

Abstract
Native-state hydrogen exchange experiments under EX1 conditions can distinguish partially unfolded intermediates by their formation rates and identify the amide hydrogens exposed and protected in each. Results obtained define a cytochrome c intermediate seen only poorly before and place it early on the major unfolding pathway. Four distinct unfolding steps are found to be kinetically ordered in the same pathway sequence inferred before.
Keywords: protein folding, unfolding rollover, alkaline transition
Thermodynamic principles require that protein molecules must continually explore all possible higher-energy conformational states, even under native conditions. These partially unfolded forms (PUFs) are the intermediates through which the protein population folds, regardless of whether the PUFs are distinctly structured or well populated in the usual folding experiment. These forms, although infinitesimally populated under native conditions, can be studied by hydrogen exchange methods.
An equilibrium native-state HX exchange (eNHX) method can under favorable conditions distinguish different PUFs by their free-energy level and surface exposure, and identify their protected and exposed residues (refs. 1 and 2; Fig. 1). PUFs so far found in this way for a number of proteins resemble partially formed native structures (1, 3–7). Results for cytochrome c (Cyt c) identified four unfolding units (1, 2, 8), color-coded in Fig. 1A. The structural elements are referred to as the Red and Yellow Ω loops, the Green Ω loop and helix, and the Blue bihelix. Equilibrium NHX experiments (1, 2) and a related stability labeling experiment (9) suggested an unfolding sequence in the order N(RYGB) to Red unfolded(rYGB) to Red + Yellow unfolded(ryGB) to Red + Yellow + Green unfolded(rygB) to Blue unfolded(rygb = U) (U, unfolded; N, native). If this is correct, then the same sequence in reverse order must represent the major refolding pathway, because these experiments were done under equilibrium native conditions where each unfolding reaction must be matched by an equal and opposite refolding reaction.
Fig 1.

Unfolding by native-state HX. (A) Cyt c with color-coded unfolding units and its suggested folding/unfolding pathway. (B) Equilibrium vs. kinetic NHX. Equilibrium NHX, done under EX2 conditions, can distinguish intermediates (I but not I′) by their equilibrium ΔG and by their mo, ΔHo, or ΔVo in denaturant, temperature, or pressure-dependent HX experiments, respectively. The kinetic NHX method, done under EX1 conditions, can distinguish intermediates (I or I′) by their unfolding rates and their sensitivity to the high pH necessarily used. Unfolding barriers may be detected independently of whether the barrier is seen (rate-limiting) in the usual folding experiment or whether the intermediate formed is stable relative to U, unlike eNHX. Both methods identify the amino acids protected and exposed in each PUF by their HX labeling.
and by their mo, ΔHo, or ΔVo in denaturant, temperature, or pressure-dependent HX experiments, respectively. The kinetic NHX method, done under EX1 conditions, can distinguish intermediates (I or I′) by their unfolding rates and their sensitivity to the high pH necessarily used. Unfolding barriers may be detected independently of whether the barrier is seen (rate-limiting) in the usual folding experiment or whether the intermediate formed is stable relative to U, unlike eNHX. Both methods identify the amino acids protected and exposed in each PUF by their HX labeling.
However, equilibrium data alone cannot define a kinetic sequence (10, 11). In fact, neither can kinetic data, but the temporal order of the different steps is clearly important. Here we implement a kinetic version of the native-state HX approach (kNHX) that can provide temporal information. The kNHX experiment can distinguish different PUFs by the rate of the unfolding reactions that produce them, even though the determining barriers may not be rate-limiting in the usual kinetic folding experiment. The amino acids protected and exposed in each PUF can be identified regardless of whether the intermediates are populated or even stable relative to U (see Fig. 1B).
We applied the kNHX approach to oxidized equine Cyt c, testing two related approaches that use either time or pH as the independent variable. Both document a partially unfolded form with the Red Ω loop unfolded. This PUF is separated from the native state by an initial on-pathway unfolding barrier. Subsequent unfolding moves through the PUFs in the same order indicated before.
Theoretical Background
HX Theory.
The exchange rate of a freely exposed amide hydrogen is determined by a proton transfer reaction (12) in which the proton is transferred across an H-bond to a hydroxide ion (ref. 13; above pH ≈ 4). The chemical exchange rate (kch) is therefore determined by pH and also by an intrinsic second-order rate constant (kint), which depends on temperature, nearest neighbor inductive and blocking effects, and isotope effects (Eq. 1; refs. 14–16).
 |
Any preexisting structural H-bond must first be separated (12). In steady state, the overall exchange rate (kex) is given by Eq. 2 (17, 18), where kop and kcl are opening and reclosing rates of the protecting H-bond.
 |
When kcl ≫ kch (the EX2 bimolecular exchange condition), Eq. 2 reduces to Eq. 3. Measured exchange rate (kex) together with the known kch (Eq. 1) then provides Kop and hence ΔG .
