

Abstract
The steady-state abundance of an mRNA is determined by the balance between transcription and decay. Although regulation of transcription has been well studied both experimentally and computationally, regulation of transcript stability has received little attention. We developed an algorithm, MatrixREDUCE, that discovers the position-specific affinity matrices for unknown RNA-binding factors and infers their condition-specific activities, using only genomic sequence data and steady-state mRNA expression data as input. We identified and computationally characterized the binding sites for six mRNA stability regulators in Saccharomyces cerevisiae, which include two members of the Pumilio-homology domain (Puf) family of RNA-binding proteins, Puf3p and Puf4p. We provide computational and experimental evidence that regulation of mRNA stability by these factors is modulated in response to a variety of environmental stimuli.
Keywords: cis-regulatory element, gene expression, microarray, mRNA decay, Puf protein
Most genomic studies of gene expression regulation focus on transcription rather than on mRNA decay. However, recent years have witnessed increasing interest in the regulation of mRNA stability (1, 2). In Saccharomyces cerevisiae, transcriptional arrest microarray experiments have been used to infer gene-specific mRNA decay rates (3–5); similar studies were performed in a variety of other organisms (6–11). In these experiments, transcription is halted by using a heat-labile mRNA polymerase or a chemical treatment, and genome-wide concentrations of mRNAs are measured over time. These studies have provided insight into general trends, such as coordinated decay rates between functionally related genes. However, condition-specific regulation of mRNA stability was not addressed in any of the yeast studies (3–5). García-Martínez et al. (12) used a genomic run-on method to infer genome-wide mRNA decay rates in S. cerevisiae from the discrepancies between the steady state mRNA abundances and their transcription rates. Although these authors provided evidence for condition-specific regulation of mRNA stability, they did not identify any responsible transfactors or regulatory sequence elements. Gerber et al. (13) measured the genome-wide targets of five yeast proteins from the Pumilio-homology domain (Puf) family, a group of RNA-binding proteins that posttranscriptionally regulate their targets by binding to 3′ UTRs and are found in a variety of eukaryotes (14). Although they provided no evidence of regulation of mRNA stability by the Pufs, their results demonstrated that the targets of the Pufs have clear biases for specific functional categories of genes. Kellis et al. (15) were not studying regulation of mRNA stability per se, but they did identify six 3′ cis-regulatory elements that were conserved between four species of Saccharomyces.
In this study, we took a different approach that takes advantage of the fact that information about mRNA stability is implicitly represented in steady-state mRNA abundances. Using only steady state gene expression data and nucleotide sequence data, we were able to show that Puf3p, Puf4p, and several unidentified factors, whose sequence specificities we nevertheless could infer, alter the stability of their target genes in response to specific environmental conditions. Our findings suggest that such dynamic regulation of mRNA stability is not a special-case phenomenon but rather a pervasive regulatory mechanism that is used to rapidly adapt cellular processes to a changing environment.
For a given gene g, the transcription rate αg and the mRNA decay rate τg together determine the steady-state transcript concentration [mRNA]g
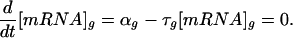 |
Therefore, in a standard comparative microarray hybridization, the log-ratio of mRNA concentrations between the two experimental conditions “red” and “green” is given by
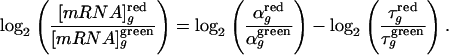 |
In other words, any change observed in the abundance of a transcript could be the result of a change in its transcription rate, a change in its decay rate, or both. We previously used regression analysis to model the change in transcription rate αg in terms of binding sites in the gene's promoter and changes in the post translational activity of the corresponding transcription factors (16). In this study, we successfully modeled changes in decay rate τg in terms of inferred binding sites in the 3′ UTR of the mRNA transcript and changes in activity of RNA-binding factors.
We developed an algorithm, MatrixREDUCE, and applied it to a set of ≈750 publicly available microarray hybridizations for S. cerevisiae and the 200 nt downstream of each yeast ORF (Fig. 1). Our algorithm discovered the nucleotide binding specificities, in the form of position-specific affinity matrices (PSAMs), and condition-specific activities for transfactors that dynamically regulate mRNA stability. These inferred activities were combined into transfactor activity profiles (TFAPs) across all ≈750 conditions, which enabled us to identify the physiological states in which each putative transfactor (de-)stabilizes its target mRNAs. We showed that two of the discovered PSAMs correspond to the RNA-binding proteins Puf3p and Puf4p.
Fig. 1.

MatrixREDUCE data flow. Using microarray data for 758 pairs of experimental conditions and the downstream 200 nt of every yeast ORF as input to MatrixREDUCE, we identified PSAMs corresponding to putative mRNA stability regulators and inferred TFAPs across all microarray conditions. For each discovered PSAM, “responder analysis” (see Methods) produced a list of putative target genes that are likely to contain functional binding sites for each transfactor modeled by a PSAM.
