

Abstract
HPr kinase/phosphorylase (HprK/P) controls the phosphorylation state of the phosphocarrier protein HPr and regulates the utilization of carbon sources by Gram-positive bacteria. It catalyzes both the ATP-dependent phosphorylation of Ser-46 of HPr and its dephosphorylation by phosphorolysis. The latter reaction uses inorganic phosphate as substrate and produces pyrophosphate. We present here two crystal structures of a complex of the catalytic domain of Lactobacillus casei HprK/P with Bacillus subtilis HPr, both at 2.8-Å resolution. One of the structures was obtained in the presence of excess pyrophosphate, reversing the phosphorolysis reaction and contains serine-phosphorylated HPr. The complex has six HPr molecules bound to the hexameric kinase. Two adjacent enzyme subunits are in contact with each HPr molecule, one through its active site and the other through its C-terminal helix. In the complex with serine-phosphorylated HPr, a phosphate ion is in a position to perform a nucleophilic attack on the phosphoserine. Although the mechanism of the phosphorylation reaction resembles that of eukaryotic protein kinases, the dephosphorylation by inorganic phosphate is unique to the HprK/P family of kinases. This study provides the structure of a protein kinase in complex with its protein substrate, giving insights into the chemistry of the phospho-transfer reactions in both directions.
Protein phosphorylation plays a central role in signal transduction and cellular regulation (1). In bacteria, Ser/Thr kinases are involved in the regulation of a large number of catabolic genes optimizing the use of carbon, nitrogen, and sulfur sources (2). Carbon catabolite repression is a response to metabolic and environmental conditions (3, 4), which stimulates the expression of glycolytic enzymes and represses enzymes catalyzing the metabolism of less efficient carbon sources. In Bacillus subtilis, the expression of ≈10% of the genome is regulated by carbon catabolite repression (5, 6).
In Gram-positive bacteria, the first step of the carbon catabolite repression response is the phosphorylation on residue Ser-46 of the histidine phosphocarrier protein (HPr). HPr can be phosphorylated also on His-15 by the phosphoenolpyruvate:sugar phosphotransferase system, a general system for sugar transport in bacteria. His-15 phosphorylation is phosphoenolpyruvate-dependent, and the phosphate is transferred to the transported carbohydrate (7). In contrast, the serine modification is ATP-dependent and plays a regulatory role. Serine-phosphorylated HPr (P-Ser-HPr) interacts with the catabolite control protein A, forming a complex that binds to DNA at the catabolite response elements cre (8, 9). Phosphorylation is achieved by a specific kinase that also is able to dephosphorylate Ser-46 (10).
In the accompanying paper (11), we show that the dephosphorylation of Ser-46 involves the phosphorolysis of the phosphoserine rather than its hydrolysis. Inorganic phosphate (Pi) is a substrate and pyrophosphate (PPi) is a product of this reaction. Thus, we now call the bifunctional enzyme HPr kinase/phosphorylase (HprK/P). The kinase activity is activated by fructose-1,6-bisphosphate and inhibited by Pi (10). In starved cells, where the Pi concentration is high and that of fructose-1,6-bisphosphate is low, HPr is dephosphorylated. In the presence of glucose, it is phosphorylated.
HprK/Ps from various bacteria have a highly conserved C-terminal catalytic domain that carries both enzymatic activities and responds to the effectors. The x-ray structure of the catalytic domain of the Lactobacillus casei enzyme (12) shows that it forms a hexamer with a subunit fold unrelated to that of eukaryotic Ser/Thr protein kinases. The subunit contains an ATP-binding subdomain with a phosphate-binding loop (P loop) similar to that of phosphoenolpyruvate carboxykinase (PCK; refs. 13 and 14). A x-ray structure is available also for the full-length Staphylococcus xylosus HprK/P (15). This protein forms the same hexamer as in L. casei, with N-terminal domains that are connected loosely to the catalytic domains.
We now describe a 2.8-Å x-ray structure of the complex obtained by cocrystallizing the L. casei catalytic domain and B. subtilis HPr. Six HPr molecules bind to the hexamer at sites that overlap two of its subunits. We also describe a complex where HPr was phosphorylated on Ser-46 by the simple addition of PPi to the crystallization mixture. A Pi ion, product of the reaction, is located in the P loop in the proper position for the reverse reaction to take place. These results demonstrate that both phosphorylation and phosphorolysis are carried out by the same active site and suggest a common mechanism for both reactions.