.
 |
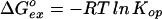 |
When kcl ≪ kch (the EX1 monomolecular exchange limit), exchange rate is given by Eq. 5, allowing the measurement of structural opening rates (18).
 |
The EX1 limit exploited here can be reached by increasing pH so that kch = kint[OH−] > kcl, or by using denaturant to make kcl slower. The transition from EX2 to EX1 behavior with pH is illustrated in Fig. 2 A and B.
Fig 2.

Simulated HX results. (A and B) At low pH where exchange is in the EX2 limit, HX rate increases 10-fold for each pH unit. At high pH, HX rate limits at kop (EX1 limit). (C) Simulation of expected curves when the HX exposure time is kept constant and pH is varied (kop = 10 s−1, kcl = 104 s−1, kint = 108 s−1). The arrow shows the EX2 to EX1 transition point (kch = kcl). An experimental curve in the gray region most sensitively measures the opening/closing parameters. At high pH, a different EX1 pattern can be produced when kop increases with pH (black line in Fig. 5).
Native-State HX.
Any protected hydrogen may exchange through opening reactions that represent the transient global unfolding (gl), a subglobal cooperative unfolding (sg), or more local fluctuations (lf) (1, 8, 19–23), as in Eq. 6, where each rate constant has the form in Eq. 2. The fastest pathway will determine the rate.
 |
We want to study the subglobal unfoldings, which may identify folding pathway intermediates. Subglobal unfoldings are often not observed in HX experiments because exchange through local fluctuations is faster. A native-state HX strategy (1, 2) uses mildly destabilizing conditions to selectively promote the larger unfolding reactions so that each unfolding comes to dominate the exchange of the hydrogens that it exposes. HX measurements can then detect and identify the PUFs.
In the equilibrium mode, NHX measurements are made under EX2 conditions (Eq. 3), where the HX rate of the residues exposed in each PUF is determined by the equilibrium constant, Kop, and ΔG of the PUF (Eqs. 3 and 4; Fig. 1B). In the kinetic mode, measurements are made under EX1 conditions where HX rates and PUF recognition depend on the rate of PUF formation (Eq. 5; Fig. 1B).
of the PUF (Eqs. 3 and 4; Fig. 1B). In the kinetic mode, measurements are made under EX1 conditions where HX rates and PUF recognition depend on the rate of PUF formation (Eq. 5; Fig. 1B).
Kinetic Native-State HX.
To reach the EX1 condition, pH 10 or higher will be necessary to make kch larger than kcl. At pH 10 and 20°C, kch for an exposed amide is about 104 s−1. Exchange can then be too fast for straightforward NMR measurement, even for protected hydrogens. A useful strategy is to expose proteins to the high pH exchange condition for only a short time period (pulse) (24, 25).
The kNHX experiment can be done in two different modes, with increasing pulse time at constant pH or with constant pulse time at increasing pH. With pulse time as the variable, labeling is mono-exponential (Eq. 7, Fig. 2B), if the population is homogeneous.
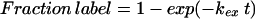 |
Fig. 2C simulates curves obtained with constant exposure time at increasing pH. The arrow at pH 10.2 marks the point where kch = kcl. Higher pH provides the desired EX1 condition, where kex = kop = 1/τop. Exposure time between 0.3 and 1.5 τop then produces a fractional labeling plateau that is very sensitive to opening rate (Eq. 7). A shorter pulse time achieves little labeling because opening hardly occurs during the labeling pulse. A longer pulse causes full labeling already in the EX2 region, before the EX1 condition is reached.
With a pulse time near τop, an experimental series done as a function of pH can measure the entire transition between EX2 and EX1 conditions. A fit of the transition curve (Eq. 7 with Eqs. 2 and 1) then provides all of the kinetic and thermodynamic parameters. The EX1 plateau level specifies kop, whereas the EX2 data at lower pH determine Kop and ΔG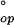 , and then kcl.
, and then kcl.
For Cyt c, an additional EX1 opening process with kop dependent on pH becomes significant above pH 12. Increasing pH promotes unfolding to a recognizable high pH intermediate and then to the unfolded state. Here measured kex gives kop directly (26–28).
Materials and Methods
Materials.
Horse heart Cyt c was from Sigma, ultra pure urea from ICN, 99.9% D2O from Isotec, and chemicals from Fisher. All experiments used 0.5 M KCl at 20°C unless otherwise specified.
Equilibrium and Kinetic Melting.