Not every mRNA that contains a match to a particular PSAM will necessarily be regulated by the corresponding transfactor. However, as previously observed by Gao et al. (17), TFAPs can be used to enhance the predictions of functional target genes. Individual transcripts that not only contain a good match to a PSAM but also have an mRNA expression profile across the ≈750 conditions that correlates well with the TFAP were identified by our “responder analysis” method as the best candidates for genes whose expression is actually regulated by the corresponding transfactor. Additional biological annotation of the discovered PSAMs was provided by scoring these targets for enrichment in Gene Ontology (18) functional categories. Finally, we provided experimental evidence that Puf3p acts as a condition-specific regulator of mRNA stability, which validated our computational method and further demonstrated the reliability of our computational predictions.
Methods
Sequence Data. Yeast genomic sequence and annotation were downloaded from the Saccharomyces Genome Database (19). The average length of 3′ UTRs in fungi based on known sequences is about 200 nucleotides (20). Thus, we retrieved 200bp downstream of every yeast ORF to approximate the 3′ UTRs. The downstream sequence was truncated if it overlapped with a neighboring ORF. To avoid biases, redundant sequences were removed by using blastn (21) to compare the sequence set to itself. For groups of genes linked by E values of 10–10 or smaller, one gene was randomly chosen to represent the group. This resulted in exclusion of ≈5% of the genes in the genome. The same procedure was used to obtain a nonredundant set of 600 base pairs upstream of every yeast ORF for the 5′ sequence analysis.
Expression Data. Data from 804 gene expression microarrays was gathered from publication supplements. All data were analyzed as the log2 ratio of background-corrected spot intensities between two experimental conditions in a single hybridization. To eliminate the deleterious effects of outliers and skewed expression data on later analysis, all values for a given set were first subjected to Grubbs' test for outliers (P value = 10–6; ref. 22). A given data set was only used for further analysis if, after being purged of outliers, it was roughly nonskewed (index of skewness <1.0) and roughly normally distributed (Kolmogorov–Smirnov test statistic <0.1). A total of 758 of 804 data sets passed these filters.
MatrixREDUCE. Regulatory element detection using correlation with expression (REDUCE; ref. 16) fits a multivariate model to gene expression where the explanatory variables are occurrences of transfactor binding sites within regulatory regions
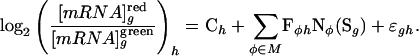 |
[1] |
Here, φ is one of the transfactors in the model M; Fφh is a regression coefficient that quantifies the change in posttranslational activity of the factor φ in hybridization h; Nφ(Sg) scores the occurrences of binding sites for factor φ in the regulatory sequence Sg of gene g; εgh represents the residuals; and Ch is an intercept term for hybridization h.
Whereas, in the original MotifREDUCE (16), Nφ(Sg) was simply the count of a particular oligonucleotide motif in the regulatory region of gene g, MatrixREDUCE uses a more powerful representation of the binding specificity of the transfactor in the form of a PSAM
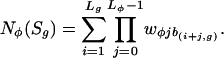 |
[2] |
For each nucleotide position j in the PSAM for factor φ, there is a weight wφjb for each nucleotide b. The weights range between zero and one. The score for a given position i in the sequence for gene g represents the binding affinity relative to the optimal binding sequence and equals the product of the weights w for the nucleotides found at positions (i + j) with 0 ≤ j < Lφ, where Lφ is the length of the PSAM; b(k,g) represents the base at position k in the sequence Sg. The score Nφ(Sg) for the entire regulatory sequence Sg equals the sum of the scores for each position i in the sequence of length Lg.
MatrixREDUCE performs a forward variable selection on all hybridizations in the expression data set simultaneously. At each iteration, the correlation is calculated between the log2 expression ratios for each hybridization and the occurrences of each dyad motif from a large dictionary (from all possible pairs of trimers with gaps from zero to eleven nucleotides) in the 3′ sequence associated with each gene. Then, the combination of the best-correlating motif and hybridization is identified. This dyad motif plus three flanking N′s on each side (12 ≤ Lφ ≤ 23; for example, Puf3p element: NNNGTANATANNN) is converted into a seed matrix by replacing nucleotides with weights of ones (acceptable) and zeros (not acceptable). The seed matrix serves as the starting point for the PSAM discovery, which is run on the 3′ sequence and the expression data for the hybridization in which the seed motif scored best. The PSAM discovery procedure uses a conjugate gradient numerical minimization algorithm to find the best fit parameters (Ch, Fφh, and the set of all wφjb) for a given hybridization h and transfactor φ by minimizing the χ2φh sum of squared deviations between the model and the expression data
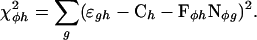 |
[3] |
Once the parameter fit converges, the new PSAM is added to the multivariate model M. The updated model is then fit separately to every hybridization, and the residuals are used for selecting the next seed motif. A Bonferroni-corrected P value cutoff equal to 0.05 (partial F test) is used as the threshold significance for the final PSAM added to the model.
