

Abstract
Phytocannabinoids, such as the principal bioactive component of marijuana, Δ9-tetrahydrocannabinol, have been used for thousands of years for medical and recreational purposes. Δ9-Tetrahydrocannabinol and endogenous cannabinoids (e.g., anandamide) initiate their agonist properties by stimulating the cannabinoid family of G protein-coupled receptors (CB1 and CB2). The biosynthesis and physiology of anandamide is well understood, but its mechanism of uptake (resulting in signal termination by fatty acid amide hydrolase) has been elusive. Mounting evidence points to the existence of a specific anandamide transport protein; however, no direct evidence for this protein has been provided. Here, we use a potent, competitive small molecule inhibitor of anandamide uptake (LY2318912, IC50 7.27 ± 0.510 nM) to identify a high-affinity, saturable anandamide transporter binding site (LY2318912; Kd = 7.62 ± 1.18 nM, Bmax = 31.6 ± 1.80 fmol/mg protein) that is distinct from fatty acid amide hydrolase. Systemic administration of the inhibitor into rodents elevates anandamide levels 5-fold in the brain and demonstrates efficacy in the formalin paw-licking model of persistent pain with no obvious adverse effects on motor function. Identification of the anandamide transporter binding site resolves a missing mechanistic link in endocannabinoid signaling, and in vivo results suggest that endocannabinoid transporter antagonists may provide a strategy for positive modulation of cannabinoid receptors.
Keywords: anandamide, fatty acid amide hydrolase, cannabinoid, marijuana, transporter
Endocannabinoids are recognized as significant intracellular lipid signaling molecules in the central nervous system with extensive control of physiological and behavioral mood and affect. Increases in endocannabinoid neurotransmission have broad therapeutic potential, including reduction of nausea and emesis (1), appetite stimulation (2), analgesia (3), anxiolytic activity (4), antispasmodic activity (5), and lowering of intraocular pressure in glaucoma (6). Identification of a specific binding site for the phytocannabinoid, Δ9-tetrahydrocannabinol (Δ9-THC) (7), cloning of the cannabinoid receptors (CB1 and CB2) (8, 9), and the identification of an endogenous ligand, anandamide (N-arachidonoylethanolamide) (10), provided evidence of an endogenous cannabinoid system. Anandamide represents a class of lipid neurotransmitters that stimulate not only presynaptic and postsynaptic CB1 receptors but also TRPV1 ion channels (11, 12), 5-hydroxytryptamine receptors (13-16), and possibly other receptors, as well as CB2 receptors in the periphery (10, 17-19). More recently, the enzymes that are responsible for anandamide synthesis (phospholipase D) and catabolism (fatty acid amide hydrolase, FAAH) have been identified and characterized (20, 21). Unlike typical neurotransmitter molecules, anandamide is synthesized in the membrane bilayer, resulting in the phospholipid precursor of anandamide, N-arachidonoylphosphatidylethanolamine (22-25). Calcium-activated phospholipase D then releases anandamide into the synapse from either the presynaptic or postsynaptic plasma membranes (26-28).
Termination of anandamide signaling appears to involve a two-step process, transport across the plasma membrane followed by enzymatic hydrolysis by FAAH (20, 22, 29-31). For anandamide to be metabolized by FAAH, it first must be transported through the plasma membrane to the cellular compartments where FAAH is localized (32). Although the exact molecular nature of this transport process is still under debate, it is agreed that anandamide movement across the plasma membrane is saturable, rapid (33, 34), temperature-dependent (33), and enantioselective (35), and it does not appear to require an ion gradient or chemical energy supply (ATP) (31). Several hypotheses for the mechanism of anandamide uptake have been considered, including passive diffusion, endocytosis, facilitative transport, and FAAH-mediated mechanisms (36, 37). It has been suggested that anandamide accumulation depends solely on diffusion and that FAAH-mediated enzymatic cleavage of anandamide maintains the inward driving concentration gradient (38). Others have hypothesized the involvement of an endocytic process (39) and/or the existence of a cellular compartment that sequesters anandamide upon transport (40). The cellular accumulation profile of anandamide, along with the observation that anandamide uptake can be inhibited specifically by compounds that are not substrates for, or inhibitors of, FAAH (41-43) suggests that anandamide transport occurs by a carrier-mediated process in which a protein carrier binds and translocates anandamide from one side of the plasma membrane to the other. Although controversy remains regarding the exact molecular mechanism of anandamide uptake, mounting evidence points to the existence of a specific anandamide transporter protein that is distinct from FAAH enzyme. However, no direct evidence for this protein has been provided.