Methods
Protein Expression, Purification, and Crystallization.
The His-tagged catalytic domain (residues 128–319) of L. casei HprK/P (12) and His-tagged B. subtilis HPr (16) were used for cocrystallization by the hanging-drop vapor diffusion method by using the JBScreen sparse matrix kit (JenaBioScience, Jena, Germany). Crystals grew at 18°C within 3 days over wells containing 28% polyethylene glycol 400, 100 mM Hepes-Na (pH 7.5), and 200 mM CaCl2. The drops were prepared by mixing 2 μl of the well mixture with 2 μl of a solution containing 0.25 mM HprK/P monomer and 0.5 mM HPr in the presence or absence of 10 mM PPi.
X-Ray Diffraction Data Collection.
Data collection was performed at 100 K on flash-frozen crystals at European Synchrotron Radiation Facility (Grenoble, France) on stations ID14-H2 for the HPr complex and BM14 for the P-Ser-HPr complex. Images were processed with DENZO, and data were scaled with SCALEPACK (17). Statistics are given in Table 1. The crystals of the two complexes are isomorphous and belong to space group P3212 with half a HprK/P hexamer and three HPr or P-Ser-HPr molecules per asymmetric unit.
Table 1.
Data collection and refinement statistics
| Complex with: | HPr | P-Ser-HPr |
|---|---|---|
| Data collection | ||
| Space group | P3212 | P3212 |
| Unit cell, Å | a = b = 81.135 c = 253.192 | a = b = 80.832 c = 252.488 |
| Resolution, Å | 2.8 | 2.8 |
| Measurements | 392,766 | 274,803 |
| Unique reflections | 23,732 | 23,597 |
| Completeness, % | 99.7 (99.5) | 99.1 (99.7) |
| Rsym, % | 6.0 (15.0) | 4.0 (14.6) |
| I/σ | 13.6 | 26.3 |
| Refinement | ||
| Rcryst, % | 21.3 | 20.0 |
| Rfree, % | 26.4 | 25.7 |
| Mean β factor, Å2 | 52 | 53 |
| Reflections used | 23,554 | 23,472 |
| No. of atoms | ||
| Protein | 5,688 | 5,877 |
| Ca2+ | 3 | 3 |
| Phosphates | — | 27 |
| Water | 179 | 171 |
| rms deviation | ||
| Bond, Å | 0.007 | 0.007 |
| Angle, ° | 1.29 | 1.37 |
Numbers in parentheses are for the last resolution shell.
Structure Determination and Refinement.
Phases were obtained by molecular replacement with the AMORE package (18). The starting model was a trimer of the L. casei catalytic domain (PDB code ; ref. 12). In the initial density map derived from rigid-body refinement (R = 46.3%, Rfree = 47.2%), electron density on the surface of the HprK/P trimer could be interpreted as three HPr molecules. These molecules were built by using the B. subtilis HPr model (PDB code ; ref. 19). After rigid-body refinement, simulated annealing refinement with CNS (20) and manual rebuilding yielded the final models (Table 1). Local threefold symmetry was imposed on the cores of both HprK/P and HPr during refinement. In the final model, minor differences are observed between the three HPr molecules and three HprK/P subunits present in the asymmetric unit, with root-mean-square (rms) deviations less than 0.35 Å for Cα positions. Accessible and buried surface areas were evaluated with the ASA program (A. Lesk, University of Cambridge Clinical School, Cambridge, U.K.) implementing the Lee and Richards algorithm (21).
Results and Discussion
The Overall Structure.
Three HPr molecules bind on top of the HprK/P hexamer (Fig. 1), and three bind underneath, respecting its D3 symmetry. Binding induces little change in HPr, the Cα rms deviation being ≈0.5 Å relative to the free molecule (19). Similarly, the quaternary structure and subunit conformation of the enzyme are close to those of the free protein (12). The rms deviation is 0.7 Å for 127 of the 191 Cα excluding the C-terminal helix α4 and a surface loop formed by residues 236–258. Helix α4, which is in contact with HPr, rotates by ≈30° relative to the free L. casei protein. Because the helix has the same orientation in substrate-free S. xylosus HprK/P (15), its movement is unlikely to be caused by HPr binding. The 236–258 loop is disordered in the free enzyme and in the complex with HPr, but it has interpretable electron density in the complex with P-Ser-HPr.