High pH equilibrium melting used an Aviv CD 202 spectrometer (Aviv Associates, Lakewood, NJ). An increasing amount of 10 μM Cyt c in 2 M NaOH was added to 10 μM Cyt c manually. CD and fluorescence (Fl) were recorded. Fl was normalized by NATA and corrected for high pH fluorophore changes.
Kinetic HX studies used a Biologic SFM4 stopped-flow instrument (Fl, averaging six shots). Cyt c was diluted into CAPS buffer (Sigma), incrementing pH 9 and 12 syringes to obtain different final pH. Output was collected for pH measurement. Urea was at 0 and 1.3 M.
Kinetic unfolding experiments used 695 nm absorbance (0.3 mM Cyt c) and Fl (50 μM Cyt c) with incremented GdmCl. The pH 10 rollover studies started with protein at pH 6 to avoid histidine to heme misligation. At high GdmCl, the unfolding rate is greater than the misligation rate.
kNHX(t).
Time-dependent HX labeling used a Biologic SFM4 in three syringe quench mode. Protein (in syringe 2, 6 mM in D2O, 1.3 M urea, 10 mM KPO4 at pDr 6, previously deuterated at 55°C for 3 hr) was mixed with high pH buffer (in syringe 1, 50 mM CAPS, pH 10.5 or 11.8, 1.3 M urea, H2O), passed through a delay line varied to adjust the pulse time, and then quenched (syringe 3, 0.6 M citrate, 0.35 M ascorbate) to pH 5.3, optimized for H-retention during sample work-up. Mixing ratio was 1:5:2. Samples were concentrated, run into analysis buffer (D2O, 10 mM ascorbate, 100 mM acetate, pDr 5, + TSP) and stored at −80°C pending NMR analysis. Pulse pH was checked by manual mixing and corrected for the fractional (F) D2O content (pHcorr = pDr + 0.4F). A no-pulse control (manual mix of d-Cyt c with high pH and quench buffers) provided the NMR baseline for residue-specific D to H leakage during sample work-up and analysis.
kNHX(pH).
The pH-dependent D to H labeling was done as above except that pH rather than time was varied. Mixing buffers were 50 mM of either Hepes, EPPS (Sigma), Bicine, CHES (Sigma), CAPS, or K3PO4 at the different pH values used. To set the 100% labeling level and control for H loss during analysis, a protonated control was prepared (d-Cyt c mixed with the high pH and exchanged to equilibrium; 83% H).
NMR.
NMR analysis for H-labeling used correlated spectroscopy (COSY) 1H–1H spectra acquired with a 500 MHz Varian INOVA spectrometer (2,048 time domain points, spectral width 10 kHz, 512 blocks with 16 transients, ≈3 hr; FELIX 2.3). The average of heme bridge 4 and Tyr-97 5H–6H nonexchanging cross-peaks served as internal standards.
Results
Cyt c at High pH.
Studies were done to determine the effects of the high pH condition necessary for kNHX experiments (Fig. 3).
Fig 3.

High pH denaturation of Cyt c (no urea). (A) Above pH 12, denaturation occurs in two steps with a populated intermediate. A partial unfolding removes 45% of the CD222 of the native protein and recovers all of the fluorescence of the U state (corrected for Trp pH titration and quenching). Both probes show the same midpoint (pK 12.8) and slope (1.5). Equilibrium unfolding to U occurs above pH 13. (B) The kinetics of intermediate formation. The black curve in Fig. 5A is constructed from Fl data in 1.3 M urea. The zero urea data are shown here because CD222 is obscured in high pH urea.
Cyt c undergoes an alkaline transition with pK 9.3, in which the Met-80 whose sulfur is ligated to heme Fe is replaced by a neutral lysine (29). This transition has almost no effect on the equilibrium Fl or CD222 (Fig. 3A). Kinetically, the major ligand replacement reaction occurs with rate of 0.6 s−1 at pH 10 and increases with pH to a maximum rate (30, 31) of 8 s−1, which matches the unfolding rate of the Red Ω loop discussed below.
At pH above 12, a partially unfolded equilibrium intermediate begins to populate (pK 12.8, Δlog Ku/ΔpH ≈ 1.5; Fig. 3A). When formed it recovers all of the unfolded state fluorescence (fully quenched in the native state by transfer to the heme) while retaining ≈55% of the native helical CD222 (Fig. 3A). HX data (below) show that much of the N- and C-terminal helices, which account for 61% of the native helical residues, are maintained in the high pH intermediate. Therefore, the initial CD222 loss must be mainly accounted for by unfolding of the 60s helix. To account for the fluorescence, (much of) the Yellow loop (residues 37–60), which buries the fluorescent Trp-59 residue close to the quenching heme, must be unfolded. In this case, the Red Ω loop (residues 71–85), with no supporting interactions (Fig. 1A), is also very likely to be unfolded. Thus, the high pH intermediate appears to have unfolded the Red and Yellow Ω loops, the Green 60s helix, and the Green Ω loop (see below). A later phase (pH > 13) removes the remaining CD.