Transporter-mediated movement of anandamide across the plasma membrane is a key regulation point for anandamide signaling and, therefore, represents an essential component of signal termination. Identification of a specific anandamide binding site has proven to be elusive because of the relative lack of potency of arachidonic acid-based inhibitory compounds, as well as their lipophilic nature. This article describes a radioligand, [125I]LY2318912, which potently inhibits functional anandamide uptake and exhibits saturable high-affinity binding to RBL-2H3 cell plasma membranes. The parent compound of LY2318912, LY2183240, increases anandamide levels in rat cerebellum and displays efficacy in persistent pain models. Together, these results suggest that [125I]LY2318912 and structurally related molecules are potent anandamide uptake inhibitors that can be used to characterize a binding site that is pharmacologically consistent with an anandamide transport protein.
Materials and Methods
Reagents. LY2183240 (5-biphenyl-4-ylmethyl-tetrazole-1-carboxylic acid dimethylamide) and LY2318912 (5-[(4-azido-3-iodo-benzoylamino)-methyl]-tetrazole-1-carboxylic acid dimethylamide) were synthesized at Eli Lilly and Company. Fatty-acid-free BSA was obtained from Serologicals Corp. (Norcross, GA). RBL-2H3 cells were obtained from the American Type Culture Collection. HeLa cells were a gift from Eric Barker (Purdue University, West Lafayette, IN).
Transfections. HeLa cells (passes 7-25) were plated at a concentration of 7.5 × 106 cells per T75 flask 24 h before transfection. Cells were transfected with wild-type human FAAH gene in pcDNA3.1(+) plasmid. DNA and Lipofectamine 2000 mixtures were combined in OptiMEM-1 reduced-serum media (Invitrogen) for 20 min. The DNA/Lipofectamine 2000 transfection mixture was added to growth medium lacking antibiotics and cells were incubated in transfection mixture for 6 h. The mixture was replaced with normal growth media and cells were harvested ≈48 h after transfection, allowing sufficient time for the expression of FAAH.
Anandamide Uptake Assays. Experiments were performed with RBL-2H3 cells (passes 3-20). Cells were plated at 2.5 × 104 cells per well in 96-well Cytostar-T plates (Amersham Biosciences) and incubated overnight. Uptake buffer was prepared containing 25 mM Hepes, 125 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.3 mM CaCl2, and 5.6 mM glucose (pH 7.4). Growth medium was aspirated, and cells were washed once with uptake buffer. Inhibitor y compounds and substrate (anandamide or LY2318912) were diluted in uptake buffer containing 2% fatty-acid-free BSA (final concentration, 1% BSA). Cells were incubated with inhibitory compounds for 10 min before addition of 5 μM[14C]anandamide or 5 nM [125I]LY2318912. After addition of substrate, cells were incubated at room temperature, and total substrate accumulation was determined by counting for 1 min per well with a Trilux Microbeta counter (PerkinElmer). Kinetic constants of [14C]anandamide uptake did not differ over temporal assay conditions of minutes to hours; therefore, incubation times were optimized for convenience and robust signal window (e.g., uptake-inhibition assays, 16-20 h, Fig. 1b; kinetic assays, 1 h, Fig. 2). All inhibition curves were fit to a single site model.
Fig. 1.

Identification of a high-affinity anandamide (N-arachidonoylethanolamide; AEA) transporter binding site with LY2318912. (a) Chemical structures. (b) Anandamide uptake in RBL-2H3 cells is inhibited by LY2318912 and LY2183240. (c) Binding of [125I]LY2318912 to P2 membranes prepared from RBL-2H3, wild-type HeLa (FAAH-/-), and human FAAH-transfected HeLa cells. (d) Competitive inhibition of [125I]LY2318912 binding to RBL-2H3 membranes by LY2183240, LY2318912, and anandamide. (e) Effect of FAAH expression on anandamide uptake and FAAH enzymatic activity. (f) Uptake of [14C]anandamide or [125I]LY2318912 by RBL-2H3 cells as a function of time. All data represent the mean of duplicate or triplicate determinations and are representative of at least three separate experiments, with the exception of saturation binding data (n = 2). Error bars for triplicate determinations indicate SEM.