Fig 1.

The HPr–HprK/P complex. The top half of the L. casei HprK/P hexamer is viewed along its threefold axis. The three subunits colored in red, green, and blue bind three B. subtilis HPr molecules, drawn with their molecular surface in gold.
Protein–Protein Interaction.
In the complex with HPr, enzyme–substrate contacts bury 1,400 Å2 and create six hydrogen bonds per HPr molecule. In the complex with P-Ser-HPr, an additional 600 Å2 are buried as a result of contacts made by the 236–258 loop. These interface areas are comparable to those observed in other stable, specific complexes (22). Each HPr molecule interacts with two adjacent subunits of the enzyme (colored in green and red in Fig. 2). The interface with the green subunit makes up 60% of the buried area. This interface is of functional relevance, because it involves the active site of the enzyme and Ser-46, located at the N terminus of helix αB of HPr. Helix αB and the preceding β-strand provide most of the contacts. On the enzyme side, this interface involves strand βA, the P loop connecting βC to helix α1, and the tip of the βD–βE hairpin (Fig. 2B). A separate interface involves the C-terminal helix α4 of the red subunit, which is in contact with helix αB of HPr and also with the first turn of helix αA N-capped by His-15. Interestingly, the same region of HPr also interacts with proteins EIIAGlc and EI of the phosphoenolpyruvate:sugar phosphotransferase system (23) and with the catabolite control protein A (24). In the complex with P-Ser-HPr, an additional contact is formed between Arg-245 of the red subunit and the phosphoserine. This interaction helps to stabilize the 236–258 loop, which is ordered only in this complex.
Fig 2.

The two interface regions of the HPr–HprK/P contact. Enzyme subunits are in red and green as described for Fig. 1. HPr (in gold) is phosphorylated on Ser-46. (A) Stereoview of the binding site shared by the red and green subunits. (B) The Ser-46 region of HPr interacts with the active site of the green subunit and with Arg-245 of the red subunit. The 236–258 loop bearing the arginine is ordered in the phosphorylated complex. (C) The His-15 region of HPr interacts with helix α4 of the red subunit.
Specificity.
The crystalline complex contains L. casei HprK/P and B. subtilis HPr. L. casei and B. subtilis HPr have 67% sequence identity and differ by only 2 of 20 residues in contact with HprK/P. The Gram-negative organism Escherichia coli is more distantly related to B. subtilis. Although the overall structure of HPr is very similar in these two bacteria (25), E. coli HPr cannot be phosphorylated on Ser-46 (26). Experiments on chimeric HPr indicate that residues 43–57 are important for specific recognition by HprK/P. Replacing residues 48, 49, 52, or 53 in Mycoplasma capricolum HPr by the corresponding E. coli residues abolished Ser-46 phosphorylation (27). The inactivity of the mutants at positions 48, 49, and 52 is explained easily by the structure of the complex. Met-48 is at the center of the interface and makes the largest contribution to the buried area (Fig. 2B). Gly-49 is a lysine in E. coli with a bulky side chain that cannot be fitted easily at the interface. Ser-52 makes a side chain-to-side chain hydrogen bond with Arg-180 of HprK/P. On the other hand, mutation effects observed at position 53 remain unexplained, because this residue is leucine in both B. subtilis and E. coli but Met in M. capricolum HPr.
HPr Phosphorylation in the Crystal.
HprK/P can phosphorylate HPr with either ATP or PPi as the phosphate donor (11). In the crystal, phosphorylation by PPi was almost complete as indicated by the electron density near Ser-46 (Fig. 3). This modification induces no conformational change in bound HPr. This observation is in agreement with the x-ray structure of (free) P-Ser-HPr (28), which is very similar to free HPr. Next to the phosphoserine, a peak of electron density indicates the presence of a Pi ion, presumably the product of the reaction with PPi. Residues of the enzyme that interact with either the phosphoserine or the Pi ion are likely to play a major role in catalysis. Among these, Asp-179 is conspicuous (Fig. 3). Before phosphorylation, its carboxylate hydrogen bonds with the Ser-46 hydroxyl. Remarkably, the interaction is maintained after phosphorylation. The short oxygen–oxygen distance (2.8 Å) between Asp-179 and the phosphoserine implies that Asp-179 now is protonated, with the negative charge of the phosphoserine helping to lower the pKa of the carboxyl. Several other polar side chains of the enzyme are implicated also in hydrogen bonds. They belong to His-140, Ser-157, and Lys-161 of the green subunit and to Arg-245 of the red subunit. Furthermore, the phosphoserine is within hydrogen-bonding distance of the Pi ion, which may be PO4H2− rather than PO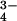 . The Pi ion interacts with the side chains of Lys-161 and Ser-157 and with main-chain NH groups of the P loop. Thus, both phosphate groups make extensive polar interactions with protein groups.
