

Abstract
We used a combination of bioinformatics, electron cryomicroscopy, and biochemical techniques to identify an oxidoreductase-like domain in the skeletal muscle Ca2+ release channel protein (RyR1). The initial prediction was derived from sequence-based fold recognition for the N-terminal region (41–420) of RyR1. The putative domain was computationally localized to the clamp domain in the cytoplasmic region of a 22Å structure of RyR1. This localization was subsequently confirmed by difference imaging with a sequence specific antibody. Consistent with the prediction of an oxidoreductase domain, RyR1 binds [3H]NAD+, supporting a model in which RyR1 has a oxidoreductase-like domain that could function as a type of redox sensor.
During excitation–contraction coupling in skeletal muscle, Ca2+ is released from the lumen of the sarcoplasmic reticulum (SR) via the Ca2+ release channel, also known as the ryanodine receptor, RyR1. In skeletal muscle, the Ca2+ release channel is physically coupled to the L-type voltage dependent Ca2+ channel dihydropyridine receptor (DHPR), such that a depolarization induced change in the conformation of DHPR induces the opening of ryanodine receptor 1 (RyR1). This leads to an increase of cytoplasmic Ca2+, triggering a sequence of events that lead to muscle contraction. RyR1 is a homotetramer (1) whose subunits are ∼565 kDa (e.g., human, 5,038 residues; rabbit, 5,037 residues) (2, 3). Mutations in three domains of this protein, one of which is between amino acids 35 and 614, have been implicated in the pathogenesis of two human diseases, malignant hyperthermia and central core disease (4, 5).
The Ca2+ release channel exists in at least two functional states, opened and closed (6), which likely have conformational differences. The low-resolution structures of the Ca2+ release channel in different functional states have been studied extensively by electron cryomicroscopy (7–9). On opening, a number of structural changes occur in several regions of the channel, including both the clamp-like domains in the cytoplasmic region and the transmembrane domain. The clamp domains are the most likely candidates for interaction with DHPR (8) and must, therefore, be allosterically coupled to the transmembrane domain in order for DHPR to induce the opening of the Ca2+ permeable pore of RyR1. Here we describe a unique approach for identification of new functional and structural domains of this complex protein.
Methods
Sequence Analysis.
Initial motif searching in the primary sequence of rabbit RyR1 (P11716) was done by using proscan (10) with a threshold of 70%. Subsequently, 500-residue consecutive, serial sequence segments of the RyR1 were submitted to the University of California, Los Angeles–Department of Energy (UCLA–DOE) Fold recognition server (11). Primary sequence alignments were performed by using CLUSTALW (Gonnet weight matrix) with a Gonnet Pam250 positive-value similarities scoring system (12, 13). Additionally, multiple sequence alignments were done with other RyR sequences. As sequence identity in this region is extremely high, only rabbit RyR1 is shown.
Sample Preparation.
RyR1 was purified from the rabbit skeletal muscle SR membranes as described (14).
Electron Cryomicroscopy and Image Processing.
Purified RyR1 in the “closed conformation” in the presence of 1 mM EGTA (free Ca2+ < 10 nM) was prepared for electron cryomicroscopy (15) and examined in a JEOL1200 electron microscope operated at 100 kV (9). Images were recorded on Kodak SO-163 film at a nominal magnification of ×40,000. The micrographs were digitized by using a Zeiss SCAI scanner with a step size of 14 μm. A total of 7,300 particle images were selected from 10 micrographs. Single particle reconstruction with complete amplitude and phase correction of the contrast transfer function was performed in EMAN (16). A resolution of ∼22 Å using the standard 0.5 Fourier shell correlation criterion was calculated. Note that our earlier publications (7–9) used the 3σ resolution criteria which would have yielded ∼19 Å.
Homology Modeling.
A homology model of the N-terminal domain of RyR1 (residues 41–420) was constructed in INSIGHTII with the Homology and Modeler packages (Accelrys, San Diego) using 4ICD and three related oxidoreductases, 9ICD (18), 1IDE (19), and 1GRO (20). rms deviation (rmsd) between the homology models and templates were calculated (<1 Å rmsd) using the “magic fit” option in the SWISSPDB VIEWER (21). A 20 Å resolution density model (1283 voxel map, 5.25 Å per pixel) of the homology modeled domain was created by using pdb2mrc (16).
Fold Localization.
FOLDHUNTER was initially run to localize the modeled domain to RYR1 with an angular step size of 10°, where a minimum of ∼12° is required for accurate localization at 22 Å. A refinement of the FOLDHUNTER, using the “smart” option, was done in a section corresponding to one of the four equivalent subunits of the closed state structure. Visualization of the fitting was done by using IRIS EXPLORER (NAG, Downers Grove, IL).