Fig. 2.

Competitive nature of LY2318912 vs. AM404 for inhibition of anandamide uptake. (a) Effect of LY2318912 on anandamide uptake saturation curve in RBL-2H3 cells. (b) Lineweaver-Burk double-reciprocal plots depicting the competitive nature of anandamide uptake inhibition by free LY2318912. (c) Effects of AM404 on anandamide uptake saturation curve in RBL-2H3 cells. (d) Lineweaver-Burk double-reciprocal plots depicting the noncompetitive nature of anandamide uptake inhibition by AM404. All data represent the mean of duplicate determinations and are representative of at least three separate experiments (error bars indicate SEM). AEA, anandamide (N-arachidonoylethanolamide).
Ligand Binding Assays. Ligand binding assays were performed in uptake buffer in the presence of 0.3% fatty-acid-free BSA at 30°C in polypropylene 96-well blocks. Except for the kinetic assays, all incubations were allowed to equilibrate for 30 min. Incubations were started by addition of [125I]LY2318912 into triplicate wells containing 200 μg of P2 membrane protein. Nonspecific binding was determined by preincubation of the membranes with 10 μM LY2183240. After incubation, P2 membranes were harvested by using a 9600 Harvester (Tomtec) onto a glass fiber printed filtermat that had been soaked in 0.3% polyethyleneimine in 50 mM Tris at 4°C for at least 2 h. After harvest of membranes, filter mats were washed with 50 mM ice-cold Tris buffer (pH 7.4). Filters were dried at room temperature, infused with melt-on scintillant sheets, and counted for 30 s per well on a 1205 Betaplate scintillation counter (PerkinElmer). Specific binding was defined as the difference between total binding and binding in the presence of 10 μM LY2183240.
FAAH Enzymatic Assays. Assays were performed by a modification of the method described in ref. 44. Briefly, P2 plasma membrane preparations were incubated with 5 nM [ethanolamine-1-3H] anandamide (American Radiolabeled Chemicals, St. Louis) for various times in the presence or absence of 100 μM AM404. Reactions were terminated by extraction in two volumes of chloroform/methanol (1:1, vol/vol). Nonspecific cpm was defined with 1 μM PMSF. Production of [3H]ethanolamine was determined by liquid scintillation counting of aqueous phase.
Statistical Analysis of in Vitro Data. IC50 values, error analysis, and statistical significance were calculated by using prism (Version 4.03, GraphPad, San Diego). Data are given as the mean ± SEM of at least three independent determinations (with the exception of saturation binding assays), each in duplicate.
Brain Tissue Anandamide Measurements (Rat). Anandamide in cerebellar tissue samples from male Sprague-Dawley rats (Harlan Labs, Indianapolis; weight, 200-250 g) (n = 4-8 per group) was measured 90 min after i.p. drug injection by using a liquid chromatography/tandem MS (LC/MSMS) analysis system (triple quadrapole mass spectral detector; API 3000, Applied Biosystems) with fast gradient elution and a C18 column. Results are expressed as mean ± SEM nanograms of anandamide per millgrams of protein.
Formalin Pain Model (Rat). Male Sprague-Dawley rats (Harlan Labs; weight: 200-250 g) were injected s.c. into the plantar surface of the right hind paw with 50 μl of 5% formalin (n = 8 per group). Paw-licking behavior was measured from 0 to 60 min in 5-min blocks after formalin injection, and data are expressed as mean ± SEM paw-licking events or total paw-licking events in late phase (11-40 min).
Rotorod Test (Rat). Motor control and coordination were tested by using an automated accelerating rotorod (Omnitech Electronics, Columbus, OH). Rats (see above) (n = 8 per group) were trained (three times) to perform on the rotorod at 24 h before drug testing. Data are expressed as the mean ± SEM time on rotorod.