The high pH intermediate begins to reach detectable amplitude at pH ≈ 12 (Fig. 3A), where kobs ≈ 10 s−1 so kop ≈ 1 s−1 (from Fig. 3B because kobs = kop + kcl and Kop = kop/kcl ≈ 0.1). Therefore, below pH 12, the slow pH-dependent structural transition will not affect HX behavior during the 75-ms HX pulse time used here. At higher pH, unfolding becomes faster and begins to affect measured HX. From the kinetic data in Fig. 3B, a reference kinetic curve can be drawn (in Fig. 5A) that predicts the EX1 HX behavior that would be mediated by the high pH unfolding.
Fig 5.

Kinetic NHX as a function of pH, and protein unfolding. (A) Measured amide hydrogens in the four unfolding units of Cyt c (1.3 M urea; data without urea are very similar), with constant pulse labeling time of 75 ms. Dotted lines show hydrogens previously found to exchange by way of local fluctuations; solid lines show previously identified marker hydrogens. The reference black curve shows the HX labeling that would be determined by the formation of the high pH intermediate, assuming EX1 exchange. Data for each hydrogen are fit by Eqs. 7, 1, and 2 plus the high pH intermediate formation. Black points for the Yellow unit are from Fig. 4B. Residues 70 and 91 at the two ends of the Red segment (71–85; 91 H-bonds to 87) begin to show plateauing, like the Red unit. His-33 is protected in the U state (1). (B) The individual residues in the Red unit without and with added urea. All exchange as markers, with the subglobal Red loop unfolding, because of the high pH destabilizing condition. Different hydrogens plateau earlier or later depending on their kint value. Labeling rate increases sharply at pH 12 and above because of formation of the high pH intermediate. Black points (Lower) are from the time-dependent HX data in Fig. 4C. (C) Rollover of the Cyt c unfolding chevron compared with HX unfolding rates for the Red unit. The unfolding arm at pH 7 and the rollover at pH 7 and 10 are shown. (At lower denaturant, unfolding at pH 10 becomes biphasic as in Fig. 3; data shown are monophasic.) Back extrapolation of the pH 10 rollover rate is shown at ±1σ. To convert 1.3 M urea to equivalent GdmCl, a factor of 2.5 was used (41); the stronger GdmCl denaturant was necessary to access the rollover region. (D) Fractional H-labeling at the given pH (75 ms). Bars represent the average H-label of the marker residues for each unfolding unit, taken from A. The sequential decrease in opening rates is best explained by a sequential unfolding pathway, as in E. (E) Classical stepwise folding/unfolding pathway for Cyt c. The diagram illustrates barrier heights and well depths that will generate the Red, Yellow, and Green unfoldings indicated by the kNHX results. [In this case the relationship τobs = Στi closely holds, where the rate of first passage opening (τ) is measured by EX1 HX and the summation is over all prior openings.]
kNHX as a Function of Exchange Time.
Fig. 4 shows kNHX data for the marker hydrogens in Cyt c, recorded as a function of exchange time at pH 10.16 and 11.24. Marker hydrogens, previously identified, are so well protected that they exchange only when the segment that they occupy concertedly unfolds (1, 2, 8).
Fig 4.

Kinetic NHX as a function of time. HX behavior is shown for marker residues in the Blue and Green units (A) and the Yellow unit (B), and for all of the protected residues in the Red unit (C). Conditions were pH 10.14 (open symbols, dashed lines) and pH 11.24 (closed symbols, bold lines) in 0.5 M KCl at 20°C with 1.3 M urea to slow reclosing.
Markers for the Blue and Green helical unfolding units (Fig. 4A) do not exchange at all because they open much less than once over the 400-ms time scale. The Yellow loop markers (Fig. 4B) begin to approach EX1 behavior (like Fig. 2B). In the Red Ω loop, all of the protected residues in 100% of the molecules approach (Fig. 4C) and reach (see Fig. 5B) the same limiting EX1 opening rate. This shows that the entire protein population experiences the same whole-Red-loop unfolding.
kNHX as a Function of pH.
Kinetic NHX experiments were done as a function of pH, using a pulse labeling time of 75 ms to specifically target the opening of the Red unit (τop ≈ 100 ms). Fig. 5A shows results for all of the measured amide hydrogens in the four known unfolding units (1.3 M urea; similar but less complete data without urea not shown). Hydrogens previously identified as markers and nonmarkers are separately indicated.