It may be possible to detect PSAMs within 3′ UTRs that are not involved in regulation of mRNA stability, such as mRNA trafficking signals. Such a feature could still have a correlation with gene expression that would be caused by frequent occurrence in genes controlled by a particular transcription factor. Thus, to increase the likelihood that any discovered 3′ PSAM was indeed involved in regulation of mRNA decay, PSAM discovery was first performed on upstream sequences (results not shown here). Then, downstream sequences were analyzed by using the residuals εgh of the model based on 5′ PSAMs.
MatrixREDUCE was implemented in perl and c. Software and additional supplementary information are available for download upon request (see supporting information, which is published on the PNAS web site).
TFAP Generation. One of the outputs of MatrixREDUCE is the set of regression coefficients Fφh for each PSAM corresponding to factor φ in the multivariate linear model fit to each microarray hybridization h. MatrixREDUCE also reports a t value for each regression coefficient, which measures the deviation of Fφh from zero in units of its standard deviation. A TFAP was defined as the set of t values for a PSAM across all hybridizations. TFAPs were derived for each identified PSAM (Fig. 1). Our graphical representation of TFAPs was created with the help of treeview (23).
Matrix Logo Generation. We developed a graphical logo representation that highlights the most discriminative nucleotides and positions in the PSAM matrices. Because the parameters in our matrices represent physical interactions, the height of the nucleotides in the logo is not based on the usual information-theoretical measure. Instead, the height of each nucleotide is determined by subtracting the smallest weight for any nucleotide at that position and then dividing by the sum of all four weights. This method ensures that uninformative positions have zero letter height. However, the logos are only a visual aide; consult the supporting information for the actual PSAM weights.
Responder Analysis. Information for determining the most likely target genes for a given transfactor is provided by their sequence scores for the PSAM (Eq. 2) and the correlation between their mRNA expression profiles and the TFAP. We calculated Pearson correlations between PSAM regression coefficients Fφh and mRNA expression levels for all conditions. To keep the scale consistent between the two quantities, TFAPs composed of regression coefficients rather than t values were used. All genes were then ranked by their expression-TFAP correlation and their sequence scores Nφ(Sg). The optimal rank thresholds were determined by calculating the hypergeometric P value for every combination of ranks between the two metrics as follows
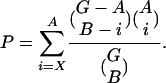 |
[4] |
Here, G is the number of genes in the genome; A is the number of genes with a rank equal to or higher than the rank being tested for the first metric; B is the number of genes with a rank equal to or higher than the rank being tested for the second metric (B ≥ A); and X is the number of genes in the overlap between both gene sets. Once the optimal P value was determined, genes were considered target genes if they had ranks above threshold for both the sequence score and the TFAP correlation.
Gene Ontology (GO) Analysis. GO annotations for yeast genes were retrieved from the Saccharomyces Genome Database (19) and the GO Consortium web site (18). For each identified PSAM, GO categories that contained more than one gene were scored for enrichment among its target genes by using the cumulative hypergeometric distribution. The resulting P values were Bonferroni-corrected for the number of categories scored. A corrected P value threshold of 10–3 was used to identify significantly regulated functional categories for each PSAM.
Identifying PSAMs as Puf Protein-Binding Sites. Although the PSAMs that MatrixREDUCE discovers reflect the binding affinity of transfactors, it is impossible to identify the corresponding transfactors without additional information. One such kind of additional information is protein binding microarray data. Gerber et al. (13) measured the genome-wide binding of the five yeast Puf proteins. It is expected that mRNAs that are more tightly bound by a Puf protein will be enriched for the specific binding sites for that Puf. We calculated correlations between the genome-wide downstream sequence match scores for each PSAM and the genome-wide log2-binding-ratios for each Puf. A strong positive correlation between sequence matches for a PSAM and the Puf binding data suggests that the PSAM is the binding site for the Puf.
Identifying PSAMs as Conserved Motifs. Although it is possible to compare the novel 3′ motifs from Kellis et al. (15) and our PSAMs at the sequence level, we chose to use the cumulative hypergeometric distribution to score the overlap between the lists of genes with downstream matches to the 3′ motifs from Kellis et al. (15) and the target lists for each of our PSAMs. If the number of shared genes between a Kellis et al. (15) motif and a PSAM was much larger than expected (P value < 10–10), then the motif and the PSAM were considered to represent the same cis-regulatory element.
Yeast Strains. The genotypes of the three S. cerevisiae strains used in the experimental studies are as follows: yWO7 (24) (yRP693), MATα, leu2-3,112, ura3-52, rpb1-1; yWO43 (24) (yRP1360), MATα, his4-539, leu2-3,112, trp1-1, ura3-52, cup1:LEU2/PM, rpb1-1, puf3:Neor; yWO50 (24) (yRP1546), MATa, his3-1,15, his4-539, leu2-3,112, trp1-1, ura3, rpb1-1, cox17:TRP1.