Statistical Analysis of in Vivo Data. Statistical analysis was performed with one-way ANOVA and Duncan's or Dunnett's post hoc tests where appropriate.
Results and Conclusions
To identify an anandamide transporter binding site, we first developed a class of structurally similar nonaliphatic ligands that potently inhibit [14C]anandamide uptake. Functional [14C]anandamide uptake is inhibited in a live cell assay by 5-biphenyl-4-ylmethyl-tetrazole-1-carboxylic acid dimethylamide (LY2183240, Fig. 1a; IC50 = 270 ± 29.4 pM, Fig. 1b). To create a radioligand and photoaffinity probe, iodo and azido groups were added to LY2183240, forming 5-[(4-azido-3-iodo-benzoylamino)-methyl]-tetrazole-1-carboxylic acid dimethylamide (LY2318912, Fig. 1a), which resulted in a modest shift of uptake inhibition potency (LY2318912, IC50 = 7.27 ± 0.510 nM, Fig. 1b) from the parent compound, LY2183240. These results indicate that LY2183240 and LY2318912 are highly potent inhibitors of anandamide uptake that are structurally distinct from the anandamide-like molecules that are commonly used to probe uptake (e.g., AM404, Fig. 1a; IC50 = 14.9 ± 1.73 μM, Fig. 1b) (30).
The radioligand, [125I]LY2318912, was used to characterize high-affinity binding sites that were detected in RBL-2H3 (Kd = 7.62 ± 1.18 nM, Bmax = 31.6 ± 1.80 fmol/mg protein), wild-type HeLa (FAAH-/-) (Kd = 7.06 ± 1.69 nM, Bmax = 32.2 ± 2.98 fmol/mg protein), and human FAAH-transfected HeLa (Kd = 11.2 ± 3.34 nM, Bmax = 24.3 ± 3.08 fmol/mg protein) P2 membranes (Fig. 1c). Binding of [125I]LY2318912 is temperature-sensitive (data not shown), rapid (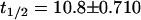 min), and fully displaceable (Fig. 1d), consistent with a plasma membrane protein binding site. The observation that the binding affinity of [125I]LY2318912 to these membranes is positively associated with the potency of functional [14C]anandamide uptake inhibition of the unlabeled congener (IC50 = 7.27 ± 0.510 nM) suggests that this binding site is involved directly in anandamide uptake. The fact that neither the binding affinity (Kd) nor the Bmax change significantly between FAAH-negative and FAAH-expressing cell types indicates that this binding site is independent of FAAH.
min), and fully displaceable (Fig. 1d), consistent with a plasma membrane protein binding site. The observation that the binding affinity of [125I]LY2318912 to these membranes is positively associated with the potency of functional [14C]anandamide uptake inhibition of the unlabeled congener (IC50 = 7.27 ± 0.510 nM) suggests that this binding site is involved directly in anandamide uptake. The fact that neither the binding affinity (Kd) nor the Bmax change significantly between FAAH-negative and FAAH-expressing cell types indicates that this binding site is independent of FAAH.
To eliminate the possibility that FAAH contributes to [125I]LY2318912 binding, we verified the presence or absence of FAAH activity in RBL, HeLa (FAAH-/-) and FAAH-transfected HeLa cells by means of [3H]-anandamide hydrolysis. These studies confirmed that wild-type HeLa (FAAH-/-) cells contain no measurable FAAH activity, whereas RBL-2H3 and human FAAH-transfected HeLa cells display robust FAAH activity (Fig. 1e). These results are consistent with findings (45, 46) demonstrating no detectable FAAH expression or anandamide hydrolysis activity in wild-type HeLa (FAAH-/-) cells.
If this binding site were indeed an anandamide transport protein, then [125I]LY2318912 should be displaceable with its endogenous agonist, anandamide. The rank order of potency (Ki) for [125I]LY2318912 binding is as follows: LY2183240, Ki = 540 ± 170 pM; LY2318912, Ki = 3.73 ± 1.18 nM; and anandamide, Ki = 23.4 ± 0.401 μM (Fig. 1d). These affinities of LY2183240 and LY2318912 are consistent with the functional inhibition (IC50) of [14C]anandamide uptake shown in Fig. 1b. Also, the relatively low anandamide binding affinity (Ki) is similar to reports of anandamide uptake affinity (Km) in RBL-2H3 cells (47, 48) (Fig. 2a). The positive association between the functional inhibition of anandamide uptake (IC50) and the binding characteristics of LY2318912 (Kd), as well as its displacement with anandamide (Ki), further strengthen the argument that this high-affinity binding site is an anandamide transporter.