The nonmarker hydrogens all exchange by way of fast local fluctuations at very close to the EX2 rate expected from earlier results at pH 7. This is not unexpected, because local fluctuations are likely to preserve EX2 HX behavior even at high pH and to be insensitive to pH-dependent structure destabilization.
We are more interested here in the subglobal unfolding reactions. In Fig. 5A, all of the Red loop hydrogens reach an EX1 plateau. The Yellow markers approach the same EX1 plateau (kop), but at higher pH because their kcl is faster. The Blue and Green markers open much less than once on the 75-ms time scale (except at very high pH).
The black curve at high pH benchmarks the HX labeling that would be produced by the pH-dependent formation of the high pH intermediate (from Fig. 3B). This exchange is in the EX1 limit because at pH 12 and higher, kch > 106 s−1, faster than any reasonable refolding.
Cooperative Unfolding of the Red Loop.
Fig. 5B displays the Red loop hydrogens in more detail. They all reach an EX1 plateau at about 50% labeling during the limited 75-ms HX pulse (like Fig. 2C), indicating that the entire loop unfolds as a cooperative unit. Opening rate for the Red loop averages 9.1 s−1 without urea and 11.7 s−1 with 1.3 M urea, in agreement with the 9.7 s−1 value found from HX as a function of time (all ± 20% total range; mu ≈ 0.1 kcal/mol per M urea). At higher pH, labeling rate increases again, incremented by the kop of the high pH intermediate (black curve in Fig. 5A).
Interestingly, the well known Cyt c alkaline transition reaches this same limiting rate (30, 31) at high pH (8 s−1), suggesting that both are limited by the same barrier. At lower pH, Red unfolding maintains the same rate while the alkaline transition is much slower (unfavorable pH-dependent preequilibrium), indicating that the Red unfolding does not depend on the alkaline transition.
Reclosing rates for the Red loop hydrogens are ≈7000 s−1 in 1.3 M urea and slightly higher without urea (≈3-fold total range). The opening rate is better defined, as has been seen for other proteins (24, 32, 33). In the EX1 region, the HX labeling plateau is likely to show the correct opening rate for all of the hydrogens even when some have residual protection because, at high pH, exchange will tend to occur before the loop recloses (34).
The Red loop hydrogens suggest a ΔG of 3.7 kcal/mol in urea, less by 2 kcal/mol than at pH 7. The destabilization promotes the subglobal unfolding and causes it to dominate exchange, in competition with local fluctuations, fortunately generating the condition necessary for native-state HX experiments.
of 3.7 kcal/mol in urea, less by 2 kcal/mol than at pH 7. The destabilization promotes the subglobal unfolding and causes it to dominate exchange, in competition with local fluctuations, fortunately generating the condition necessary for native-state HX experiments.
The Yellow Loop, and ryGB.
Above pH 10, the Yellow loop markers (Fig. 5A) begin to exchange within the 75-ms pulse, pass through an EX2 range, and approach a limiting EX1 plateau, indicating an unfolding rate that is nearly identical to the Red loop (12 ± 2 s−1; 1.3 M urea), perhaps limited by the same barrier. The Green and Blue units remain folded, pointing to ryGB. This form was demonstrated before by a stability labeling method (9) and is rationalized by the structural observation that the Red and Yellow loops are in intimate multiatom contact.
The Yellow loop recloses faster than the Red (kcl = 4 × 104 s−1; from ΔG ≈ 4.7 kcal/mol at EX2 conditions, less than at pH 7 by ≈3 kcal/mol). This leaves Red independently open, as rYGB, a form that has also been demonstrated before (1, 9).
≈ 4.7 kcal/mol at EX2 conditions, less than at pH 7 by ≈3 kcal/mol). This leaves Red independently open, as rYGB, a form that has also been demonstrated before (1, 9).
Above pH 12, the onset of high pH intermediate formation (black curve) sweeps up both the Red and Yellow loops.
The Green and Blue Units, and rygB.
At pH 10–11.5 where the Red and Yellow loop hydrogens largely exchange, the Green unit opens much less than once and no exchange occurs. Above pH 11.5, the Green unit begins to unfold significantly within the 75-ms pulse. The same exchange rate and pH dependence is shown (Fig. 5A) by Leu-32, the Green loop marker, by Leu-68, the Green helix marker, and by Met-65 and Glu-69, the other slowest hydrogens in the Green helix. The Blue unit remains closed.
The rate and pH dependence for unfolding of the Green unit are identical to the high pH intermediate (the black curve in Fig. 5A, drawn for EX1 exchange). The Fl and CD of the high pH intermediate, and the fact that the Blue unit remains closed, seems to require that the Red and Yellow loops, as well as the Green unit, are unfolded (see before), pointing to rygB. The rygB intermediate was seen before in kinetic folding (35) and stability labeling (9) experiments, and it was shown to be on the folding pathway (36, 37).