In Vivo mRNA Decay Analysis. Steady-state transcriptional shut-off experiments were performed essentially as described in Caponigro et al. (25) on strains yWO7, yWO43, and yWO50, which contain the temperature-sensitive rpb1-1 allele for RNA polymerase II (26). For carbon source analysis, yWO7 or yWO43 was transformed with plasmids expressing MFA2 RNA (pWO68) or the hybrid MFA2/COX17 3′-UTR RNA (pWO69) under the control of the constitutive GPD promoter. pWO68 and pWO69 were created by inserting SacI–HindIII fragments from either pRP485 containing MFA2 (27), or from pWO25 containing MFA2/COX17 3′-UTR (28), respectively, into pWO67. A 662-bp PCR product containing the GPD promoter was inserted between the EcoRI sites on pRP22 (27) to create pWO67. Transformed strains were grown in yeast peptone (YP) media supplemented with 2% glucose or 2% ethanol as a carbon source. A transcriptional shutoff was performed by expressing the MFA2 or MFA2/COX17 mRNAs to steady-state levels under the control of the constitutive GPD promoter, then transcription was rapidly repressed by a shift to high temperature. Northern blots were probed for the plasmid-derived MFA2 mRNA using oRP140 (29) or the MFA2/COX17 hybrid mRNA using oWO303 (5′-GTCAGTAAGATCGATCTAGAGGATCTCTTGGTTGTCG). For rapamycin treatment analysis, strain yWO50, which is deleted for the endogenous COX17 gene, was transformed with plasmids expressing MFA2 RNA (pRP485) or the hybrid MFA2/COX17 3′-UTR RNA (pWO25) under the control of the GAL1 UAS. Transformed strains were grown in selective media with 2% galactose. Rapamycin (Sigma), when used, was added to a final concentration of 0.2 μg/ml when the culture reached an OD600 of 0.3, then the cells were incubated a further 60 min before the temperature shift. Northern blots were probed for MFA2 mRNA using oRP140 or MFA2/COX17 hybrid mRNA using oWO2 (oCOX17-P; ref. 24). All Northern blots were quantified with a Molecular Dynamics PhosphorImager, and the signal for each RNA normalized for loading to the stable scRI RNA, an RNA polymerase III transcript (30).
Results and Discussion
MatrixREDUCE discovered eight PSAMs from the expression data set of ≈750 hybridizations and the downstream sequences of every gene. Six of the PSAMs showed significant correlation with differential expression (Bonferroni-corrected P value < 0.05) in >10% of the hybridizations, and the environmental conditions for those hybridizations suggested coherent biological functions. The other two PSAMs are not discussed here, but their TFAPs and target genes are available in the supporting information as unknown 1 and 2. Responder analysis was performed for each of the discovered PSAMs. Of those genes with predicted high-affinity binding sites (high PSAM sequence scores), only between 32.5% and 53.3% had TFAP correlations above threshold and were thus considered functional targets, which highlights the utility of responder analysis.
Puf3p Element (P3E). The first PSAM that we discovered using MatrixREDUCE, the P3E (Fig. 2) represents the binding specificity of the RNA-binding protein Puf3p. Our expression data-derived P3E weight matrix agrees with a described P3E (13), and P3E matches in downstream sequences strongly correlate with Puf3p binding microarray data (P value ≅ 10–291). When the P3E targets are scored for overlap with genes that have downstream matches to the conserved 3′ motifs reported by Kellis et al. (15), P3E targets are overwhelmingly overrepresented (P value < 10–10) among the gene lists for motifs 6 and 59. Upon visual inspection, motif 6 (WTATWTACADG) is indeed a close reverse complement of the P3E PSAM, and the P3E PSAM also fits within motif 59 (TRTAMATAKWT). The list of P3E targets with annotation as to whether they are bound by Puf3p (13) or contain motifs 6 and 59 is available in the supporting information.
Fig. 2.

Discovered 3′ PSAMs logos. The PSAMs were discovered by MatrixREDUCE using a large set of steady-state mRNA expression data and the 200 nt downstream of every ORF.
Gerber et al. (13) showed that Puf3p primarily binds to transcripts of proteins involved in mitochondrial function, but our results present evidence that Puf3p is a condition-specific regulator of mRNA stability. Specifically, Puf3p destabilizes mitochondrion-related transcripts when sugars such as glucose or fructose are present. The list of P3E targets is significantly enriched with mitochondrion-related transcripts, especially transcripts coding for components of mitochondrial ribosomes (Fig. 3), which is consistent with previous GO analysis of Puf3p targets (13). Fig. 4 A and B summarizes the condition-specific activities of the P3E. Transcripts containing a P3E match were up-regulated when a nonrepressing carbon source was present, which includes the late time points from a diauxic shift time course (31), the middle time points from stationary phase time courses (32, 33), and an ethanol carbon source condition (32). Concordantly, transcripts containing a P3E match were down-regulated when a repressing carbon source was present such as fructose, glucose, or sucrose conditions (32). However, not all strongly correlated conditions fit within the carbon source theme: for example, P3E matches positively correlated with expression under oxidative stress conditions (peroxide or menadione; refs. 32, 34, 35), proteosome inhibition experiments (36), and heat shock and diamide conditions (32).