To determine the nature of anandamide transporter inhibition by LY2318912, functional anandamide uptake competition assays were performed. The Michaelis constant (Km = 4.69 ± 0.460 μM) (Fig. 2a) of anandamide uptake saturation is lower than any reported value for RBL-2H3 cells, which range from 9.3 (47) to 33 (48) μM. Similar to other studies, anandamide uptake is saturable, as well as time- and temperature-dependent (34, 43, 45, 48, 49). When [14C]anandamide uptake is measured in the presence of LY2318912 (vehicle, 5 and 10 nM), Km is shifted to the right (4.69 ± 0.460, 13.4 ± 2.23, and 26.6 ± 5.24 μM, respectively), with little change in Vmax (2.15 × 10-17, 1.91 × 10-17, and 1.83 × 10-17 mol/min per cell, respectively) (Fig. 2 a and b), indicating that LY2318912 acts as a competitive antagonist at the anandamide binding site of the transporter. Moreover, LY2318912 is not accumulated by RBL-2H3 cells, suggesting that LY2318912 is not a transporter substrate (Fig. 1f). In contrast, AM404 (vehicle, 5 and 10 μM) does not shift Km (5.96 ± 0.612, 5.92 ± 0.809, and 6.58 ± 1.16 μM, respectively) (Fig. 2 c and d) yet decreases Vmax (2.23 × 10-17, 1.99 × 10-17, and 1.55 × 10-17 mol/min per cell, respectively), suggesting that it acts as a noncompetitive inhibitor of anandamide uptake in RBL-2H3 cells. Although these results contrast a previous report in RBL-2H3 cells in which the Km was right shifted by AM404 (34), they are consistent with a molecule that is a substrate for transport (35) and an inhibitor of FAAH activity. Together, the results shown in Figs. 2 and 1f indicate that LY2318912 competitively inhibits the anandamide binding site of the transporter and is not a substrate of the transporter, suggesting that it is not only structurally distinct from anandamide-like inhibitors but also functionally unique.
If the described binding site was an anandamide transporter, then administration of selective binding site inhibitors would elevate endocannabinoid levels and generate a relevant physiological and behavioral response. The i.p. administration of LY2183240 results in a dose-dependent increase in anandamide concentrations in rat cerebellum (ED50 = 1.37 ± 0.980 mg/kg) (Fig. 3a), consistent with the action of an anandamide uptake blocker in vivo. Given the importance of cannabinoids in pain control, analgesia was used as an anandamide-relevant biological endpoint with which to test our hypothesis. LY2183240 (i.p.) dose-dependently attenuates formalin-induced paw-licking pain behavior in the formalin model of persistent pain mechanisms (Fig. 3 c and d). Also, combined subthreshold doses of LY2183240 (3 mg/kg) and anandamide significantly decrease formalin-induced late-phase pain behavior (64.6 ± 15.5% reduction from vehicle control). These data suggest that blockade of the anandamide transporter binding site with LY2183240 generates physiologically and behaviorally relevant cannabimimetic responses.
Fig. 3.

Effects of LY2183240 administration (i.p.) on rats. (a) Concentration of anandamide in rat brain cerebellum. (b) Rotorod performance 30 and 90 min after administration of LY2183240. (c) Formalin-induced paw-licking pain behavior, expressed as mean paw-licking events. Data were collected at 5-min intervals over a 50-min time period, after the administration of intraplantar formalin. (d) Formalin-induced paw-licking pain behavior from c expressed as total paw-licking events in the early (0-5 min) and late (11-40 min) phases. All in vivo studies were performed in male Sprague-Dawley rats (200-250 g) and are representative of at least three separate experiments (error bars indicate SEM). *, P < 0.05, compared with vehicle group (Dunnett's test).
