Arg-212 CZ), compared with 11.2 Å for this human topoisomerase I structure (Tyr-723-Phe CZ—Arg-488 CZ) (ref. 13; Fig. 4B). Such docked orientations are likely to represent inactive conformations used to limit catalytic activity until suitable substrates are bound. Other members of the tyrosine recombinase family dock their catalytic tyrosines away from their active sites at a range of distances: Flp recombinase at 27.2 Å, bacteriophage HP1 integrase at 11.2 Å, and XerD recombinase at 10.2 Å (Tyr CZ–Arg CZ) (11, 12, 14). It is expected that both human topoisomerase I and the tyrosine recombinases would have to reorganize their active sites to generate active conformations for catalysis. The use of a docked, inactive tyrosine conformation may represent a common regulatory mechanism for both type IB topoisomerases and tyrosine recombinases.
Discussion
We have shown a crystal structure of a DNA topoisomerase in the presence of a site of DNA damage. The structure reveals a unique conformation at the human topoisomerase I active site: the catalytic Tyr-723–Phe residue in this noncovalent complex is rotated 64° away from the scissile phosphate and is docked into an interior binding pocket (Figs. 3B and 4A). In addition, the side chain of Arg-590 is disordered (Fig. 2A), suggesting that when the catalytic Tyr-723–Phe residue is rotated away this adjacent active-site residue cannot adopt a stable conformation. The tyrosine recombinase family of bacterial phage integrases and recombinases also positions its catalytic tyrosines up to 27 Å away from its remaining active-site residues (11–14). These enzymes share a similar structure, active site, and catalytic mechanism of single-stranded DNA cleavage with the eukaryotic type I topoisomerases (7–10). Thus, the positioning of a catalytic tyrosine away from the active site may be a common mechanism used by both the type IB topoisomerases and the tyrosine recombinases to regulate catalytic activity. These results expand our understanding of the roles active-site flexibility play in regulating DNA manipulation and metabolism.
Pourquier et al. (16) have shown that the presence of an 8-oxoG base at the scissile +1 position decreases DNA cleavage by human topoisomerase I in single-turnover studies. We can explain this observation based on our crystal structure. The inactive docking of the catalytic residue away from the site of catalysis, as well as disorder in the active site, are likely to slow the DNA cleavage activity of the enzyme. The O8 atom of the 8-oxoG base also appears to clash sterically with Thr-718 in the position this residue occupies in all of the previous human topoisomerase I structures reported to date (Fig. 4A), forcing the enzyme to favor its inactive conformation in the presence of an 8-oxoG base. The CAT and C-terminal regions change in structure to accommodate the conformation of the Tyr-723–Phe residue, which fits within an interior pocket and could be stabilized by two hydrogen bonds (Figs. 3B and 4A). Thus, the presence of the 8-oxoG base appears to limit the ability of human topoisomerase I to assemble its active site before catalysis, slowing the observed rate of DNA cleavage by the enzyme.
Pourquier et al. (16) also report that human topoisomerase I has increased affinity for a DNA duplex containing 8-oxoG at the scissile +1 position. We find that the Asn-722 side chain of human topoisomerase I can be modeled to form a potential hydrogen bond with the O8 atom of the 8-oxoG base (although in the current structure this hydrogen bond is not made). It is likely that at the DNA binding stage of the topoisomerase I catalytic cycle Asn-722 hydrogen bonds with the DNA phosphate backbone or with an 8-oxoG base in a damaged DNA molecule. Mutagenesis studies support the importance of polar interactions between the side chain immediately N-terminal to the catalytic tyrosine and the DNA substrate. An Asn-726–His mutation in Saccharomyces cerevisiae topoisomerase I (equivalent to Asn-722 in the human enzyme) or Asn-722–His in human topoisomerase I generate enzymes with increased equilibrium cleavage activity relative to wild-type protein on nondamaged DNA substrates (16, 31). The yeast Asn-726–His mutant topoisomerase I shows a further 5-fold increase in cleavage activity in the presence of an 8-oxoG at the scissile +1 position (16). Thus, the amino acid adjacent to the catalytic tyrosine in topoisomerase I is likely to form critical interactions with the DNA that enhance catalysis.
