

The term “error catastrophe,” originally introduced in the theory of molecular evolution (1), has become fashionable among virologists. In a recent paper in PNAS (2), it was suggested, on the basis of quantitative sequence studies, that ribavirin, a common antiviral drug, by its mutagenic action drives poliovirus into an error catastrophe of replication, thereby turning a productive infection into an abortive one. Previous studies by Loeb and his group (3, 4) on the AIDS virus (HIV) and by Domingo, Holland, and coworkers (5, 6) on foot-and-mouth disease virus (FMDV) have led to similar conclusions, suggesting a paradigm shift in antiviral strategies (7). A recent issue of PNAS presents a paper by Grande-Pérez et al. (8), which deals with the “molecular indetermination in the transition to error catastrophe,” shedding light on the complexity of the mechanisms involved in virus infection and stressing the need for a careful molecular analysis of the detail, which may differ greatly from one virus to another. Because of its practical relevance for developing potent antiviral drugs and, beyond that, its general importance for an understanding of molecular evolution, this commentary will highlight the theoretical basis and point out the kind of conclusions that can be drawn in discussing experimental results.
The term error catastrophe is of a descriptive nature and lacks a clear-cut definition. A catastrophe is usually triggered if certain tolerances are exceeded. For replication, there is indeed such a limiting value of error or mutation rate that must not be surpassed if the wild type is to be kept stable. We call this limit the “error threshold.” Why is it a sharply defined limit? Why does the efficiency of replication not vary monotonically with the error rate? The information stored in the genomic sequence melts like ice at 0°C. This comparison is indeed a very apt one. The information melts away in a process that has all the physical characteristics of a first-order phase transition requiring cooperative behavior with unlimited coherence lengths, as we encounter in the melting of a solid or the evaporation of a liquid at its boiling point. The error threshold is caused by the inherent autocatalytic nature of replication, which represents not only the transfer of information from one generation to the next, as would be the case for a message sent through a transmission channel. Rather, replication provides an exponential proliferation of the information contained in the sequence as a whole. In the population formed, this results in competition among the various slightly differing sequences, which behave as cooperative units. Natural selection is a direct consequence of this competitive replication. It presupposes differences in efficiency of replication without excluding neutral mutants. Neutral copies, all belonging to the group of best-adapted ones, are selected against the rest, but because of their inherently reproductive behavior, they continue to compete with one another in a stochastic manner. Kimura and Ohta (9) called this nondeterministic fluctuating selection “non-Darwinian,” although Darwin himself anticipated it. Kimura and Ohta's stochastically fluctuating selection reminds us of “critical phase transitions,” as found in ferro- or antiferromagnetism or liquid-gas transformation near the critical point where, in analogy to neutrality among replicative units, the densities of the liquid and gaseous phases become equal, with the consequence of density fluctuations on all scales of spatial dimensions manifesting themselves in the phenomenon of “critical opalescence.”
However, note that these phase transitions associated with natural selection do not take place in the space–time coordinates of our physical space. They refer rather to an abstract “information space” and are therefore not easy to visualize, because they may appear scattered in physical space and over extended periods of time. Information space is a discrete point space with a metric named after Richard Hamming (10). Each of the possible 4N sequences of length N is assigned to one and only one point, with all neighborhoods among sequences correctly ordered according to their kinship distances. This “spatial” order requires a 22N-dimensional Hamming space. The dynamical equations of the rise and fall of populations can be written in a fairly general phenomenological form, yielding the quasispecies model (11, 12). A quasispecies is a population structure in information space and is the “condensed” mutant distribution that results from the phase transition representing natural selection. It has been termed “quasispecies” because the whole distribution behaves “quasi” as a single species, because it is determined by one (namely the largest) eigenvalue of its system of dynamical equations. The eigenvalues, being invariants of the equations, are determined as soon as the mutant spectrum is defined, regardless of whether the final stationary population structure is achieved. Rather than elaborating on further details of theory, I shall now discuss the important parameters that determine selection and hence also the behavior of virus populations, as expressed in the work this commentary refers to.