Cannabinoid receptor agonists, such as Δ9-THC, have unwanted side effects, including dulling of short-term memory, psychotropic disturbances (50, 51), dependence liability (52), suppression of fertility and the immune system (53, 54), and hypolocomotion (55). In contrast, a 30 mg/kg dose of LY2183240 shows no overt behavioral changes and no performance deficits in the rotorod model in rats (Fig. 3b), indicating that LY2183240 has minimal impact on motoric responses in rodents. The lack of effects of LY2183240 on motor performances suggests that anandamide transport blockade may be associated with a reduced side-effect profile compared with ligands that directly impact CB1 receptor sites. In contrast to indiscriminate activation of cannabinoid receptors by direct agonists, anandamide transport antagonists would hypothetically increase the signaling intensity of anandamide and related endocannabinoids only at active sites of release, impacting the endocannabinoid system in its tonically active state rather than perturbing the system in a nonphysiological manner. This strategy has proven to be highly successful for other neurotransmitter systems, including therapies used for depression and pain that inhibit biogenic amine transport proteins.
By using a selective ligand, a high-affinity anandamide transporter binding site was identified in RBL-2H3 cells with binding characteristics that positively associate with the pharmacological properties of anandamide uptake. Observation of indistinguishable binding characteristics in FAAH-/- HeLa cells confirms that the binding site is distinct from FAAH. There has been much debate as to the existence of a specific anandamide transport protein, making the identification of this binding site a significant advancement in the understanding of endocannabinoid signaling. The pharmacological properties of this binding site could possibly facilitate the testing of alternative anandamide transport hypotheses, including diffusional, endocytic, and FAAH-mediated mechanisms (37). Ultimately, cloning and expression of this transport protein should best define its role and regulation.
Acknowledgments
We thank Dr. Jeffrey Witkin for help preparing the manuscript, Dr. Paul Emmerson for providing significant intellectual and technical expertise on the topic of receptor pharmacology, Amy A. Webster for technical assistance in the rotorod and formalin paw-licking assays, and Brad Wainscott for assistance with Lineweaver-Burk analysis.
Author contributions: S.A.M., A.K.D.-C., B.-P.Y., R.M.A.S., and D.L. performed experiments; C.C.F., J.M.S., S.I., Y.-C.X., L.P., D.A.S., and G.G.N. contributed intellectual/technical input; and C.C.F., S.A.M., and A.K.D.-C. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Δ9-THC, Δ9-tetrahydrocannabinol; FAAH, fatty acid amide hydrolase.
See Commentary on page 17541.
References
- 1.Kwiatkowska, M., Parker, L. A., Burton, P. & Mechoulam, R. (2004) Psychopharmacology 174, 254-259. [DOI] [PubMed] [Google Scholar]
- 2.Mechoulam, R. & Hanus, L. (2001) Pain Res. Manag. 6, 67-73. [DOI] [PubMed] [Google Scholar]
- 3.Walker, J. M. & Huang, S. M. (2002) Pharmacol. Ther. 95, 127-135. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez de Fonseca, F., Carrera, M. R., Navarro, M., Koob, G. F. & Weiss, F. (1997) Science 276, 2050-2054. [DOI] [PubMed] [Google Scholar]