Antibody Labeling and Difference Imaging.
The sequence-specific antibody against synthetic peptide with sequence KGLDSFSGKPRGSGPPAGP corresponding to residues 416–434 of the RyR1 coupled to keyhole limpet hemocyanin was produced in rabbits by Pel Freeze Biologicals (Rogers, AR). The antibody was purified using a protein A affinity column (Pierce Endogen) according to manufacturer's protocol and an antigenic peptide-affinity column (22). The antibodies were characterized by ELISA assays and Western blotting analysis with SR membranes and purified RyR1.
RyR1/antibody immunocomplexes were prepared by incubating purified RyR1 (0.2 mg/ml) with purified IgG (0.1 mg/ml) at 1:12 molar ratio in the presence of EGTA, to minimize possible functional transitions of the channel and to stabilize the resultant complexes, predominantly in closed conformation (7). The mixture was allowed to incubate overnight at 4°C. Only images with the defocus values within the narrow range of 2.0–2.2 μm underfocus as determined by EMAN (16) were used for image processing.
Approximately 1,000 antibody-labeled particle images were boxed and analyzed. Only “top views” of RyR1 (views along the 4-fold axis) were extracted from the data set and subjected to an iterative procedure comprised of multivariate statistical analysis followed by classification and image-averaging. Projection images were generated from our previously determined three-dimensional reconstruction of the RyR1 in its closed state and were used as references in the multireference alignment procedure using IMAGIC. The top views were extracted by using projection matching techniques in EMAN. Images of RyR1 in the presence of only EGTA were processed the same way as images of RyR1/IgG complexes. A difference map was calculated by subtracting densities in the average image of the control sample from the average image of the RyR1/antibody complex. The position of the bound antibody was identified from the positive density differences.
To assess the statistical significance of differences between two averaged images of RyR1/antibody and RyR1, t values associated with each picture element in the difference map were calculated by using Eq. 1, where d̄ is the mean density of RyR1 with and without antibody (±), N is the number of images in each set, S is the standard deviation (i refers to the picture element).
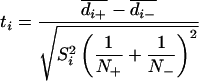 |
This t map was contoured and interpreted with reference to a table of t distribution critical values. The differences were considered to be significant at the confidence level greater than 98% (i.e., random chance is <0.02) (23–25).
[3H]NAD+ Binding.
SR membranes [20 μg in 200 μl of 300 mM NaCl/1 mM EGTA/1.2 mM CaCl2/50 mM Mops, pH 7.4/100 μg/ml BSA/0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)] were incubated with 50 nM [3H]NAD+ and with increasing concentrations of unlabeled NAD+ ranging from 0 to 1 mM to determine maximal [3H]ryanodine binding to RyR1. The samples were incubated for 15 h at room temperature, and bound radiolabel was determined by filtration using Whatman GF/F filters. The data were transformed to generate a Scatchard plot. The dissociation rate constant was determined by a plot of ln(B/B0), where B is the amount bound at a given time t and B0 is the amount bound before initiation of dissociation by the addition of 1 mM unlabeled NAD+ to samples that had been incubated for 15 h with [3H]NAD+. For measuring the rate of association, [3H]NAD+ samples were incubated with 50 nM [3H]NAD+ and filtered at the indicated times after the addition of radioligand. The association rate constant was calculated from the equation kon = (kobs − koff)/[L], where kon is the association rate constant, koff is the dissociation rate constant, [L] is the concentration of [3H]NAD+ and kobs is the slope obtained from a plot of ln(Be/(Be − Bt)) where Be is the amount bound at equilibrium and Bt is the amount bound at time t.
In the FKBP12-pulldown assays, multiple replicate tubes containing either CHAPS solubilized membranes (80 μg protein or purified RyR1 (<1 μg) and 20 nM [32P]NAD+ (30 Ci/mmol, New England Nuclear; 1 Ci = 37 GBq) in the above buffer were incubated for 5 h at room temperature. Nonlabeled NAD+ (1 mM) was used to define nonspecific binding. GST fused FKBP12 protein bound to glutathione affi-beads was added to the samples and the bound was separated from free by sedimentation. Samples were washed twice with 1 ml of 1.5% CHAPS/300 mm NaCl/50 mm Mops (pH 7.4), and counted in 5 ml of Beckman scintillation fluid
Results
Because of its large size, it is reasonable to expect a single subunit of RyR1 to be composed of multiple domains with distinct folds.
Sequence Analysis.