. The Pi ion interacts with the side chains of Lys-161 and Ser-157 and with main-chain NH groups of the P loop. Thus, both phosphate groups make extensive polar interactions with protein groups.
Fig 3.

Stereoview of the HprK/P active site in the HPr and P-Ser-HPr complexes. (A) The complex with HPr. Dashes indicate possible hydrogen bonds. A Ca2+ ion and a water molecule are seen in the P loop formed by residues 157–162 of the enzyme. (B) The complex with Ser-46-phosphorylated HPr. The electron density around the phosphoserine and the phosphate ion is from an Fo-Fc omit map contoured at 2σ. The gray sphere is Ca2+, and Arg-245 is from a neighboring subunit.
Metal Ions.
The crystallization mixture contained Ca2+ at high concentration. Electron density at the active site indicates that the divalent cation is present at the active site, presumably taking the place of Mg2+, which is required for both the phosphorylation and dephosphorylation reactions (29). Ca2+ ligates the Glu-204 and Ser-162 side chains of HprK/P and also the phosphoserine in the complex with P-Ser-HPr. Water molecules must complete the coordination shell of the cation, but they cannot be identified reliably at the present 2.8-Å resolution.
Catalytic Mechanism.
In the complex with HPr (Fig. 3A), Asp-179 is in a position to deprotonate the Ser-46 hydroxyl, a prerequisite for its phosphorylation. Asp-179 is supported in its role as a base by His-140, to which it hydrogen-bonds. The side chain of His-140 is mobile in the x-ray structure of the free L. casei protein (12), and the interaction with Asp-179 is formed only when HPr is present. Lys-161 and Arg-245, both strictly conserved residues, are likely to play a part in catalysis by neutralizing the negative charge that develops on the phosphate group during the transfer reaction and lowering the energy of the transition-state. The complex of HprK/P with P-Ser-HPr (Fig. 3B) gives additional information on the dephosphorylation mechanism. The geometry of the reactive groups is such that the phosphoserine can readily undergo a nucleophilic attack by Pi, yielding free serine and PPi. Asp-179, which is protonated but still hydrogen-bonded to His-140, acts as an acid catalyst by giving its proton to the serine moiety of the phosphoserine, whereas Lys-161 and Arg-245 play the same role as during phosphorylation.
Comparison with PCK.
PCK and HprK/P have similar ATP-binding domains and active sites (14, 15). All residues of the HprK/P active site mentioned above have their equivalent in PCK. This set of residues includes Arg-245, which is equivalent to Arg-333 in PCK but comes from an adjacent subunit in HprK/P. The two phosphate groups seen in the complex with P-Ser-HPr occupy the position of the γ and β phosphates of ATP bound to PCK. The catalytic mechanism of this enzyme (30, 31) therefore must be conserved in HprK/P. Up to now, only two of the HprK/P active site residues have been tested by site-directed mutagenesis: Asp-178 and Asp-179, equivalent to Asp-268 and Asp-269 in PCK. Residues 178–179 form the tip of the βD–βE hairpin, where the polypeptide chain forms a turn with an unusual main-chain conformation (12). Catalytic activity is lost after substitution of either aspartate (14). The x-ray structure suggests that Asp-179 is the catalytic base, but the role of Asp-178 is less obvious. This residue interacts with Ser-162 and Glu-204, both Ca2+ ligands, and may help in forming the metal-binding site. In PCK (32), two divalent cations interact with the bound ATP. One is at the site of Ca2+ in our structure, but the second has no equivalent and would overlap with the Ser-46 side chain.
Comparison with Eukaryotic Ser/Thr Kinases.