- 5.Baker, D., Pryce, G., Croxford, J. L., Brown, P., Pertwee, R. G., Huffman, J. W. & Layward, L. (2000) Nature 404, 84-87. [DOI] [PubMed] [Google Scholar]
- 6.Hepler, R. S. & Frank, I. R. (1971) J. Am. Med. Assoc. 217, 1392 (lett.). [Google Scholar]
- 7.Devane, W. A., Dysarz, F. A., III, Johnson, M. R., Melvin, L. S. & Howlett, A. C. (1988) Mol. Pharmacol. 34, 605-613. [PubMed] [Google Scholar]
- 8.Matsuda, L. A., Lolait, S. J., Brownstein, M. J., Young, A. C. & Bonner, T. I. (1990) Nature 346, 561-564. [DOI] [PubMed] [Google Scholar]
- 9.Munro, S., Thomas, K. L. & Abu-Shaar, M. (1993) Nature 365, 61-65. [DOI] [PubMed] [Google Scholar]
- 10.Devane, W. A., Hanus, L., Breuer, A., Pertwee, R. G., Stevenson, L. A., Griffin, G., Gibson, D., Mandelbaum, A., Etinger, A. & Mechoulam, R. (1992) Science 258, 1946-1949. [DOI] [PubMed] [Google Scholar]
- 11.Akerman, S., Kaube, H. & Goadsby, P. J. (2004) Br. J. Pharmacol. 142, 1354-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross, R. A. (2003) Br. J. Pharmacol. 140, 790-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barann, M., Molderings, G., Bruss, M., Bonisch, H., Urban, B. W. & Gothert, M. (2002) Br. J. Pharmacol. 137, 589-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan, P. (1995) J. Neurophysiol. 73, 907-910. [DOI] [PubMed] [Google Scholar]
- 15.Oz, M., Zhang, L. & Morales, M. (2002) Synapse 46, 150-156. [DOI] [PubMed] [Google Scholar]
- 16.Kimura, T., Ohta, T., Watanabe, K., Yoshimura, H. & Yamamoto, I. (1998) Biol. Pharm. Bull. 21, 224-226. [DOI] [PubMed] [Google Scholar]
- 17.Felder, C. C., Briley, E. M., Axelrod, J., Simpson, J. T., Mackie, K. & Devane, W. A. (1993) Proc. Natl. Acad. Sci. USA 90, 7656-7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pertwee, R. (1993) Gen. Pharmacol. 24, 811-824. [DOI] [PubMed] [Google Scholar]
- 19.Mackie, K., Devane, W. A. & Hille, B. (1993) Mol. Pharmacol. 44, 498-503. [PubMed] [Google Scholar]
- 20.Cravatt, B. F., Giang, D. K., Mayfield, S. P., Boger, D. L., Lerner, R. A. & Gilula, N. B. (1996) Nature 384, 83-87. [DOI] [PubMed] [Google Scholar]
- 21.Cadas, H., Schinelli, S. & Piomelli, D. (1996) J. Lipid Mediat. Cell Signal. 14, 63-70. [DOI] [PubMed] [Google Scholar]
- 22.Di Marzo, V., Fontana, A., Cadas, H., Schinelli, S., Cimino, G., Schwartz, J. C. & Piomelli, D. (1994) Nature 372, 686-691. [DOI] [PubMed] [Google Scholar]
- 23.Di Marzo, V., De Petrocellis, L., Sepe, N. & Buono, A. (1996) Biochem. J. 316, 977-984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cadas, H., Gaillet, S., Beltramo, M., Venance, L. & Piomelli, D. (1996) J. Neurosci. 16, 3934-3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cadas, H., di Tomaso, E. & Piomelli, D. (1997) J. Neurosci. 17, 1226-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson, R. I. & Nicoll, R. A. (2001) Nature 410, 588-592. [DOI] [PubMed] [Google Scholar]
- 27.Wilson, R. I., Kunos, G. & Nicoll, R. A. (2001) Neuron 31, 453-462. [DOI] [PubMed] [Google Scholar]
- 28.Wilson, R. I. & Nicoll, R. A. (2002) Science 296, 678-682. [DOI] [PubMed] [Google Scholar]
- 29.Deutsch, D. G. & Chin, S. A. (1993) Biochem. Pharmacol. 46, 791-796. [DOI] [PubMed] [Google Scholar]
- 30.Beltramo, M., Stella, N., Calignano, A., Lin, S. Y., Makriyannis, A. & Piomelli, D. (1997) Science 277, 1094-1097. [DOI] [PubMed] [Google Scholar]
- 31.Hillard, C. J., Edgemond, W. S., Jarrahian, A. & Campbell, W. B. (1997) J. Neurochem. 69, 631-638. [DOI] [PubMed] [Google Scholar]