Fig. 3.

Gene Ontology analysis. The enrichment of the PSAM targets in each category was quantified by using the cumulative hypergeometric distribution. The color scale corresponds to Bonferroni-corrected P values. The ontology to which a category belongs is indicated by C (subcellular component), P (biological process), or F (molecular function). Only a representative selection of all significant functional categories is shown. The significantly enriched functional categories for each PSAM were generally consistent with their inferred activity profiles (Fig. 4).
Fig. 4.

Transfactor activity profiles (TFAPs). (A–D) The direction and degree of the correlation between PSAM match score and mRNA expression data are represented by a color scale ranging from bright blue (strong negative correlation) to bright yellow (strong positive correlation). These correlations correspond to changes in activity of the mRNA stability-regulating transfactors whose binding specificities are modeled by the PSAMs. The triangles represent the progression of time within time course experiments. References for the microarray data sets used for the displayed analysis are as follows: diauxic shift (31); stationary phase, ethanol, fructose, glucose, sucrose, menadione, heat shock, diamide, nitrogen depletion, DTT, and amino acid and adenine starvation (32); peroxide (35); proteosome inhibition by PS-341 (36); H-2k(b) expression and tunicamycin (38); rapamycin (39); and puf4 deletion transcriptional arrest (5).
Experimental Validation of Puf3p Condition-Specific Activity. We experimentally verified that the ability of Puf3p to destabilize mitochondrion-related transcripts depends on the available carbon source. We have previously shown that COX17 is a target of Puf3p regulation, with Puf3p binding directly to the COX17 3′ UTR and promoting rapid deadenylation and decay of this transcript (24). We have also shown that the COX17 3′ UTR is sufficient to direct Puf3p decay regulation when attached to the ORF of MFA2 (28). Thus, transformed strains expressing either wild-type MFA2 mRNA or the MFA2/COX17 3′ UTR hybrid mRNA were grown in media containing either glucose (repressing) or ethanol (nonrepressing) as the carbon source. A transcriptional shut-off was performed (see Methods), and as expected, wild-type MFA2 mRNA decays with a half-life of 4 min in both growth conditions (Fig. 5). In contrast, the Puf3p-regulated MFA2/COX17 mRNA decays rapidly with a half-life of 2.5 min in the glucose media, but is stabilized ≈4-fold in the ethanol media to a half-life of 10.5 min (Fig. 5). To be certain that it was indeed Puf3p that was mediating the altered stability between the two media conditions, we repeated the above experiment in a puf3 deletion strain. As expected, in the absence of Puf3p, the MFA2/COX17 mRNA is stable, and the half-life is unaltered by media conditions (Fig. 5). There is a large difference between the decay rates of the wild-type MFA2 transcript and the MFA2/COX17 transcript in the absence of Puf3p activity. However, it is only important that the COX17 3′ UTR confers differential stability on the MFA2 ORF between glucose and ethanol-containing media conditions in the presence of Puf3p. The native MFA2 3′ UTR has properties that cause the transcript to be rapidly degraded in all conditions yet tested (37).
Fig. 5.

Regulation of Puf3p activity in response to a change in carbon source. Shown are Northern blot analyses of the decay of MFA2 mRNA or the hybrid MFA2/COX17 mRNA expressed from wild-type or puf3Δ yeast grown in media containing 2% glucose or 2% ethanol. Minutes after transcriptional repression are indicated above the set of blots, with the half-lives (t1/2) as determined from multiple experiments.
Puf4p Element (P4E). The P4E corresponds to the binding specificity of Puf4p (Fig. 2). Sequence matches to the P4E PSAM correlate well with binding by Puf4p (P value ≅ 10–15, t test). A binding site for Puf4p has been identified (13), and deletion of puf4 was shown to affect the stability of transcripts encoding ribosomal proteins and ribosome biogenesis factors (5); however, as with Puf3p, no previous evidence suggested that Puf4p controls mRNA stability in a condition-specific manner. Our analysis suggests that Puf4p regulates ribosomes in response to starvation. The top P4E targets were enriched with transcripts for ribosomal proteins and nucleolar proteins (Fig. 3), which is consistent with a previous analysis of the targets of Puf4p (13). Furthermore, P4E matches are associated with a decrease in transcript abundance for many late stationary phase time points (32, 33), the late time points from a diauxic shift time course (31), and the late time points from a nitrogen depletion time course (32). In addition to starvation conditions, P4E matches corresponded to decreased expression in heat shock conditions (32, 33) (Fig. 4A).