We also compared HprK/P with eukaryotic protein kinases. In the cAMP-dependent protein kinase A, Asp-166 is reported to be the catalytic base (33). When we superimposed the active site regions of the HPr–HprK/P and the protein kinase A–inhibitor complexes (PDB code ; ref. 34) based on the positions of the serine phosphate acceptor, the phosphate groups of ATP in PCK, and the catalytic aspartate, other catalytic groups did not superimpose. Thus, the arrangement of the catalytic groups is different in the eukaryotic and bacterial protein kinases, although they probably use the same chemical mechanism in which the activated hydroxyl of the protein substrate performs an in-line attack on the γ phosphate of the donor nucleotide (35). With an active site that can accept PPi as phosphoryl donor and carry out phosphorolysis of the phosphoserine, HprK/P belongs to a family of kinases that is unrelated to eukaryotic Ser/Thr protein kinases. The HprK/P family is part of the larger family of P-loop enzymes, which has many members in both prokaryotes and eukaryotes and includes kinases that phosphorylate nucleosides, nucleotides, and other small molecules. It remains to be determined whether some of the eukaryotic members of the P-loop family of kinases also accept proteins as substrates and whether they can carry out dephosphorylation along with phosphorylation.
Acknowledgments
We thank the staff of European Synchrotron Radiation Facility beamlines for help during data collection and C. Habegger-Polomat (Laboratoire d'Enzymologie et Biochimie Structurales) for critically reading the manuscript. Thanks also to Bill Weiss (Stanford University, Stanford, CA) for refinement parameters of the phosphoserine. This work was supported by Association pour la Recherche Contre le Cancer. S.F. is grateful to La Ligue Nationale de Lutte Contre le Cancer for a personal fellowship.
Abbreviations
HPr, histidine phosphocarrier protein
P-Ser-HPr, serine-phosphorylated HPr
HprK/P, HPr kinase/phosphorylase
P loop, phosphate-binding loop
PCK, phosphoenolpyruvate carboxykinase
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes and ).
References
- 1.Hunter T. (2000) Cell 100, 113-127. [DOI] [PubMed] [Google Scholar]
- 2.Saier M. H., Wu, L. F. & Reizer, J. (1990) Trends Biochem. Sci. 15, 391-395. [DOI] [PubMed] [Google Scholar]
- 3.Stülke J. & Hillen, W. (2000) Annu. Rev. Microbiol. 54, 849-880. [DOI] [PubMed] [Google Scholar]
- 4.Deutscher J., Galinier, A. & Martin-Verstraete, I. (2001) in Bacillus subtilis and Its Closest Relatives: From Genes to Cells, eds. Sonenschein, A. L., Hoch, J. A. & Losick, R. (Am. Soc. Microbiol., Washington, DC), pp. 129–150.
- 5.Miwa Y., Nakata, A., Ogiwara, A., Yamamoto, M. & Fujita, Y. (2000) Nucleic Acids Res. 28, 1206-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moreno M. S., Schneider, B. L., Maile, R. R., Weyler, W. & Saier, M. H. (2001) Mol. Microbiol. 39, 1366-1381. [DOI] [PubMed] [Google Scholar]
- 7.Postma P. W., Lengeler, J. W. & Jacobson, G. R. (1993) Microbiol. Rev. 57, 543-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujita Y., Miwa, Y., Galinier, A. & Deutscher, J. (1995) Mol. Microbiol. 17, 953-960. [DOI] [PubMed] [Google Scholar]
- 9.Galinier A., Deutscher, J. & Martin-Verstraete, I. (1999) J. Mol. Biol. 286, 307-314. [DOI] [PubMed] [Google Scholar]
- 10.Kravanja M., Engelmann, R., Dossonnet, V., Blüggel, M., Meyer, H. E., Frank, R., Galinier, A., Deutscher, J., Schnell, N. & Hengstenberg, G. (1999) Mol. Microbiol. 31, 59-66. [DOI] [PubMed] [Google Scholar]