- 32.Oddi, S., Bari, M., Battista, N., Barsacchi, D., Cozzani, I. & Maccarrone, M. (2005) Cell. Mol. Life Sci. 62, 386-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maccarrone, M., Bari, M., Lorenzon, T., Bisogno, T., Di Marzo, V. & Finazzi-Agro, A. (2000) J. Biol. Chem. 275, 13484-13492. [DOI] [PubMed] [Google Scholar]
- 34.Rakhshan, F., Day, T. A., Blakely, R. D. & Barker, E. L. (2000) J. Pharmacol. Exp. Ther. 292, 960-967. [PubMed] [Google Scholar]
- 35.Piomelli, D., Beltramo, M., Glasnapp, S., Lin, S. Y., Goutopoulos, A., Xie, X. Q. & Makriyannis, A. (1999) Proc. Natl. Acad. Sci. USA 96, 5802-5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glaser, S. T., Kaczocha, M. & Deutsch, D. G. (2005) Life Sci. 77, 1584-1604. [DOI] [PubMed] [Google Scholar]
- 37.McFarland, M. J. & Barker, E. L. (2004) Pharmacol. Ther. 104, 117-135. [DOI] [PubMed] [Google Scholar]
- 38.Glaser, S. T., Abumrad, N. A., Fatade, F., Kaczocha, M., Studholme, K. M. & Deutsch, D. G. (2003) Proc. Natl. Acad. Sci. USA 100, 4269-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McFarland, M. J., Porter, A. C., Rakhshan, F. R., Rawat, D. S., Gibbs, R. A. & Barker, E. L. (2004) J. Biol. Chem. 279, 41991-41997. [DOI] [PubMed] [Google Scholar]
- 40.Hillard, C. J. & Jarrahian, A. (2003) Br. J. Pharmacol. 140, 802-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ortar, G., Ligresti, A., De Petrocellis, L., Morera, E. & Di Marzo, V. (2003) Biochem. Pharmacol. 65, 1473-1481. [DOI] [PubMed] [Google Scholar]
- 42.Fegley, D., Kathuria, S., Mercier, R., Li, C., Goutopoulos, A., Makriyannis, A. & Piomelli, D. (2004) Proc. Natl. Acad. Sci. USA 101, 8756-8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ligresti, A., Morera, E., van der Stelt, M., Monory, K., Lutz, B., Ortar, G. & Di Marzo, V. (2004) Biochem. J. 380, 265-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Omeir, R. L., Chin, S., Hong, Y., Ahern, D. G. & Deutsch, D. G. (1995) Life Sci. 56, 1999-2005. [DOI] [PubMed] [Google Scholar]
- 45.Day, T. A., Rakhshan, F., Deutsch, D. G. & Barker, E. L. (2001) Mol. Pharmacol. 59, 1369-1375. [DOI] [PubMed] [Google Scholar]
- 46.Wilson, S. J., Lovenberg, T. W. & Barbier, A. J. (2003) Anal. Biochem. 318, 270-275. [DOI] [PubMed] [Google Scholar]
- 47.Jacobsson, S. O., Wallin, T. & Fowler, C. J. (2001) J. Pharmacol. Exp. Ther. 299, 951-959. [PubMed] [Google Scholar]
- 48.Bisogno, T., Maurelli, S., Melck, D., De Petrocellis, L. & Di Marzo, V. (1997) J. Biol. Chem. 272, 3315-3323. [DOI] [PubMed] [Google Scholar]
- 49.Jacobsson, S. O. & Fowler, C. J. (2001) Br. J. Pharmacol. 132, 1743-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hill, M. N., Froc, D. J., Fox, C. J., Gorzalka, B. B. & Christie, B. R. (2004) Eur. J. Neurosci. 20, 859-863. [DOI] [PubMed] [Google Scholar]
- 51.Harder, S. & Rietbrock, S. (1997) Int. J. Clin. Pharmacol. Ther. 35, 155-159. [PubMed] [Google Scholar]
- 52.Le Foll, B. & Goldberg, S. R. (2005) J. Pharmacol. Exp. Ther. 312, 875-883. [DOI] [PubMed] [Google Scholar]
- 53.Dalterio, S., Badr, F., Bartke, A. & Mayfield, D. (1982) Science 216, 315-316. [DOI] [PubMed] [Google Scholar]
- 54.Pacifici, R., Zuccaro, P., Pichini, S., Roset, P. N., Poudevida, S., Farre, M., Segura, J. & De la Torre, R. (2003) J. Am. Med. Assoc. 289, 1929-1931. [DOI] [PubMed] [Google Scholar]
- 55.Prescott, W. R., Gold, L. H. & Martin, B. R. (1992) Psychopharmacology 107, 117-124. [DOI] [PubMed] [Google Scholar]