Grigull et al. (5) performed a microarray time course that compared genome-wide mRNA levels after a transcriptional arrest between a puf4 deletion strain and a wild-type strain. This experiment was performed in non-stress-inducing rich media conditions. Based on the results for P4E presented in Fig. 4A, Puf4p must either be a stabilizer active in non-stress conditions or a destabilizer active in stress conditions. If Puf4p acts as a stabilizer, we would expect to see a negative correlation with mRNA levels over the time course, because the Puf4p targets would be less stable in the mutant than in the wild type. If Puf4p acts as a destabilizer, we would expect to see no correlation over the time course because there would be no active Puf4p in either the wild type or the mutant. Fig. 4D shows that the P4E does not correlate with the differential mRNA levels between the puf4 mutant and wild type at 0 min, but only has a strong positive correlation in the later time points. Therefore, we posit that Puf4p is a destabilizer and is activated by the stress of a transcriptional arrest, causing the observed positive correlation.
Negative Response to Starvation Elements (NRSE1 and NRSE2). The NRSE1 and NRSE2 (Fig. 2) are PSAMs bound by unknown transfactors. The target mRNAs for the NRSE1 and NRSE2 have strong overlaps (P value < 10–10) with the genes hit by motif 60, TtTATAnTATATAnA, from Kellis et al. (ref. 15; see supporting information). NRSE1 and NRSE2 matches were both negatively correlated with expression for late stationary phase (32, 33) and diauxic shift (31) time points as well as several heat shock conditions (32, 33) (Fig. 4A). Targets of the NRSE1 and NRSE2 were enriched for transcripts encoding cytosolic ribosomal proteins and proteins involved in ribosome biogenesis (Fig. 3). Thus, the factors that recognize the NRSE1 and NRSE2 seem to play roles similar to Puf4p in regulating ribosomes. Additionally, the targets of the NRSE1 are enriched for transcripts encoding proteins involved in cytoskeleton biogenesis and cell polarity.
Positive Response to Starvation Element (PRSE). The PRSE (Fig. 2) is another PSAM corresponding to an unknown transfactor. The PRSE has a strong similarity in sequence to motif 66 (AATATTCTT) from Kellis et al. (ref. 15; see supporting information), but the P value is less extreme than the best target overlaps for other PSAMs (P value = 0.0004). Like Puf3p, the factor that binds the PRSE seems to regulate mitochondrial transcripts in response to the available carbon source, but unlike the P3E, it seems to target the electron transport chain rather than mitochondrial translation (Fig. 3). Transcripts containing PRSE matches were down-regulated when a repressing carbon source was present such as glucose, fructose, and sucrose (32) and were up-regulated when a nonrepressing carbon source was present as in the late time points from the stationary phase (32, 33) and diauxic shift (31) time courses. The PRSE was also associated with increased expression in nitrogen starvation (32) and heat shock conditions (32, 33) (Fig. 4A).
Response to Unfolded Protein Element (RUPE). Although the other PSAMs are involved in the response to starvation, the RUPE is involved in adapting to conditions that would cause misfolded proteins. RUPE matches are positively associated with gene expression in treatments with tunicamycin (38), DTT, and diamide (32). The RUPE also corresponds to increased expression under amino acid and adenine starvation conditions (32) (Fig. 4C). The automated GO scoring of RUPE targets did not reveal any significantly enriched functional categories of genes. However, a manual inspection of the annotations for RUPE targets shows a number of ER and Golgi-associated genes.
The Target of Rapamycin (TOR) Pathway. All of the PSAMs we identified except the RUPE are involved in the response to starvation and carbon source, but they have another common theme: The P3E, P4E, PRSE, NRSE1, and NRSE2 all correlated with expression during rapamycin treatments and, therefore, are likely to be downstream of the TOR pathway. Our analysis of rapamycin treatment data for several strains (39) showed that the matches to the P3E and PRSE positively correlated with expression, whereas the matches to the P4E, NRSE1, and NRSE2 negatively correlated with expression (Fig. 4A). This finding is consistent with the inferred activities for these PSAMs under starvation conditions and with the observation that rapamycin-treated yeast cells are indistinguishable from starved cells (40).
Experimental Validation of the Effect of Rapamycin on Puf3p Activity. As shown in Fig. 6A and in previous work (41), MFA2 mRNA decays rapidly with a half-life of 3.5 min with or without rapamycin treatment. In contrast, rapamycin treatment stabilizes the MFA2/COX17 mRNA by 2-fold, with the half-life increased from 2 min in the nontreated strain to 4 min in the rapamycin-treated strain (Fig. 6B). These results provide evidence that rapamycin treatment reduces the ability of Puf3p to destabilize target mRNAs, and support the prediction that Puf3p is downstream of the TOR signaling pathway.
Fig. 6.