To identify potential domains within the sequence of RyR1, a search for related PROSITE sequence motifs, which uniquely identify a class or activity of proteins, was done by using proscan (10). A threshold of 70% was chosen which was set to detect distantly related PROSITE sequence motifs. All nonanimal motifs and motifs with high probability of random occurrences were excluded. Several dehydrogenase and NAD+/NADH oxidoreductase signatures, were identified in the N-terminal 1,300 residues of RyR1 (Table 1). These signatures, primarily localized into two regions, 50–500 and 700-1200, encompass both catalytic residues and binding sequences common to dehydrogenases and oxidoreductases, suggesting that RyR1 contains the necessary elements for enzymatic activity. These two regions could be two separate, autonomous domains; however, it is also possible that these two domains make up a large multidomain oxidoreductase, as in the case of xanthine oxidase (26) and bacterial methanol dehydrogenase (27). It is worth noting that, although not as diverse and densely packed, a few other dehydrogenase signatures are located in the last ∼2,000 aa of RyR1. Although the N-terminal residues likely correspond to the cytoplasmic region of RyR1, the C-terminal signatures are likely to be within the transmembrane and/or luminal portions of the channel.
Table 1.
Results of a motif search in the N-terminal region of RyR1
| Sequence motif | RyR1 residues | Similarity, % |
|---|---|---|
| Iron-containing alcohol dehydrogenase signature | 86–106 | 72 |
| Acyl–CoA dehydrogenases signature | 120–132 | 70 |
| Aldeyde dehydrogenase cys active site | 246–257 | 70 |
| Short chain dehydrogenase/reductase family signature | 398–426 | 70 |
| Short chain dehydrogenase/reductase family signature | 484–512 | 71 |
| Short chain dehydrogenase/reductase family signature | 699–727 | 70 |
| Acyl–CoA dehydrogenases signature 2 | 700–719 | 75 |
| Copper amine oxidase copper binding site signature | 731–744 | 72 |
| 2-Oxo acid dehydrogenases acyltransferase component lipoyl binding site | 936–965 | 71 |
| Zinc containing alcohol dehydrogenases signature | 1135–1149 | 74 |
| D isomer specific 2-hydroxyacid dehydrogenases NAD binding signature | 1191–1218 | 75 |
Structure Prediction.
A sequence-based fold-recognition (11) search was performed against known structures from the Protein Data Bank to identify potential structural homologues for these domains in RyR1. By screening large, serial, overlapping segments of the RyR1 primary sequence, a region near the N terminus of RyR1, amino acids 41–420, was found to have significant structural homology, a z score of 12.12 and 4.83 with the old and new UCLA–DOE fold recognition server, respectively, to phosphorylated isocitrate dehydrogenase, 4ICD (28). In both versions of the fold recognition server, it should be noted that 4ICD is the top scoring fold and above the threshold z score (>4.0 for the old scoring system). The N-terminal domain had a sequence identity of 19% and similarity of 51% to 4ICD over the aligned sequences (Fig. 1A). No other significant fold similarity was recognized in the regions where oxidoreductase signatures were found in PROSITE.
Fig 1.

N-terminal Domain of RYR1. (A) Sequence alignment between the 4ICD sequence and residues 41–420 of RyR1. The similarities are highlighted in red, and the green shading illustrates identical residues. The four prosite motifs (Table 1) are boxed in the sequence alignment. The IDH motif is also demarcated by a bar above the sequence alignment. The purple triangles represent residues involved in NADP+ binding of 4ICD. (B) Homology model for the RyR1 oxidoreductase domain and corresponding 20-Å density map. The rmsd between the 4ICD template and the model was less than 1 Å. (Scale bar represents ≈10 Å.)
4ICD is a α+β protein that belongs to the SCOP (29) family of isocitrate and isopropylmalate dehydrogenases. The members of this family belong to a larger group of oxidoreductases, which vary considerably in size and structure and generally use either NADP+ or NAD+ as the cofactor with a wide variety of substrates. Although identifications of sequence motifs and structure folds are independent of each other, some of the observed motifs (i.e., alcohol dehydrogenase, aldehyde reductase) and 4ICD have the same α/β/α core structure. A structural similarity to an oxidoreductase may suggest that RyR1 has a similar associated activity.
A second region of RyR1 sequence (residues 547-1192) was found to have a similar fold as 1B2N, methanol dehydrogenase (z score of 5.5), suggesting possible structural similarity to another oxidoreductase. Sequence similarity with this region is extremely low (<25%) and thus, no further analysis of this structure was done.