Fig. 1 shows a computer simulation of a model case that is representative of the phenomenon of error catastrophe. Such simulations were first performed by Schuster and Swetina (13). The present example was computed by Tarazona (14). It shows the stationary structure of a population consisting of binary sequences of length N = 20, in which all sequences have equal values of all their replication rates except for one sequence, which shows a 10-fold higher rate. The error rate (1 − q), i.e., the relative number of misincorporations per site, has been assumed to be uniform for all sequences in the distribution. Fig. 1 shows a plot of the relative population number of the steady-state population against the error rate (1 − q), the numbers 0, 1, 2, etc., referring to 0 errors (= master sequence) and the sums of all of the 1, 2, 3, error sequences, respectively. The error threshold is seen clearly at 1 − q ≈ 0.11. Although the individual curves vary quite markedly with the error rate, the order of the quasispecies, represented by the consensus sequence, is clearly conserved up to the “melting point,” i.e., the error threshold. Above the error threshold, each of the ≈106 (i.e., 2N) possible individual sequences occurs with the same probability of ≈10−6. Because the distribution was centered around the master sequence (0 errors), the sum of all k-error sequences is given by the binomial coefficient (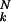 ), which has its maximum at k = N/2. That is why the sum curve for 10 errors shows the most frequent representation (corresponding to about 17.6% of the ≈106 sequences). Above the threshold, no memory of the former wild type remains.
), which has its maximum at k = N/2. That is why the sum curve for 10 errors shows the most frequent representation (corresponding to about 17.6% of the ≈106 sequences). Above the threshold, no memory of the former wild type remains.
Fig 1.

Relative population numbers of binary sequences Sk (ordinate) as functions of single-digit error rate (1 − q) (abscissa). The length of all binary sequences is N = 20. All 2N ≈ 106 sequences are degenerate in their reproductivity except for one “master” sequence Sm, which reproduces 10 times more efficiently than the rest. The resulting quasispecies distribution is centered at the master sequence (“0” errors). The numbers 1, 2, … 20 refer to the sum of all 1-, 2-, … 20-error mutants. The red curve refers to the consensus sequence, which shows a sharp first-order phase transition at the error threshold.
The model simulated in Fig. 1 suffices to show the clear analogy to a first-order phase transition (14) applying to the information content of the quasispecies as a whole. The transition would be even sharper for (more realistic) longer sequences, reaching a slope of ∞ for N → ∞, whereas smaller selective advantages of the wild type would just cause a shift of the transition point along the abscissa to lower error rates without much changing the overall shapes of the curves. What is essential is that the population of the master and of individual mutant types already changes quite conspicuously below the error threshold but is “all or none” above the threshold. However, the model chosen is entirely unrealistic if we want to apply it to our discussion of virus quasispecies. Let me discuss the parameters that determine the shape of the curves, and we shall see more clearly what to expect in situations closer to those of real viruses.