Regulation of Puf3p activity in response to rapamycin. Data from Northern blot analyses of MFA2 (A) or the hybrid MFA2/COX17 (B) mRNA decay are plotted, with minutes following transcriptional repression on the x axis and the fraction of RNA remaining as compared to the steady-state RNA level at time 0 on the y axis. Decay was monitored with or without rapamycin treatment for 60 min before transcriptional repression as follows. (A) MFA2 without rapamycin (filled square) and MFA2 with rapamycin (open square). (B) MFA2/COX17 without rapamycin (filled circle) and MFA2/COX17 with rapamycin (open circle). Data points are averages of multiple experiments.
Previous Observations Explained. Our global approach to studying mRNA stability transfactors also allowed us to reconcile some seemingly disparate prior observations about whether transcripts are stabilized or destabilized upon diauxic shift (41, 42). Our results suggest that both are occurring: Whereas the factors that bind the P3E and the PRSE are involved in stabilizing transcripts upon diauxic shift, those that bind the P4E, NRSE1, and NRSE2 are involved in destabilizing transcripts. Thus, the latter three transfactors may be playing a critical role in the down-regulation of ribosomal mRNAs upon the entry and maintenance of stationary phase (43). In addition, if the factors that bind the P4E, NRSE1, and NRSE2 are downstream of the TOR pathway, as our results suggest, it explains the destabilization of some transcripts upon rapamycin treatment (41).
Conclusion. Conceptually, if transcriptional arrest microarray experiments (3–11) were performed under varying environmental conditions, they could be used to infer activities and binding sites for additional mRNA stability regulators. However, interpretation of the data may be muddled if the same means used to stop transcription also changes the activities of mRNA decay regulators, as seems to be the case for Puf4p in the data from Grigull et al. (5). The study by Garcia-Martinez et al. (12) did not have the complication of a transcriptional arrest but required the development of a new microarray method. By contrast, our methods use physiological, steady-state mRNA abundances measured from standard comparative hybridization microarray experiments. As long as transcript nucleotide sequence and steady-state microarray data are available, our methods allow one to discover the PSAMs of mRNA stability regulators, determine which mRNAs are most likely targeted by the transfactors, and identify the conditions under which the activities of these factors are modulated.
Supplementary Material
Acknowledgments
We are grateful to Alex Tzagoloff for experimental support and Jim Manley and Larry Chasin for critical readings of the manuscript. This work was supported by National Institutes of Health Grants GM008798, LM007276, GM63759 (to W.M.O.), and HG003008 (to H.J.B.).
Author contributions: B.C.F., W.M.O., and H.J.B. designed research; B.C.F. and S.S.H. performed research; B.C.F. contributed new reagents/analytic tools; B.C.F. and S.S.H. analyzed data; and B.C.F., W.M.O., and H.J.B. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PSAM, position-specific affinity matrix; TFAP, transfactor activity profile; GO, Gene Ontology; P3E, Puf3p element; PRSE, positive response to starvation element; RUPE, response to unfolded protein element.
References
- 1.Keene, J. D. & Tenenbaum, S. A. (2002) Mol. Cell 9, 1161–1167. [DOI] [PubMed] [Google Scholar]
- 2.Hieronymus, H. & Silver, P. A. (2004) Genes Dev. 18, 2845–2860. [DOI] [PubMed] [Google Scholar]
- 3.Holstege, F. C., Jennings, E. G., Wyrick, J. J., Lee, T. I., Hengartner, C. J., Green, M. R., Golub, T. R., Lander, E. S. & Young, R. A. (1998) Cell 95, 717–728. [DOI] [PubMed] [Google Scholar]
- 4.Wang, Y., Liu, C. L., Storey, J. D., Tibshirani, R. J., Herschlag, D. & Brown, P. O. (2002) Proc. Natl. Acad. Sci. USA 99, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grigull, J., Mnaimneh, S., Pootoolal, J., Robinson, M. D. & Hughes, T. R. (2004) Mol. Cell Biol. 24, 5534–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lam, L. T., Pickeral, O. K., Peng, A. C., Rosenwald, A., Hurt, E. M., Giltnane, J. M., Averett, L. M., Zhao, H., Davis, R. E., Sathyamoorthy, M., et al. (2001) Genome Biol. 2, research0041.1–0041.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S. & Cohen, S. N. (2002) Proc. Natl. Acad. Sci. USA 99, 9697–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutierrez, R. A., Ewing, R. M., Cherry, J. M. & Green, P. J. (2002) Proc. Natl. Acad. Sci. USA 99, 11513–11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raghavan, A., Ogilvie, R. L., Reilly, C., Abelson, M. L., Raghavan, S., Vasdewani, J., Krathwohl, M. & Bohjanen, P. R. (2002) Nucleic Acids Res. 30, 5529–5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M. & Rosenow, C. (2003) Genome Res. 13, 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang, E., van Nimwegen, E., Zavolan, M., Rajewsky, N., Schroeder, M., Magnasco, M. & Darnell, J. E., Jr. (2003) Genome. Res. 13, 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Martinez, J., Aranda, A. & Perez-Ortin, J. E. (2004) Mol. Cell 15, 303–313. [DOI] [PubMed] [Google Scholar]