Of the identified N-terminal PROSITE sequence motifs, four motifs were contained within the 4ICD-like region of RyR1 (Fig. 1A). The first signature motif, the iron-containing alcohol dehydrogenase signature 2 (excluding the gapped region) is more than 50% similar. Although the gap within this region is substantial, the sequences are highly variable among ryanodine receptors and are also moderately variable within the members of isocitrate dehydrogenase/isopropylmalate dehydrogenases. Both the aldehyde dehydrogenase and short chain dehydrogenase signatures are ∼50% similar. Additionally, 12 of the 20 residues in the isocitrate and isopropylmalate dehydrogenase (IDH_IMDH) PROSITE pattern (Fig. 1A, bar) are similar to RyR1.
Further sequence analysis was done by analyzing the residues involved in cofactor binding of 9ICD, a structural isoform of 4ICD complexed with NADP+. Eleven residues, marked by triangles in Fig. 1A, show the residues in IDH responsible for interaction with the cofactor. Six of the eleven residues that coordinate NADP+ binding are similar between 4ICD and RyR1. It should be noted, however, that the family of IDH and IMDH binds multiple cofactors through interactions with varied residues, suggesting that the exact residues responsible for cofactor binding are not well conserved even within this family. Thus, the rudimentary similarity of this region of RyR1 to 4ICD and the associated sequence motifs may suggest RyR1 is capable of binding a cofactor, similar to NAD+/NADP+, and act as an oxidoreductase.
22 Å Structure of RyR1.
The structure of RyR1 in the closed state was determined to ∼22 Å resolution (Fig. 2A) by using electron cryomicroscopy (16). This map represents a slightly higher resolution structure than previous maps (7, 9, 30) and also includes the contrast transfer function correction. The map has the same overall shape and general dimensions as previously published lower-resolution structure of RyR1. However, the structural features are better refined, exhibiting a less hollow appearance and smaller protrusions in the clamp-shaped cytoplasmic subdomains.
Fig 2.

Computational localization of the N-terminal domain. (A) Top view of the ∼22 Å map of RyR1 tetramer. A clamp domain of one subunit is boxed. (Scale bar represents ∼100 Å.) (B) Localization of the homology model. The top four correlation peaks assigned by foldhunter (red) are found in the four clamp domains of the RyR1 reconstruction with identical angular positioning. The remaining top peaks, localized in equivalent positions, have less <5° angular difference from the top peaks. (C) Refined fitting of the N-terminal density (red) and model (ribbon) within the quarter of RyR1 tetramer (blue cage). A star indicates the C terminus of the modeled structure. (D) Side view of C.
Constructing a Homology Model.
In an attempt to translate the sequence information to the structure of RyR1, a model for the N-terminal region of RyR1 was generated based on the 4ICD structure as well as related members of the isocitrate/isopropylmalate SCOP family. As this model would be used to probe a relatively low-resolution electron cryomicroscopy map of RyR1 for a similar domain, no additional refinement and analysis was performed. A density model for the N-terminal domain with equivalent resolution was also generated (Fig. 1B).
Localizing the Structural Model.
A six-dimensional fitting program, FOLDHUNTER (31), was used to probe the entire channel structure for the best fit of the RyR1 N-terminal domain model. It assigned the N-terminal domain to the clamp region in the three-dimensional structure (8, 9) (Fig. 2B). This modeled domain also localized to the clamp domain of the previously determined (7) structure of open state RyR1 (not shown). A further refinement of the position of the N-terminal domain in the closed state was done by restricting the localization of the model domain to a single quarter of the channel. This fitting led to the final placement of the model in the clamp domain with its C terminus facing the cytoplasmic side of the closed channel (Fig. 2 C and D).
Antibody Labeling.
To confirm the location of this N-terminal region of RyR1, a sequence specific antibody to amino acids 416–434 was prepared. As seen in the computational localization of the ICD-like domain, these residues are likely exposed and thus amenable to antibody labeling. The antibody binds to both the full length RyR1 and the calpain cleaved N-terminal fragment, but not to the 410-kDa C-terminal fragment (Fig. 3A), demonstrating the specificity of the antibody. The intact channel was labeled with the antibody and imaged by using electron cryomicroscopy. The molecular envelope of the antibody is not fully resolved in the images of the labeled channel. A difference map, derived by subtracting the top views of the average image of RyR1 (control) from the average top view image of the RyR1/antibody complexes, shows regions of positive density within the clamp domain (Fig. 3C). Only the well-ordered densities are detectable in the difference map, probably because of conformational flexibility of the intact IgG molecule. However, we cannot exclude the possibility of only partial occupancy of antibody in the available RyR1 binding sites because of the binding conditions and interference of the detergent at a relatively high concentration (∼0.4% CHAPS).
Fig 3.