In a final phase at even higher pH, the marker residues for the Blue bihelix unit begin to open significantly and exchange, all with a common EX1 rate, increasing with pH. Opening of the Blue unit is known to represent final unfolding to the U state (21). This step would quantitatively account for the CD loss in the second unfolding phase seen in Fig. 2.
Unfolding Pathway: The Red Loop.
Fig. 5C shows the unfolding arm of a Cyt c folding chevron. Rollover occurs when a limiting unfolding barrier is replaced by a newly limiting barrier earlier in unfolding (like Hammond behavior) with less dependence on denaturant (less new surface exposed).
The rollover unfolding rate at pH 10 extrapolates back to the rate for unfolding of the Red loop at near-zero denaturant. Evidently both are limited by the same early on-pathway unfolding barrier (as are also the alkaline transition and formation of the high pH intermediate).
In summary, results found for the Red Ω loop show that the entire loop acts as a concerted unfolding unit, that its unfolding represents an early step on the major Cyt c unfolding pathway, and that all of the Cyt c molecules experience the same, apparently obligatory, on-pathway barrier.
Unfolding Pathway: Kinetic Order.
Although quantitative unfolding rates are distorted by the high pH, these data qualitatively order several kinetic unfolding steps. Unfolding of the Red loop is limited by an early barrier that appears to be on the unfolding pathway (rYGB). At higher pH, the Yellow unit unfolds at close to the same rate as the Red unit, whereas Green and Blue remain closed (ryGB). At still higher driving pH, Green can be additionally forced to open (rygB). Still higher pH forces the Blue unit to open (rygb = U). This progression provides one measure of kinetic order.
Parallel information is obtained at pH 11.3 and 12.0 where some degree of opening-dependent labeling occurs for all of the units during the 75-ms pulse. Fig. 5D shows that at each given pH the EX1 unfolding rates for the different units are detectably different. This fine discrimination is made possible by the sensitive vertical scale provided by the brief labeling experiment. At each pH, unfolding order is the same as was indicated before. Fig. 5E shows a set of pH-dependent wells and barriers that will generate the HX results found. They also appear to account for the alkaline transition (first barrier) and formation of the high pH intermediate (Green unfolding).
These results show that information on the unfolding order of the different intermediates can be provided by their EX1 labeling plateaus, by their positioning on the pH axis under EX1 conditions, and by the labeling sequence of the different units at a given pH. The results obtained suggest the same intermediate structures seen before (rYGB, ryGB, rygB) and place in kinetic order the unfolding reactions that produce them. The major result is that the same sequence previously inferred on other grounds is found.
Discussion
Native-State HX.
Equilibrium NHX done under EX2 conditions detects hidden intermediates by their equilibrium parameters. Intermediates so far found with a number of proteins are constructed by using the protein's intrinsically cooperative secondary structural elements as building blocks. Kinetic NHX, done under EX1 conditions, can detect hidden intermediates by their formation rates, can show the order in which the different PUFs unfold, and can indicate whether unfolding and refolding occur through one or multiple pathways. Both modes use mildly destabilizing conditions so that, although the native state is still the abundant form, large transient unfoldings come to dominate the exchange of the hydrogens that they expose and can be identified thereby.
In kNHX experiments, pathway order must be defined by measuring unfolding rather than refolding rates. This is so because the native state is the abundant form. A consequence is that measured reclosing rates do not relate to pathway order. Transiently formed PUFs reprotect their exposed hydrogens at a rate that neither depends on nor reveals their position in the pathway (consider Fig. 5E). In the unfolding direction, this ambiguity is removed. A unit that unfolds earlier must precede, or accompany, a unit that unfolds later. Identification of the structure in the different intermediates provides complementary pathway insight.
The Units of Folding.
Earlier equilibrium native-state HX exchange (eNHX) results for Cyt c showed that all of the residues in the N/C-terminal bihelical unit (Blue), and all in the 60s helix (part of the Green unit), reversibly and cooperatively unfold and refold, indicating their unit cooperative nature. In helices, almost all of the amide hydrogens are protected and can be easily measured. This is not so for Ω loops. The conditions used here (brief HX exposure time, sample analysis at low pH) made it possible to measure all of the protected hydrogens distributed through the Red Ω loop. Both kinetic approaches used show that all of the protected Red loop hydrogens exchange by way of the same whole-loop unfolding event. All have essentially the same kop, Kop, and kcl. Other experiments, to be described elsewhere, depict cooperative unfolding behavior within the Yellow Ω loop.