(i) The Most Obvious Parameter Is the Error Rate
First, a uniform fidelity q for all positions, and hence a uniform error rate 1 − q, is absolutely fictitious. Grand-Pérez et al. (8) emphasize that different regions of the virus sequences must have different error rates, and it has long been known that it is not only the kind of base and its nearest neighbors that make individual positions more or less variable, ranging from exceedingly conservative positions to “hot spots.” This is the more so if mutations are enhanced by using drugs that resemble base analogues. Because the probability of mutants is sequence-specific, we must introduce two indices: one for the particular sequence Sk and another one for the positions “i ” within each sequence Sk. Hence the fidelity of a position thus characterized is qik and its corresponding error rate (1 − qik). The overall fidelity of reproducing any given sequence Sk is then the product q1kq2k … qNk or ∏ 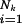 qik ≡ q̂
qik ≡ q̂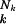 ,where q̂k is the geometric mean of all qik of sequence k, whereas Nk is its length expressed as the number of nucleotides. The geometric mean differs from the arithmetic by weighting more sensitively individual elements. Although some elements can reach zero without much changing the value of the arithmetic mean, they must not do so for the geometric mean, where the product becomes zero if one of its elements is zero. This property of the geometric mean has important consequences for the possible relevance of certain singular mutations. The theory, on the other hand, does not change its formal structure by the introduction of averages. Instead of a uniform fidelity q or error rate (1 − q) (abscissa in Fig. 1), we now use the averages q̂k and call the sequence-specific overall fidelity q̂
,where q̂k is the geometric mean of all qik of sequence k, whereas Nk is its length expressed as the number of nucleotides. The geometric mean differs from the arithmetic by weighting more sensitively individual elements. Although some elements can reach zero without much changing the value of the arithmetic mean, they must not do so for the geometric mean, where the product becomes zero if one of its elements is zero. This property of the geometric mean has important consequences for the possible relevance of certain singular mutations. The theory, on the other hand, does not change its formal structure by the introduction of averages. Instead of a uniform fidelity q or error rate (1 − q) (abscissa in Fig. 1), we now use the averages q̂k and call the sequence-specific overall fidelity q̂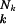 ≡Qk. Because q̂k for any realistic sequence is very close to one, meaning a small average error rate (1 − q̂k), as found between 10−3 and 10−5 for RNA viruses, a fairly precise approximation to Qk is given by e−Nk(1−q̂k). The exponent is the average number of errors per sequence and can yield Qk values appreciably smaller than one. The conclusion of this paragraph is that the probability distribution of mutants in a quasispecies is not at all uniform; single mutants may reproducibly appear orders of magnitude more frequently than others, of which some may have only a sporadic existence. This all happens below the error threshold and may produce quite nonuniform mutant distributions close to the error threshold, although the all-or-none nature of the error threshold is first realized after it is crossed.
≡Qk. Because q̂k for any realistic sequence is very close to one, meaning a small average error rate (1 − q̂k), as found between 10−3 and 10−5 for RNA viruses, a fairly precise approximation to Qk is given by e−Nk(1−q̂k). The exponent is the average number of errors per sequence and can yield Qk values appreciably smaller than one. The conclusion of this paragraph is that the probability distribution of mutants in a quasispecies is not at all uniform; single mutants may reproducibly appear orders of magnitude more frequently than others, of which some may have only a sporadic existence. This all happens below the error threshold and may produce quite nonuniform mutant distributions close to the error threshold, although the all-or-none nature of the error threshold is first realized after it is crossed.
(ii) The Error Threshold Is Not Solely Determined by the Average Error Rate
Of equal importance is the fitness landscape within the quasispecies distribution. Let us take the example represented by Fig. 1, in which one of the 106 different sequences, called Sm, is clearly distinguished by a (massive) selective advantage. At the critical error rate (1 − q̂m), the master sequence Sm has a fidelity Qm = e−Nm(1−qm) with a value well below 1, which then requires a corresponding selective advantage of the master sequence, relative to the average of its mutant distribution. Any mutation occurring in the master sequence reduces its frequency of occurrence, whereas any mutation occurring in the rest of the quasispecies (including the mutations that come about in the master sequence) produces some other member of this mutant distribution. In other words, mutations in the rest of the distribution do not lower their total number. The master sequence must therefore be at least σm times more efficient in its reproduction to make up for the loss caused by its mutation rate, such that σmQm > 1. In the above example, σm can be easily calculated. For realistic nonuniform distributions, which, in addition, may contain several neutral master copies, the σ functions, although clearly definable by the eigenvalues and eigenvectors of the exact solutions, would be calculable only if the details of the fitness landscapes are known. Experimental data, such as those presented in the papers under consideration, are therefore most important.