Localizing the N-terminal domain to RyR1. (A) SR proteins on a 5% SDS/PAGE (lane a) and Western blot of RyR1 with the antipeptide antibody (lane b). Shown are the positions of full-length RyR1 (band 1), and the two calpain-derived fragments (band 2 is the 410-kDa C-terminal fragment and band 3 is the 170-kDa N-terminal fragment). The specificity of the antibody for is shown by the lack of labeling of the 410-kDa fragment. (B) Individual channel particles labeled with the anti-peptide antibody (circled) in a representative micrograph region. The scale bar represents 300 Å. (C) Difference map between the average images of RyR1 and RyR1/antibody complex superimposed on the average image of RyR1/antibody complex. The red contour lines denote the difference map displayed at a positive density level (> 3× standard deviation of the difference, exceeding other differences by at least twofold). (Scale bar represents 150 Å.)
A statistical analysis of the difference map shows that the regions with the highest t values (significance level >98%) are located within the areas of highest positive densities in the difference map, reinforcing their statistical significance (23–25). Two other smaller regions of positive differences are detected in the central portion of the channel. Although these differences are also statistically significant, they are much smaller and are unlikely to correspond to the excess mass contributed by bound IgG. These differences may also suggest a subtle conformational change within the channel structure caused by antibody binding at the clamp. The assignment of the N-terminal domain is consistent with previous studies (32), which showed the N terminus of RyR3 tagged with GST localized to the clamp domains.
Oxidoreductase Cofactors in RyR1.
As the predicted fold for this domain suggested the possibility of enzymatic activity, a variety of potential cofactors were analyzed. NADH and NADPH had only minor effects on [3H]ryanodine binding to SR membranes. However, specific binding of [3H]NAD+ to SR membranes was detected. The inhibition of [3H]NAD+ binding with increasing concentrations of unlabeled NAD+ is shown in Fig. 4A, and a Scatchard plot is shown in Fig. 4B. [3H]NAD+ bound to SR membranes with an apparent dissociation constant (Kd) of 10 ± 2 μM and a Bmax of 650 ± 160 pmol/mg (n = 3). The rate of dissociation of [3H]NAD+ from SR membranes was extremely slow and the radioligand did not dissociate appreciably during the filtration step (Fig. 4C). Dissociation rates obtained by dilution of the bound radioligand (data not shown) were similar to those obtained by the addition of excess unlabeled ligand, suggesting an apparent lack of cooperatively in the binding. The low apparent affinity arises from a very slow rate of association such that at least 15 h were required to reach equilibrium (Fig. 4D). The calculated values for kon and koff for [3H]NAD+ were 3.2 × 104 min−1⋅M−1 and 1.7 × 10−3 min−1·M−1, to give a Kd of 53 nM. The Kd calculated from the kinetic constants is much lower than obtained from equilibrium binding, suggesting a complex interaction, possibly a ligand-induced conformational change in the binding site, which is currently being investigated. [3H]ryanodine bound to these same membranes with an apparent Kd of 9 ± 1 nM and a Bmax, the maximal amount of ligand bound, of 18 ± 4 pmol/mg (n = 3). If all of the [3H]NAD+ binding is to RyR1, there would be 37 ± 1 [3H]NAD+ binding sites per tetramer or approximately 10 sites per subunit.
Fig 4.

[3H]NAD+ binding to SR membrane. (A) The inhibition curve with membranes incubated using 50 nM [3H]NAD+ and increasing concentrations of unlabeled NAD+ ranging from 6 μM to 1 mM. (B) Scatchard plot using the plateau value as nonspecific binding (A). It was assumed that the affinity of the radioligand was identical to that of the unlabeled NAD+. (C) Dissociation initiated by the addition of 1 mM unlabeled NAD+ to samples preincubated with 50 nM [3H]NAD+. The extremely slow disassociation of [3H]NAD+ from SR membranes is shown. (D) Samples were incubated with 50 nM [3H]NAD+ and filtered at the indicated times after the addition of radioligand.