A generalization emerges. In protein molecules Ω loops and helices, or groupings thereof, tend to retain their separately cooperative nature in addition to their collective whole molecule cooperativity. (β-structures remain to be elucidated.) This provides the physical basis for stepwise folding pathways.
Pathway Information.
Earlier equilibrium NHX results (1) identified the four concerted unfolding units shown in Fig. 1. The order of increasing unfolding size (m value) and free energy was found to be Red open, Yellow open, Green open, Blue open. In agreement, a stability labeling experiment identified the partially unfolded forms as follows (9). Red open is rYGB; Yellow open is ryGB; Green open is rygB; Blue open is rygb = U. These are just the forms needed to produce a sequential unfolding pathway, and therefore an equivalent, reverse-order refolding pathway.
The present experiments add kinetic information. The same unfoldings are kinetically ordered in the same sequence. Further, other kinetic results specifically place rYGB at the beginning of the unfolding pathway (Fig. 5C) and rygB at the beginning of the folding pathway (36, 37).
The order of intermediate formation found experimentally is in striking conformity with the dictates of the native Cyt c structure (38). The Blue N/C-terminal helices contact only the Green elements. Therefore, initial folding of the most stable bihelical unit (rygB) provides a docking surface that is able to stabilize only the Green helix and loop elements (ryGB). The Green elements are needed to guide and stabilize the subsequent formation of the Yellow and Red loops. This provides a structural rationale for the Blue to Green to Yellow and Red sequence that is found.
More generally, this structural correspondence suggests that the nature of the folding pathway, linear or other, will depend on the organization of the cooperative folding units in the particular protein, operating through the principle of sequential stabilization. It is interesting that the same sequential stabilization strategy appears to extend to the first step in folding, in which a large-scale conformational search is thought to find an initial transition state that first organizes a significant part of the native topology (37–40).
Alternative interpretations for each step indicated by the kNHX data can undoubtedly be considered. Nonetheless, it seems compelling that a large amount of evidence from several kinds of independent cross-checking experimentation identifies the same pathway intermediates and places them in the same order, both structurally and kinetically. All of these results—equilibrium, kinetic, and structural—consistently support a classical, essentially sequential pathway in which the entire Cyt c population folds through a small set of defined intermediates and barriers, progressively adding cooperative native units in a sequence that is dictated by the growing native structure.
Acknowledgments
We thank the members of our lab for continuing helpful discussions, and we thank Yawen Bai, Tobin Sosnick, and Joshua Wand for expert help with a difficult manuscript. This work was supported by the National Institutes of Health and the Mathers Foundation.
Abbreviations
PUF, partially unfolded form
kNHX, kinetic native-state hydrogen exchange
Cyt c, cytochrome c
U, unfolded
N, native
Fl, fluorescence
References
- 1.Bai Y., Sosnick, T. R., Mayne, L. & Englander, S. W. (1995) Science 269, 192-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bai Y. & Englander, S. W. (1996) Proteins Struct. Funct. Genet. 24, 145-151. [DOI] [PubMed] [Google Scholar]
- 3.Chamberlain A. K., Handel, T. M. & Marqusee, S. (1996) Nat. Struct. Biol. 3, 782-787. [DOI] [PubMed] [Google Scholar]
- 4.Hiller R., Zhou, Z. H., Adams, M. W. W. & Englander, S. W. (1997) Proc. Natl. Acad. Sci. USA 94, 11329-11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuentes E. J. & Wand, A. J. (1998) Biochemistry 37, 9877-9883. [DOI] [PubMed] [Google Scholar]
- 6.Bhuyan A. K. & Udgaonkar, J. B. (1998) Proteins Struct. Funct. Genet. 30, 295-308. [PubMed] [Google Scholar]