- 13.Gerber, A. P., Herschlag, D. & Brown, P. O. (2004) PloS Biol. 2, E79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wickens, M., Bernstein, D. S., Kimble, J. & Parker, R. (2002) Trends Genet. 18, 150–157. [DOI] [PubMed] [Google Scholar]
- 15.Kellis, M., Patterson, N., Endrizzi, M., Birren, B. & Lander, E. S. (2003) Nature 423, 241–254. [DOI] [PubMed] [Google Scholar]
- 16.Bussemaker, H. J., Li, H. & Siggia, E. D. (2001) Nat. Genet. 27, 167–171. [DOI] [PubMed] [Google Scholar]
- 17.Gao, F., Foat, B. C. & Bussemaker, H. J. (2004) BMC Bioinform. 5, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C., et al. (2004) Nucleic Acids Res. 32, D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Issel-Tarver, L., Christie, K. R., Dolinski, K., Andrada, R., Balakrishnan, R., Ball, C. A., Binkley, G., Dong, S., Dwight, S. S., Fisk, D. G., et al. (2002) Methods Enzymol. 350, 329–346. [DOI] [PubMed] [Google Scholar]
- 20.Pesole, G., Mignone, F., Gissi, C., Grillo, G., Licciulli, F. & Liuni, S. (2001) Gene 276, 73–81. [DOI] [PubMed] [Google Scholar]
- 21.Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 22.Grubbs, F. E. (1969) Technometrics 11, 1–21. [Google Scholar]
- 23.Saldanha, A. J. (2004) Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- 24.Olivas, W. & Parker, R. (2000) EMBO J. 19, 6602–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caponigro, G., Muhlrad, D. & Parker, R. (1993) Mol. Cell Biol. 13, 5141–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herrick, D., Parker, R. & Jacobson, A. (1990) Mol. Cell Biol. 10, 2269–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decker, C. J. & Parker, R. (1993) Genes Dev. 7, 1632–1643. [DOI] [PubMed] [Google Scholar]
- 28.Jackson, J. S., Houshmandi, S. S., Lopez Leban, F. & Olivas, W. M. (2004) RNA 10, 1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caponigro, G. & Parker, R. (1995) Genes. Dev. 9, 2421–2432. [DOI] [PubMed] [Google Scholar]
- 30.Felici, F., Cesareni, G. & Hughes, J. M. X. (1989) Mol. Cell Biol. 9, 3260–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeRisi, J. L., Iyer, V. R. & Brown, P. O. (1997) Science 278, 680–686. [DOI] [PubMed] [Google Scholar]
- 32.Gasch, A. P., Spellman, P. T., Kao, C. M., Carmel-Harel, O., Eisen, M. B., Storz, G., Botstein, D. & Brown, P. O. (2000) Mol. Biol. Cell 11, 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Segal, E., Shapira, M., Regev, A., Pe'er, D., Botstein, D., Koller, D. & Friedman, N. (2003) Nat. Genet. 34, 166–176. [DOI] [PubMed] [Google Scholar]
- 34.Causton, H. C., Ren, B., Koh, S. S., Harbison, C. T., Kanin, E., Jennings, E. G., Lee, T. I., True, H. L, Lander, E. S. & Young, R. A. (2001) Mol. Biol. Cell 12, 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shapira, M., Segal, E. & Botstein, D. (2004) Mol. Biol. Cell 15, 5659–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fleming, J. A., Lightcap, E. S., Sadis, S., Thoroddsen, V., Bulawa, C. E. & Blackman, R. K. (2002) Proc. Natl. Acad. Sci. USA 99, 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duttagupta, R., Vasudevan, S., Wilusz, C. J. & Peltz, S. W. (2003) Mol. Cell Biol. 23, 2623–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Casagrande, R., Stern, P., Diehn, M., Shamu, C., Osario, M., Zuniga, M., Brown, P. O. & Ploegh, H. (2000) Mol. Cell 5, 729–735. [DOI] [PubMed] [Google Scholar]
- 39.Shamji, A. F., Kuruvilla, F. G. & Schreiber, S. L. (2000) Curr. Biol. 10, 1574–1581. [DOI] [PubMed] [Google Scholar]
- 40.Barbet, N. C., Schneider, U., Helliwell, S. B., Stansfield, I., Tuite, M. F. & Hall, M. N. (1996) Mol. Biol. Cell 7, 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Albig, A. R. & Decker, C. J. (2001) Mol. Biol. Cell 12, 3428–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jona, G., Choder, M. & Gileadi, O. (2000) Biochim. Biophys. Acta 1491, 37–48. [DOI] [PubMed] [Google Scholar]
- 43.Ju, Q. & Warner, J. R. (1994) Yeast 10, 151–157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.