To determine whether the [32P]NAD+ is binding directly to RyR1, FKBP12 pulldown assays with CHAPS solubilized SR membranes (Fig. 5A) and sucrose gradient purified RyR1 (Fig. 5B) were performed (35). FKBP12 binds with high affinity and specificity to RyR1 and, when immobilized, can be used to purify RyR1 (35). In the pulldown of bound [32P]NAD+ from membranes (Fig. 5A), most of the radiolabel detected by filtration (RYR1 binds to filter but other proteins do not) is pulled down by the FKBP12 beads. The pulldown of the radiolabeled RYR1 is prevented by rapamycin, a drug that blocks FKBP12 binding to RyR1, and is not seen with beads without FKBP12. In addition, purified RyR1 was incubated with [32P]NAD+ and GST–FKBP12 affi-resin was used to pull down the solubilized RyR1 (Fig. 5B). A significant fraction of the bound [32P]NAD+ is pulled down with RyR1, but no radiolabel was pulled down by GST-beads without FKBP12. The amount of radiolabel associated with the beads was consistent with apparent affinity from the membrane binding assays and with the amount of RyR1 and [32P]NAD+ used in these assays. The radiolabel associated with the beads was greatly decreased by the presence of rapamycin but not by AMP–PCP. The [32P]NAD+ is, therefore, not binding to the ATP binding site of RyR1.
Fig 5.

FKBP12 pull-down assays of [32P]NAD+ labeled RyR1. (A) Pull-down with CHAPS solubilized membranes. SR membranes (80 μg, 1 pmol in 200 μl per assay) were incubated with 20 nM [32P]NAD+ and then either filtered through Whatman GF/F filters (a) or incubated with 50 μl of FKBP12 affibeads for 30 min. Triplicate samples were also incubated with buffer alone (b), 5 μM rapamycin (c), or with beads without FKBP12 (d). (B) Pull-down with sucrose gradient purified RyR1 incubated with 20 nm [32P]NAD+ for 5 h. Radioactivity pulled down with FKBP12 affibeads (a), pulled down by FKBP12 affibeads in the presence of 50 μM rapamycin (b), pulled down with FKBP12 affibeads in the presence of 1 mM AMP-PCP (c), or pulled down with GST fused glutathione affi-beads in the absence of FKBP12 (d).
Discussion
This analysis of RyR1 raises the possibility that it has an enzymatic domain. However, the exact nature and function of this domain, including substrates and cofactors, have yet to be elucidated.
Oxidoreductase Domain in K+ Channel.
RyR1 is not the first ion channel predicted to have an oxidoreductase domain. The structure of the β subunit of Shaker voltage-gated K+ channel (1QRQ) (33) has been shown to closely resemble human aldo–keto reductase (1RAL) (34), a prototypical oxidoreductase. However, neither a substrate for the putative enzymatic activity nor a functional role for the oxidoreductase activity has yet been identified. NADPH was found in the crystal structure at a position equivalent to the position of the NADPH in the human aldo–keto reductase. Similarity between the β subunit of the K+ channel and the human aldo–keto reductase to the RyR1 N-terminal domain (44% for both) was evident, further suggesting a structural and functional similarity of the N-terminal domain of RyR1 to other oxidoreductases (not shown).
By scanning the primary sequence of RyR1 for known sequence motifs at 70% similarity, the goal was not to explicitly assign functionality based on motif similarity, but to identify candidate regions for further investigation. The occurrence of multiple oxidoreductase signatures provided a region of interest that was subsequently examined by using fold recognition. The results from fold recognition, two implementations of the UCLA fold recognition server, corroborated the motif search, in that the N-terminal domain of RyR1 indeed had structural and possibly functional similarity to an oxidoreductase. Additionally, it should be noted that using the same fold recognition methods with the sequence of the K+ channel β subunit, the structurally similar 2ALR is identified with a z score of 5.03. This finding suggests that the z score of 4.83 for the 4ICD-like domain in RyR1 represents a rather trustworthy prediction as a structural homologue. Although 4ICD is capable of binding NADP+, the degeneracy in the binding residues within this family of proteins may suggest that RyR1 does not necessarily bind NAD+ through the same residues.
Although it is likely that this N-terminal domain of RyR1 contains an oxidoreductase-like structure/function, an additional possibility is that this 4ICD-like domain only represents a portion of the protein required for oxidoreductase activity. A motif search shows that the oxidoreductase signatures actually extend beyond the first 500 residues of RyR1 to a second region from ∼700–1200. Although this second domain may represent another oxidoreductase domain, it is equally possible that this region in conjunction with the 4ICD-like domain forms a protein like xanthine oxidase, a multicomponent oxidoreductase. However, because of the large size of this domain, fold recognition is incapable of accurately predicting the entire structure of the N-terminal domain. Thus, the 4ICD-like domain may only represent a discrete structural and functional subunit.
Although the presence of a putative oxidoreductase-like domain associated with an ion channel has only been seen in RyR1 and the K+ channel β subunit, there is a possibility of similar domains in other types of ion channels. Based on the relatively high conservation and the presence of similar sequence motifs in the N-terminal domains of RyRs and IP3Rs, we surmise that other members of this family might also have a similar fold and activity. Thus, it is possible that a larger class of ion channels might be regulated through an intrinsic enzymatic domain.