We now see that it is not only the irregularity in the mutant distribution but also the contribution of each mutant to the reproductivity of the quasispecies that is rated by natural selection. A particular mutation that appears to occur very frequently may be either neutral or, if it involves a “strategic” position, deleterious, or under certain environmental changes, such as those produced by the immune response of the host, it may be advantageous for the virus. These situations cause quite dramatic differences for different viruses, as is known for polio virus and HIV (15). The equivalent importance of σm and Qm is obvious from the symmetric condition σmQm > 1. It is somewhat obscured in the standard expression defining the error threshold: (1 − q̂m) > ln σm/Nmwhich follows from σmQm if one substitutes for Qm the exponential e−Nm(1−q̂m). The logarithmic dependence on σm in the error threshold relation seems to belittle its influence, because the logarithm of numbers clearly larger than one does not depart far from one. However, σ values may be very close to one, i.e., equal to 1 + ɛ, where ɛ ≪ 1. Then the logarithm is a very small number: ln (1 + ɛ) ≈ ɛ for ɛ ≪ 1. In irregular fitness landscapes and irregular mutant distributions, σm is a complicated function of the variables concealed in the averages σm and Qm. As in the case of fidelity Qm, much of the detail appearing in the precise form of eigenvalues and eigenvectors gets lost in the averaging procedures leading to σm values. Selectivity is different for different fitness classes of mutants, the extremes being neutrals (σ → 1) and nonreproducible deleterious mutants (σ → ∞). In addition, there might be large fluctuations even for a given virus in the same environment, when very rare (but important) mutations occur stochastically in different temporal sequences.
(iii) Virus Infection Involves More Complex Operations than Just the Replication of RNA or DNA
The genomes of even the smallest viruses encode several functions, of which replication is only one, albeit a very important one. Yet what is finally weighted for selection is the virus, overall performance in the infection process. We have studied the kinetics of infection with the example of the bacteriophage Qβ [Eigen et al. (16)]. The experiments carried out by M. Gebinoga (17) involved pulse-like infection. Samples of host cells were incubated with virus for defined lengths of time, i.e., quenching the infection after 1, 2, 3, etc., minutes up to the total time interval between infection and lysis of the host cells. The quenched samples were carefully treated with toluene to remove the outer (lipid) membrane of the host cells. The remaining murein sacculus, which is not penetrable for larger molecules such as proteins and nucleic acids or for organelles, was perfused with radioactively labeled nucleoside triphosphates and amino acids. The kinetics of both RNA and protein synthesis then were recorded. The rates, extrapolated to the starting point, which reflects the profile of the process in vivo, are plotted as a function of time in Fig. 2. As seen, at the moment of infection the rate of RNA formation is zero, because specific replicase is not yet available, but protein synthesis (using the translation machinery of the host) is active right from the start. After about 10 min, enough replicase has been formed so that both replication and translation can now compete, leading to a sharp (i.e., hyperbolic rather than exponential) increase of both protein and RNA concentration, as expected for a nonlinear “hypercyclic” (18) mechanism. At the same time, replicase formation is down-regulated by the binding of coat protein (which acts as regulator) to the replicase gene. The burst of synthesis comes to an abrupt halt when the amount of RNA present has increased sufficiently to block all available binding sites provided by the host ribosome population as well as those of the (ultimately constant) replicase concentration—very much as in an “end-point titration.” From now on, both RNA and coat protein production proceeds at a constant rate until the host cell lyses, about 40 min after infection. This is a highly regulated mechanism, which yields about 10,000–20,000 complete virus particles (each consisting of one plus strand of RNA and 180 coat proteins that form its icosahedral shell). Because of error accumulation, only less than 10% of the viruses produced are viable. Nevertheless, this is more than sufficient to maintain the autocatalytic growth nature of the overall process, described by the phenomenological rate equations.
Fig 2.

Kinetic profile for the infection of E. coli by phage Qβ. The total infection cycle lasts about 40 min. The ordinate refers to the rates (i.e., the number of particles formed per minute and per host cell) that are plotted as functions of time. Red lines represent proteins (coat protein and replicase); blue lines, RNA (plus and minus strands); and the green line, complete virus particles.
What I wanted to show is the complex nature of the overall process of virus infection, which differs from one type to another but in no case is just simple replication. Coming back now to the objective of this commentary, I would like to emphasize three points:
(i) Very close to the error threshold, the mutant spectrum, as Fig. 1 demonstrates, becomes quite diverse. Before the phase transition occurs, master sequence and low-error mutants become a minority even in the unrealistic model case of uniform error rates. The wild type keeps its distance from this point of transition to maintain robust stability. The application of mutagens may change this situation in an uncontrolled way.