Based on our structural results, we propose the following model for the initial stages of human topoisomerase I catalytic activity (Fig. 5). Human topoisomerase I may exist in an inactive conformation when not bound to DNA, with its catalytic Tyr-723 residue buried in a manner similar to that observed in the 8-oxoG structure reported here. We suggest that upon binding duplex DNA human topoisomerase I remains in the catalytically inactive state with the active site Tyr-723 buried. Once the enzyme has oriented itself in a productive manner on the DNA substrate, a conformational change in the enzyme occurs in which the catalytic tyrosine rotates away from its buried position, finalizing the formation of the active site and facilitating the ordering of Arg-590. This conformation of human topoisomerase I, seen in many previously described structures, is the active state of the enzyme that is capable of catalyzing the single-strand DNA cleavage and formation of the 3′ phosphotyrosine protein–DNA intermediate. The O8 atom of 8-oxoG appears to inhibit the final reorganization of the enzyme's active site perhaps because of a steric clash with Thr-718.
Fig 5.

Proposed mechanism for the initial stages of human topoisomerase I activity. The enzyme's inactive conformation contains a docked catalytic tyrosine (Y in red), while its active conformation (Y in green) is ready for catalysis.
An alternative interpretation of our structural data is that 8-oxoG induces the formation of an inactive conformation of human topoisomerase I that is not part of this enzyme's normal catalytic cycle. However, the conservation of the two potential hydrogen bonding partners for Tyr-723 within the interior binding pocket suggests that those residues may be important for the enzyme's action within the cell. A mutation of Ser-719, one of these hydrogen bonding partners, to alanine would be useful in examining the physiological relevance of these potential interactions. If topoisomerase I only uses the interior binding pocket in the presence of 8-oxoG lesion, one would expect that a Ser-719–Ala mutation would have a greater impact on the action of the enzyme on 8-oxoG-containing DNA substrates than on nondamaged substrates.
It is likely the distortions within the DNA duplex caused by other sites of DNA damage adjacent to the strand cleavage site will cause similar structural impact on human topoisomerase I and decrease catalytic activity. Indeed, several forms of DNA damage, including etheno-adenine, Ara-C, uracil incorporation, mismatches, and abasic sites (15, 20–22), inhibit human topoisomerase I. In summary, the type IB topoisomerases appear to join the tyrosine recombinases in using docked catalytic tyrosines to regulate activity in the absence of ideal DNA substrates. Further, it may be possible to exploit this inactive form of human topoisomerase I in the development of novel therapeutics useful for treating human disease.
Acknowledgments
We thank H. Kim for help with data collection, G. Pielak for comments on the manuscript, and S. Bencharit, R. Watkins, and the members of the Redinbo Laboratory for assistance in creating figures.
Abbreviations
8-oxoG, 7,8-dihydro-8-oxoguanine
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code ).