Is RyR1 an Enzyme?
Whether RyR1 actually functions as an oxidoreductase and, if so, the functional significance of this activity is not yet known. The binding properties suggest that either other components in the cellular environment facilitate NAD+ binding or the binding of NAD+ to RyR1 serves a more regulatory role. It is interesting to speculate on the significance of a redox-sensitive domain in the clamps of RyR1, as these are likely sites of interaction with the voltage sensor (8) and a major site of conformational change associated with channel opening and closing (7). The N-terminal domain may be responsible for transducing a signal from DHPR to RyR1 or vice versa to elicit Ca2+ release. An enzyme activity or redox sensor located at this crucial site could modulate the conformation of one or both proteins and consequentially their interaction. Because this N-terminal domain is also one of the “hot-spots” for the mutations that produce malignant hyperthermia and central core disease, alterations in the enzyme activity may contribute to the heightened response of the channel to volatile anesthetics in malignant hyperthermia or to the leakiness of the channel in central core disease (36). Another possibility is that this domain allows for channel self-regulation through regulation of its redox status or closely associated modulatory proteins. It has been shown that the activity of RyR1 is regulated by oxidation (37), and this enzyme activity might in some manner control redox status and hence activity of the channel.
Conclusion
In conclusion, we have used computational methods to predict a structure and function for one domain of RyR1. This prediction was then tested by using a combination of structural and biochemical approaches, which demonstrate a potential N-terminal oxidoreductase-like domain localized within the clamp domains of RyR1. Based on the binding studies, this oxidoreductase domain likely functions more as a redox sensor than a fully functional enzyme. The possibility of a sensor domain in a functionally active channel has yet to be identified.
Acknowledgments
We thank M. Baker, J. He, R. Gereau, M. Reid, M. Schmid, D. Sweatt, W. Tang, and J.-Z. Zhang for helpful discussions. This research has been supported by grants from National Institutes of Health, the Muscular Dystrophy Association of America, the Robert Welch Foundation, the American Heart Association, and the National Center for Research Resources. M.L.B. was supported in part by BRASS and the W. M. Keck Center for Computational Biology through a training grant from the National Library of Medicine. Movies and VRML models are available online at http://ncmi.bcm.tmc.edu/∼baker/ryr/.
Abbreviations
RyR1, ryanodine receptor 1
SR, sarcoplasmic reticulum
DHPR, dihydropyridine receptor
ICD, intracellular domain
IDH, isocitrate dehydrogenase
CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Lai F. A., Erickson, H. P., Rousseau, E., Liu, Q.-Y. & Meissner, G. (1988) Nature (London) 331, 315-319. [DOI] [PubMed] [Google Scholar]
- 2.Takeshima H., Nishimura, S., Matsumoto, T., Ishida, H., Kangawa, K., Minamino, N., Matsuo, H., Ueda, M., Hanaoka, M., Hirose, T., et al. (1989) Nature (London) 339, 439-445. [DOI] [PubMed] [Google Scholar]
- 3.Zorzato F., Margreth, A. & Volpe, P. (1986) J. Biol. Chem. 261, 13252-13257. [PubMed] [Google Scholar]
- 4.McCarthy T. V., Quane, K. A. & Lynch, P. J. (2000) Hum. Mutat. 15, 410-417. [DOI] [PubMed] [Google Scholar]
- 5.Zhao M., Li, P., Li, X., Zhang, L., Winkfein, R. J. & Chen, S. R. (1999) J. Biol. Chem. 274, 25971-25974. [DOI] [PubMed] [Google Scholar]
- 6.Imagawa T., Smith, J. S., Coronado, R. & Campbell, K. P. (1987) J. Biol. Chem. 262, 16636-16643. [PubMed] [Google Scholar]
- 7.Orlova E. V., Serysheva, II, van Heel, M., Hamilton, S. L. & Chiu, W. (1996) Nat. Struct. Biol. 3, 547-552. [DOI] [PubMed] [Google Scholar]