(ii) The resulting error spectrum involves neutral, deleterious, and also—under the new conditions—advantageous mutants. In which manner they are effective, before the total information “melts away” completely, depends on the particular type of virus.
(iii) The mutant spectrum expresses itself in a spectrum of phenotypic functions that include all processes involved in the complex infection mechanism. Hence, error catastrophe is intimately linked to all functions involved, because it depends on both (realistically quite complex) parameters Q and σ.
The paper of Grande-Pérez et al. (8) shows what is to be done to cope with this situation. Theory cannot remove complexity, but it shows what kind of “regular” behavior can be expected and what experiments have to be done to get a grasp on the irregularities. This is more true in biology than in any other field of the physical sciences. The work on the error threshold opens a new paradigm for how to fight viruses, namely not by inhibiting their replication but rather by favoring it with an increased rate of mutation. At first this procedure seems to challenge the virus to escape immune protection, but at the same time, it may cause the virus to lose all its pathogenic information. The paper quoted makes it plain that a lot of experimental work has to be done for each particular type of virus, presenting what is certainly one of the great challenges of the 21st century.
See companion article on page 12938 in issue 20 of volume 99.
References
- 1.Eigen M. (1971) Naturwissenschaften 58, 465-523. [DOI] [PubMed] [Google Scholar]
- 2.Crotty S., Cameron, C. E. & Audino, R. (2001) Proc. Natl. Acad. Sci. USA 98, 6895-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loeb L. A., Essigman, J. M., Kazazi, F., Zhang, J., Rose, K. D. & Mullins, J. I. (1999) Proc. Natl. Acad. Sci. USA 96, 1492-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loeb L. A. & Mullins, J. I. (2000) AIDS Res. Hum. Retroviruses 13, 1-3. [DOI] [PubMed] [Google Scholar]
- 5.Domingo E. (2000) Virology 270, 251-253. [DOI] [PubMed] [Google Scholar]
- 6.Holland J. J. & Domingo, E. (1998) Virus Genes 16, 13-21. [DOI] [PubMed] [Google Scholar]
- 7.Eigen M. (1993) Med. Res. Rev. 4, 385-398. [DOI] [PubMed] [Google Scholar]
- 8.Grande-Pérez A., Sierra, S., Castro, M. G., Domingo, E. & Lowenstein, P. R. (2002) Proc. Natl. Acad. Sci. USA 99, 12938-12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimura M. & Ohta, T., (1971) Population Genetics (Princeton Univ. Press, Princeton, NJ).
- 10.Hamming R. W., (1980) Coding and Information Theory (Prentice–Hall, Englewood Cliffs, NJ).
- 11.Eigen M., McCaskill, J. & Schuster, P. (1988) J. Phys. Chem. 92, 6881-6891. [Google Scholar]
- 12.Eigen M., McCaskill, J. & Schuster, P. (1989) Adv. Chem. Phys. 75, 149-263. [Google Scholar]
- 13.Schuster P. & Swetina, J. (1988) Bull. Math. Biol. 50, 635-650. [DOI] [PubMed] [Google Scholar]
- 14.Tarazona P. (1992) Phys. Rev. A 45, 6038-6050. [DOI] [PubMed] [Google Scholar]
- 15.Eigen M. & Nieselt-Struwe, K. (1990) AIDS 4,(Suppl. 1), 585-593. [PubMed] [Google Scholar]
- 16.Eigen M., Biebricher, C. K., Gebinoga, M. & Gardiner, W. C. (1991) Biochemistry 30, 11005-11018. [DOI] [PubMed] [Google Scholar]
- 17.Gebinoga M., (1991) Ph.D. thesis (Technical University of Braunschweig, Braunschweig, Germany).
- 18.Eigen M. & Schuster, P., (1979) The Hypercycle: A Principle of Natural Self Organization (Springer, Berlin). [DOI] [PubMed]