- 11.Mijakovic I., Poncet, S., Galinier, A., Fieulaine, S., Monedero, V., Janin, J., Nessler, S., Marquez, J. A., Scheffzek, K., Hengstenberg, W. & Deutscher, J. (2002) Proc. Natl. Acad. Sci. USA 99, 13442-13447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fieulaine S., Morera, S., Poncet, S., Monedero, V., Gueguen-Chaignon, V., Galinier, A., Janin, J., Deutscher, J. & Nessler, S. (2001) EMBO J. 20, 3917-3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matte A., Goldie, H., Sweet, R. M. & Delbaere, L. T. (1996) J. Mol. Biol. 256, 126-143. [DOI] [PubMed] [Google Scholar]
- 14.Galinier A., Lavergne, J. P., Geourjon, C., Fieulaine, S., Nessler, S. & Jault, J. M. (2002) J. Biol. Chem. 277, 11362-11367. [DOI] [PubMed] [Google Scholar]
- 15.Marquez J. A., Hasenbein, S., Koch, B., Fieulaine, S., Nessler, S., Russell, R. B., Hengstenberg, W. & Scheffzek, K. (2002) Proc. Natl. Acad. Sci. USA 99, 3458-3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galinier A., Kravanja, M., Engelmann, R., Hengstenberg, W., Kilhoffer, M. C., Deutscher, J. & Haiech, J. (1998) Proc. Natl. Acad. Sci. USA 95, 1823-1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otwinowski Z. & Minor, W. (1997) Methods Enzymol. 276, 307-326. [DOI] [PubMed] [Google Scholar]
- 18.Navaza J. (1994) Acta Crystallogr. A 50, 157-163. [Google Scholar]
- 19.Liao D. I. & Herzberg, O. (1994) Structure (London) 2, 1203-1216. [DOI] [PubMed] [Google Scholar]
- 20.Brunger A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 21.Lee B. & Richards, F. M. (1971) J. Mol. Biol. 55, 379-400. [DOI] [PubMed] [Google Scholar]
- 22.Lo Conte L., Chothia, C. & Janin, J. (1999) J. Mol. Biol. 285, 2177-2198. [DOI] [PubMed] [Google Scholar]
- 23.Wang G., Sondej, M., Garrett, D. S., Peterkofsky, A. & Clore, G. M. (2000) J. Biol. Chem. 275, 16401-16403. [DOI] [PubMed] [Google Scholar]
- 24.Jones B. E., Dossonet, V., Küster, E., Hillen, W., Deutscher, J. & Klevit, R. E. (1997) J. Biol. Chem. 272, 26530-26535. [DOI] [PubMed] [Google Scholar]
- 25.Jia Z., Quail, J. W., Delbaere, L. T. & Waygood, E. B. (1994) Biochem. Cell Biol. 72, 202-217. [DOI] [PubMed] [Google Scholar]
- 26.Reizer J., Novotny, M. J., Hengstenberg, W. & Saier, M. H., Jr. (1984) J. Bacteriol. 160, 333-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu P. P., Herzberg, O. & Peterkofsky, A. (1998) Biochemistry 37, 11762-11770. [DOI] [PubMed] [Google Scholar]
- 28.Audette G. F., Engelmann, R., Hengstenberg, W., Deutscher, J., Hayakawa, K., Quail, J. W. & Delbaere, L. T. (2000) J. Mol. Biol. 303, 545-553. [DOI] [PubMed] [Google Scholar]
- 29.Deutscher J., Kessler, U. & Hengstenberg, W. (1985) J. Bacteriol. 163, 1203-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matte A., Tari, L. W., Goldie, H. & Delbaere, L. T. (1997) J. Biol. Chem. 272, 8105-8108. [DOI] [PubMed] [Google Scholar]
- 31.Sudom A. M., Prasad, L., Goldie, H. & Delbaere, L. T. (2001) J. Mol. Biol. 314, 83-92. [DOI] [PubMed] [Google Scholar]
- 32.Tari L., Matte, A., Goldie, H. & Delbaere, L. T. J. (1997) Nat. Struct. Biol. 4, 990-994. [DOI] [PubMed] [Google Scholar]
- 33.Zhou J. & Adams, J. A. (1997) Biochemistry 36, 2977-2984. [DOI] [PubMed] [Google Scholar]
- 34.Madhusudan M., Trafny, E. A., Xuong, N. H., Adams, J. A., Ten Eyck, L. F., Taylor, S. S. & Sowadski, J. M. (1994) Protein Sci. 3, 176-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson D. A., Akamine, P., Radzio-Andzelm, E., Madhusudan, M. & Taylor, S. S. (2001) Chem. Rev. (Washington, D.C.) 101, 2243-2270. [DOI] [PubMed] [Google Scholar]