- 8.Serysheva II, Orlova, E. V., Chiu, W., Sherman, M. B., Hamilton, S. L. & van Heel, M. (1995) Nat. Struct. Biol. 2, 18-24. [DOI] [PubMed] [Google Scholar]
- 9.Serysheva II, Schatz, M., van Heel, M., Chiu, W. & Hamilton, S. L. (1999) Biophys. J. 77, 1936-1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bairoch A., Bucher, P. & Hofmann, K. (1997) Nucleic Acids Res. 25, 217-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer D. & Eisenberg, D. (1996) Protein Sci. 5, 947-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subramaniam S. (1998) Proteins 32, 1-2. [PubMed] [Google Scholar]
- 14.Hawkes M. J., Diaz-Munoz, M. & Hamilton, S. L. (1989) Membr. Biochem. 8, 133-145. [DOI] [PubMed] [Google Scholar]
- 15.Dubochet J., Adrian, M., Chang, J. J., Homo, J. C., Lepault, J., McDowall, A. W. & Schultz, P. (1988) Q. Rev. Biophys. 21, 129-228. [DOI] [PubMed] [Google Scholar]
- 16.Ludtke S. J., Baldwin, P. R. & Chiu, W. (1999) J. Struct. Biol. 128, 82-97. [DOI] [PubMed] [Google Scholar]
- 17.van Heel M., Harauz, G. & Orlova, E. V. (1996) J. Struct. Biol. 116, 17-24. [DOI] [PubMed] [Google Scholar]
- 18.Hurley J. H., Dean, A. M., Koshland, D. E., Jr. & Stroud, R. M. (1991) Biochemistry 30, 8671-8678. [DOI] [PubMed] [Google Scholar]
- 19.Bolduc J. M., Dyer, D. H., Scott, W. G., Singer, P., Sweet, R. M., Koshland, D. E., Jr. & Stoddard, B. L. (1995) Science 268, 1312-1318. [DOI] [PubMed] [Google Scholar]
- 20.Chen R., Grobler, J. A., Hurley, J. H. & Dean, A. M. (1996) Protein Sci. 5, 287-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guex N. & Peitsch, M. C. (1997) Electrophoresis 18, 2714-2723. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y., Aghdasi, B., Dou, S. J., Zhang, J. Z., Liu, S. Q. & Hamilton, S. L. (1997) J. Biol. Chem. 272, 25051-25061. [DOI] [PubMed] [Google Scholar]
- 23.Zingsheim H. P., Barrantes, F. J., Frank, J., Hanicke, W. & Neugebauer, D. C. (1982) Nature (London) 299, 81-84. [DOI] [PubMed] [Google Scholar]
- 24.Wagenknecht T., Frank, J., Boublik, M., Nurse, K. & Ofengand, J. (1988) J. Mol. Biol. 203, 753-760. [DOI] [PubMed] [Google Scholar]
- 25.Frank J., (1996) Three-Dimensional Electron Microscopy of Macromolecular Assemblies (Academic, San Diego).
- 26.Enroth C., Eger, B. T., Okamoto, K., Nishino, T. & Pai, E. F. (2000) Proc. Natl. Acad. Sci. USA 97, 10723-10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia Z. X., Dai, W. W., Xiong, J. P., Hao, Z. P., Davidson, V. L., White, S. & Mathews, F. S. (1992) J. Biol. Chem. 267, 22289-22297. [PubMed] [Google Scholar]
- 28.Hurley J. H., Dean, A. M., Thorsness, P. E., Koshland, D. E., Jr. & Stroud, R. M. (1990) J. Biol. Chem. 265, 3599-3602. [DOI] [PubMed] [Google Scholar]
- 29.Lo Conte L., Ailey, B., Hubbard, T. J., Brenner, S. E., Murzin, A. G. & Chothia, C. (2000) Nucleic Acids Res. 28, 257-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radermacher M., Wagenknecht, T., Grassucci, R., Frank, J., Inui, M., Chadwick, C. & Fleischer, S. (1992) Biophys. J. 61, 936-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang W., Baker, M. L., Ludtke, S. J. & Chiu, W. (2001) J. Mol. Biol. 308, 1033-1044. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z., Zhang, J., Sharma, M. R., Li, P., Chen, S. R. & Wagenknecht, T. (2001) Proc. Natl. Acad. Sci. USA 98, 6104-6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gulbis J. M., Mann, S. & MacKinnon, R. (1999) Cell 97, 943-952. [DOI] [PubMed] [Google Scholar]
- 34.Hoog S. S., Pawlowski, J. E., Alzari, P. M., Penning, T. M. & Lewis, M. (1994) Proc. Natl. Acad. Sci. USA 91, 2517-2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xin H. B., Timerman, A. P., Onoue, H., Wiederrecht, G. J. & Fleischer, S. (1995) Biochem. Biophys. Res. Commun. 214, 263-270. [DOI] [PubMed] [Google Scholar]
- 36.Tong J., McCarthy, T. V. & MacLennan, D. H. (1999) J. Biol. Chem. 274, 693-702. [DOI] [PubMed] [Google Scholar]
- 37.Abramson J. J., Cronin, J. R. & Salama, G. (1988) Arch. Biochem. Biophys. 263, 245-255. [DOI] [PubMed] [Google Scholar]