- 7.Llinas M., Gillespie, B., Dahlquist, F. W. & Marqusee, S. (1999) Nat. Struct. Biol. 6, 1072-1078. [DOI] [PubMed] [Google Scholar]
- 8.Englander S. W., Mayne, L., Bai, Y. & Sosnick, T. R. (1997) Protein Sci. 6, 1101-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y., Mayne, L. & Englander, S. W. (1998) Nat. Struct. Biol. 5, 774-778. [DOI] [PubMed] [Google Scholar]
- 10.Clarke J. & Fersht, A. R. (1996) Folding Des. 1, 243-254. [DOI] [PubMed] [Google Scholar]
- 11.Clarke J., Itzhaki, L. S. & Fersht, A. R. (1997) Trends Biochem. Sci. 22, 284-287. [DOI] [PubMed] [Google Scholar]
- 12.Eigen M. (1964) Angew. Chem. Int. Ed. Engl. 3, 1-19. [Google Scholar]
- 13.Berger A. & Linderstrøm-Lang, K. (1957) Arch. Biochem. Biophys. 69, 106-118. [DOI] [PubMed] [Google Scholar]
- 14.Molday R. S., Englander, S. W. & Kallen, R. G. (1972) Biochemistry 11, 150-158. [DOI] [PubMed] [Google Scholar]
- 15.Connelly G. P., Bai, Y., Jeng, M. F. & Englander, S. W. (1993) Proteins Struct. Funct. Genet. 17, 87-92. [DOI] [PubMed] [Google Scholar]
- 16.Bai Y., Milne, J. S., Mayne, L. & Englander, S. W. (1993) Proteins Struct. Funct. Genet. 17, 75-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linderstrøm-Lang K. U. (1955) Chem. Soc. Spec. Publ. 2, 1-20. [Google Scholar]
- 18.Hvidt A. & Nielsen, S. O. (1966) Adv. Protein Chem. 21, 287-386. [DOI] [PubMed] [Google Scholar]
- 19.Kim K. S. & Woodward, C. (1993) Biochemistry 32, 9609-9613. [DOI] [PubMed] [Google Scholar]
- 20.Mayo S. L. & Baldwin, R. L. (1993) Science 262, 873-876. [DOI] [PubMed] [Google Scholar]
- 21.Bai Y., Milne, J. S., Mayne, L. & Englander, S. W. (1994) Proteins Struct. Funct. Genet. 20, 4-14. [DOI] [PubMed] [Google Scholar]
- 22.Qian H., Mayo, S. L. & Morton, A. (1994) Biochemistry 33, 8167-8171. [DOI] [PubMed] [Google Scholar]
- 23.Milne J. S., Mayne, L., Roder, H., Wand, A. J. & Englander, S. W. (1998) Protein Sci. 7, 739-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arrington C. B. & Robertson, A. D. (2000) J. Mol. Biol. 296, 1307-1317. [DOI] [PubMed] [Google Scholar]
- 25.Canet D., Last, A. M., Tito, P., Sunde, M., Spencer, A., Archer, D. B., Redfield, C., Robinson, C. V. & Dobson, C. M. (2002) Nat. Struct. Biol. 9, 308-315. [DOI] [PubMed] [Google Scholar]
- 26.Kiefhaber T. & Baldwin, R. L. (1995) Proc. Natl. Acad. Sci. USA 92, 2657-2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loh S. N., Rohl, C. A., Kiefhaber, T. & Baldwin, R. L. (1996) Proc. Natl. Acad. Sci. USA 93, 1982-1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiefhaber T. & Baldwin, R. L. (1996) Biophys. Chem. 59, 351-356. [DOI] [PubMed] [Google Scholar]
- 29.Wilson M. T. & Greenwood, C. (1996) in Cytochrome c: A Multidisciplinary Approach, eds. Scott, R. A. & Mauk, A. G. (University Science Books, Sausolito, CA), pp. 611–634.
- 30.Al-Ayash A. I. & Wilson, M. T. (1979) Biochem. J. 177, 641-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson M. T. & Greenwood, C. (1971) Euro. J. Biochem. 22, 11-18. [DOI] [PubMed] [Google Scholar]
- 32.Sivaraman T., Arrington, C. B. & Robertson, A. D. (2001) Nat. Struct. Biol. 8, 331-333. [DOI] [PubMed] [Google Scholar]
- 33.Kalodimos C. G., Boelens, R. & Kaptein, R. (2002) Nat. Struct. Biol. 9, 193-197. [DOI] [PubMed] [Google Scholar]
- 34.Bai Y. (1999) J. Biomol. NMR 15, 65-70. [DOI] [PubMed] [Google Scholar]
- 35.Roder H., Elöve, G. A. & Englander, S. W. (1988) Nature (London) 335, 700-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bai Y. (1999) Proc. Natl. Acad. Sci. USA 96, 477-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krantz B. A., Srivastava, A. K., Nauli, S., Baker, D., Sauer, R. T. & Sosnick, T. R. (2002) Nat. Struct. Biol. 9, 458-463. [DOI] [PubMed] [Google Scholar]
- 38.Rumbley J., Hoang, L., Mayne, L. & Englander, S. W. (2001) Proc. Natl. Acad. Sci. USA 98, 105-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sosnick T. R., Mayne, L. & Englander, S. W. (1996) Proteins Struct. Funct. Genet. 24, 413-426. [DOI] [PubMed] [Google Scholar]
- 40.Plaxco K. W., Simons, K. T. & Baker, D. (1998) J. Mol. Biol. 277, 985-994. [DOI] [PubMed] [Google Scholar]
- 41.Gupta R. & Ahmad, F. (1999) Biochemistry 38, 2471-2479. [DOI] [PubMed] [Google Scholar]