References
- 1.Park E.-M., Shigenaga, M. K., Degan, P., Korn, T. S., Kitzler, J. W., Wehr, C. M., Kolachana, P. & Ames, B. N. (1992) Proc. Natl. Acad. Sci. USA 89, 3375-3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruner S. D., Norman, D. P. & Verdine, G. L. (2000) Nature (London) 403, 859-866. [DOI] [PubMed] [Google Scholar]
- 3.Sugahara M., Mikawa, T., Kumasaka, T., Yamamoto, M., Kato, R., Fukuyama, K., Inoue, Y. & Kuramitsu, S. (2000) EMBO J. 19, 3857-3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fromme J. C. & Verdine, G. L. (2002) Nat. Struct. Biol. 9, 544-552. [DOI] [PubMed] [Google Scholar]
- 5.Champoux J. J. (2002) Annu. Rev. Biochem. 70, 369-413. [DOI] [PubMed] [Google Scholar]
- 6.Stewart L., Redinbo, M. R., Qiu, X., Hol, W. G. & Champoux, J. J. (1998) Science 279, 1534-1541. [DOI] [PubMed] [Google Scholar]
- 7.Stewart L., Ireton, G. C. & Champoux, J. J. (1996) J. Biol. Chem. 271, 7602-7608. [DOI] [PubMed] [Google Scholar]
- 8.Redinbo M. R., Stewart, L., Kuhn, P., Champoux, J. J. & Hol, W. G. (1998) Science 279, 1504-1513. [DOI] [PubMed] [Google Scholar]
- 9.Redinbo M. R., Stewart, L., Champoux, J. J. & Hol, W. G. (1999) J. Mol. Biol. 292, 685-696. [DOI] [PubMed] [Google Scholar]
- 10.Redinbo M. R., Champoux, J. J. & Hol, W. G. (2000) Biochemistry 39, 6832-6840. [DOI] [PubMed] [Google Scholar]
- 11.Guo F., Gopaul, D. N. & van Duyne, G. D. (1997) Nature (London) 389, 40-46. [DOI] [PubMed] [Google Scholar]
- 12.Subramanya H. S., Arciszewska, L. K., Baker, R. A., Bird, L. E., Sherratt, D. J. & Wigley, D. B. (1997) EMBO J. 16, 5178-5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwon H. J., Tirumalai, R., Landy, A. & Ellenberger, T. (1997) Science 276, 126-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y., Narendra, U., Iype, L. E., Cox, M. M. & Rice, P. A. (2000) Mol. Cell 6, 885-897. [PubMed] [Google Scholar]
- 15.Pourquier P. & Pommier, Y. (2001) Adv. Cancer Res. 80, 189-216. [DOI] [PubMed] [Google Scholar]
- 16.Pourquier P., Ueng, L. M., Fertala, J., Wang, D., Park, H. J., Essigmann, J. M., Bjornsti, M. A. & Pommier, Y. (1999) J. Biol. Chem. 274, 8516-8523. [DOI] [PubMed] [Google Scholar]
- 17.Been M. D., Burgess, R. R. & Champoux, J. J. (1984) Nucleic Acids Res. 12, 3097-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stewart L., Ireton, G. C. & Champoux, J. J. (1997) J. Mol. Biol. 269, 355-372. [DOI] [PubMed] [Google Scholar]
- 19.Stewart L., Ireton, G. C. & Champoux, J. J. (1999) J. Biol. Chem. 274, 32950-32960. [DOI] [PubMed] [Google Scholar]
- 20.Pourquier P., Ueng, L. M., Kohlhagen, G., Mazumder, A., Gupta, M., Kohn, K. W. & Pommier, Y. (1997) J. Biol. Chem. 272, 7792-7796. [DOI] [PubMed] [Google Scholar]
- 21.Pourquier P., Bjornsti, M. A. & Pommier, Y. (1998) J. Biol. Chem. 273, 27245-27249. [DOI] [PubMed] [Google Scholar]
- 22.Pourquier P., Takebayashi, Y., Urasaki, Y., Gioffre, C., Kohlhagen, G. & Pommier, Y. (2000) Proc. Natl. Acad. Sci. USA 97, 1885-1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otwinowski Z. & Minor, W. (1997) Methods Enzymol. 276, 307-326. [DOI] [PubMed] [Google Scholar]
- 24.Navaza J. (2001) Acta Crystallogr. D 57, 1367-1372. [DOI] [PubMed] [Google Scholar]
- 25.Brunger A. T., (1992) x-plor Manual (Yale Univ. Press, New Haven, CT), Version 3.1.
- 26.Laskowski R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) J. Appl. Crystallogr. 26, 283-291. [Google Scholar]
- 27.Esnouf R. M. (1999) Acta Crystallogr. D 55, 938-940. [DOI] [PubMed] [Google Scholar]
- 28.Kraulis P. J. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 29.Merritt E. A & Bacon, D. J. (1997) Methods Enzymol. 277, 505-524. [DOI] [PubMed] [Google Scholar]
- 30.Esposito D. & Scocca, J. J. (1997) Nucleic Acids Res. 25, 3605-3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woo M. H., Vance, J. R., Otero Marcos, A. R., Bailly, C. & Bjornsti, M.-A. (2002) J. Biol. Chem. 277, 3813-3822. [DOI] [PubMed] [Google Scholar]